Ab Initio Molecular Dynamics Simulation Study on the Stereo Reactions between Atomic Oxygen Anion and Methane

Key Laboratory of Life-Organic Analysis, School of Chemistry and Chemical Engineering, Qufu Normal University, Qufu 273165, China
*
Authors to whom correspondence should be addressed.
Molecules 2018, 23(10), 2495; https://doi.org/10.3390/molecules23102495
Submission received: 6 September 2018
/
Revised: 25 September 2018
/
Accepted: 27 September 2018
/
Published: 29 September 2018
(This article belongs to the Special Issue Theoretical Investigations of Reaction Mechanisms)
Abstract
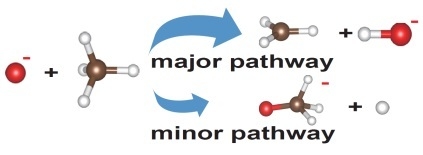
:Ion–molecule reaction between atomic oxygen anion (O−) and methane (CH4) has been systematically investigated employing the on-the-fly ab initio molecular dynamics simulations. Besides the major H-abstraction process as the exothermic reaction studied widely, an endothermic pathway to produce OCH3− and H is also observed in this study. Three typical O− attack modes with reference to the pyramid structure of CH4 fixed in space have been considered. It was found that the internal motions of the radical products are significantly dependent on the O− attack modes. As for the reaction between O− and the thermally vibrating CH4, the major pathway to produce OH− and CH3 is preferred by the direct H-abstraction and the minor pathway to produce H and OCH3− is the roaming reaction via the transient negative ion [HO-CH3]−.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
As a high clean fossil energy, how to convert methane (CH4) to high-density energy sources and high value-added chemicals has attracted interests of researchers from the fields of the energy science and coordination chemistry [1,2,3,4,5,6,7,8]. However, as the first step of the reaction, it is difficult to activate the C-H bond of CH4 due to its high stability. On the other hand, as a highly reactive free radical anion, atomic oxygen anion (O− at 2P state) can effectively activate the C-H bond, which is involved in the ion–molecule reaction. Here, ion–molecule reaction between O− and molecules has a profound significance in aspects of atmospheric chemistry, combustion, and environmental pollution control [9,10,11,12,13,14,15,16,17]. As a prototype reaction for ion–molecule reaction in hydrocarbon flames, the reaction between O− and methane (CH4) is an important example. For this reaction mechanism, Comer and Schulz reported an associative-detachment channel [18],
and it was proved to be an exothermic process (∆H = −2.43 eV [9]). In the subsequent experimental studies [19,20,21,22,23], the other reaction pathway [19,20,23,24],
was characterized as an exothermic process, in which OH− was formed via direct H-abstraction via the transition-state complex [O-H-CH3]− [21]. On the basis of the measurements of energy and angular distributions of the product OH−, Carpenter and Farr proposed two mechanisms for the above reaction, namely, one is the collisions of O− with H atom oriented essentially along the relative velocity vector and the other one is the collisions of O− with one of the three off-axis hydrogen atoms [23]. Obviously, the reaction between O− and CH4 possesses the stereodynamics features. However, the spatial properties of this reaction remain unclear to our best knowledge although the stereodynamics is one of the essential issues of molecular reactions. Theoretically, the kinetics of the reactions of O− with CH4, CH2D2, and CD4 has been explored by using the Gaussian-1, Gaussian-2, and the complete basis set extrapolation method [21]. The minimum energy reaction path for the above reaction (2) was constructed and was characteristic of the standard double minimum pathway for ion molecule reactions. In addition, the potential surface for the relevant reaction of the O(1D) atom with CH4 has also been investigated by the ab initio Multireference single and double configuration interaction (MRDCI). The proposed product channels to yield CH3 + OH and CH3O + H can give helpful clues to the title reaction [25].
O− + CH4 → CH3OH + e−
O− + CH4 → OH− + CH3 (∆H = −0.26 eV)
As for CH4, it is a tetrahedron molecule characterized by Td point group with four C3 and three C2 symmetrical axes. Therefore, three O− attack modes can take place for the reaction between O− and CH4. In details, as shown in Figure 1, (1) the O− attacks CH4 along an arbitrary C3 axis keeping O···H-C in a straight line (abbreviated as apex attack mode); (2) the O− attacks CH4 along an arbitrary C3 axis in an opposite side keeping O···C-H in a straight line (center-of-plane attack mode); (3) the O− attacks CH4 along a C2 axis of H-C-H angle bisector (center-of-edge-angle attack mode). As the doorways of the reaction, these three typical O− attack directions may lead to the OH− product with the different momentum distributions. Moreover, besides reaction (2) which was believed as a single channel reaction between O− and CH4 [19,20,21,22,23], it is still unknown about the existence of other possible reaction channels. To fully understand the reaction mechanism in detail, information concerning the possible reaction pathways for the reaction is indispensable. Therefore, in this study, the stereodynamics properties of the collision reaction between O− and CH4 have been systematically investigated employing ab initio molecular dynamics simulations with the aim to provide new insights into the reaction mechanism of the title reaction.
2. Computational Method
Firstly, the thermochemistry calculations were carried out for reaction (2) at the second-order Møller-Plesset (MP2) [26,27,28,29,30] perturbation method with 6 − 31 + G(d) basis set. The energetic release ΔE of −0.23 eV is in good agreement with the previous values [19,20,21,23,24], suggesting that the MP2/6 − 31 + G(d) level is reliable in elaborating the reaction process. Excluding the influence of any electrostatic interactions between O− and CH4 at the initial step in the dynamic simulations, we performed the interaction energy calculations for the three attack modes at the MP2/6 − 31 + G(d) level. Then, the atomic O−···C distance about 6 Å was used for the initial step in the simulations because the interaction between O− and CH4 was very weak or ignorable at this distance. In these calculations, the geometry of the CH4 target was fixed. On the other hand, two different transition-state structures were located, and their linkages with the reactants and products were verified by the intrinsic reaction coordinate calculations [31,32].
All on-the-fly trajectory calculations for the above three O− attack modes were performed by using the Born-Oppenheimer molecular dynamics (BOMD) [33,34] method at the MP2/6 − 31 + G(d) level. Two schemes have been considered in the calculations. In the first scheme, each dynamic evolution was initiated with the atomic O−···C distance about 6 Å between the O− ion with a certain velocity or kinetic energy and CH4 molecule. There are no internal motions for CH4 at the beginning. Then, the dynamic evolution for the three attack directions (apex, center-of-plane, and center-of-edge-angle) of the incident O− ion was simulated. In the other scheme, we considered the internal motions of CH4 that were mimicked with the atomic motions of CH4 at the vibrational ground state. The atomic motions or velocities of CH4 at the initial step of simulation were stochastically selected from the 400 fs BOMD simulations of the isolated CH4 at the vibrational ground state, where more details can be found in the Supporting Information (SI). In the above two schemes, the time scale of simulations was about 400 fs with a time step of 0.2 femtosecond (fs). Ten different collision energies of O− ion were selected in a range of 0.5–10.0 eV, where the translational energy of CH4 was zero. No dissociation pathways were constrained in the simulations of about 500 trajectories. All calculations were performed with Gaussian 09 program [35].
To explore the charge transfer behavior and the net charges carried on the formed fragments in the reaction process, natural bond orbital (NBO) analyses have been performed on the basis of the optimized geometries. The contour maps of the relevant NBOs were analyzed and drawn with Multiwfn program [36].
3. Results and Discussion
3.1. Intermolecular Interactions and Transition States
The rigid potential energy profiles were obtained by scanning the O−···C distance by fixing the CH4 geometry, where the O−···CH4 interaction energies were calculated including the corrections of basis set superposition errors [37,38]. As shown in Figure 1, the interaction between O− and CH4 is quite weak when the distance of O−···C is larger than 6 Å. Therefore, the following dynamics simulations will start with the distance of O−···C about 6 Å, ensuring no influences of the different strengths of the O−···CH4 interaction at the initial step. Furthermore, the minima on the reaction profiles were located around 3.4 Å. Meanwhile, as shown in Figure 1a, the strongest attraction of about 0.11 eV appears for the apex attack mode, implying the stabilization of the pre-reactive complex [O···HCH3]−.
For the O− apex attack mode, as shown in Figure 2a, a transition state (O-H-CH3)− possessing C3v symmetry has been located after the formation of the pre-reactive complex [O···HCH3]−, which is associated with the reactants and the products of OH− and CH3. To investigate the dependence of basis set for the transition state, larger basis sets 6 − 311 + G(3df,2p) and AUG-cc-pVTZ have been employed. As a result, the corresponding transition states possessing C3v symmetry can still be obtained, where the Cartesian coordinates of them have placed in Table S1 of the SI for reference.
As for the O− center-of-plane attack mode, a new reaction channel, i.e.,
has been observed. As depicted in Figure 2b, a transition state (O-CH3-H)− was located about 2.54 eV higher in energy than the reactants. Moreover, reaction (3) is an endothermic nucleophilic substitution (SN2) process (∆E = 0.20 eV), which has not been reported previously. In the transition state (O-CH3-H)−, the atomic distance between O and C and one C-H bond length are 1.725 and 1.504 Å, respectively.
O− + CH4 → H + OCH3−
As for the O− center-of-edge-angle attack mode, no corresponding transition state has been located.
3.2. Stereodynamics of the Reactions
Above quantum chemistry calculations provide us the reaction thresholds and the energy barriers on the minimum energy pathways of reactions (2) and (3). In the following BOMD simulations, the O− ion with a certain kinetic energy attacks the motionless target CH4 from a position of 6 Å away. Note that all the energies required in the reaction including the internal energies of the products and the energy barrier to be overcome must be transformed from the kinetic energy of the incident O− ion. Therefore, not all of the collisions can proceed for the O− ion with the kinetic energy in the range of 0.5–10.0 eV although reaction (2) is an exothermic process. Here, three typical trajectories have been mainly discussed, where the initial kinetic energy of the incident O− ion is 1.80 eV (corresponding to the initial velocity 4.66 × 103 m/s), 6.60 eV (8.92 × 103 m/s), and 9.20 eV (10.53 × 103 m/s) for the apex, center-of-plane, and center-of-edge-angle attack modes, respectively.
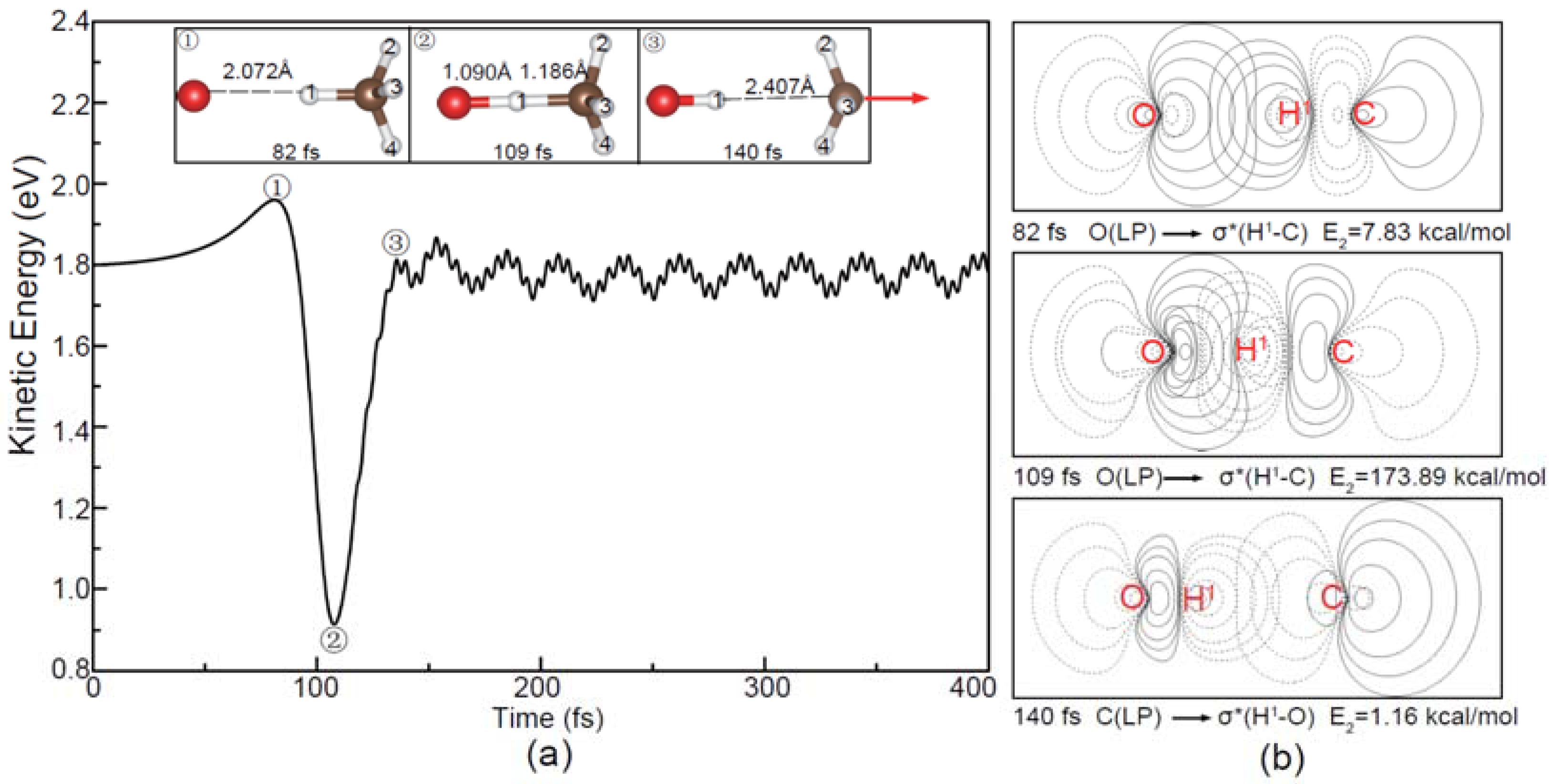
As shown in Figure 3a, the O− projectile is accelerated gradually in the apex direction from the initial kinetic energy 1.80 eV to 1.96 eV due to the attraction interaction depicted in Figure 1a. Then, the oxygen is combined with the hydrogen atom at 109 fs and the atomic distances of O-H1 and C-H1 reach the minima, indicating the lowest kinetic energy but the highest potential energy of the [O···HCH3]− complex. At 140 fs, the C-H1 bond breaks and the CH3 product moves forward. After that, two products completely depart from each other. The ripples of the total kinetic energy profile in Figure 3a are attributed to the internal energy distributions of the products. As shown in Figure 3b, the NBO contour maps show the process of the negative charge transfer from O−. Before 109 fs, the lone pair (LP) electron of O− transfers to the virtual antibonding orbital of the C-H bond followed by the formation of the O-H1 bond. For the sake of simplicity, the selected charge distributions of the reactants and the products in the dynamic process have been given in Table S2 of the SI for reference.
In the center-of-plane and center-of-edge-angle attack processes, as shown in Figure 4 and Figure 5, the O− is directly combined with C atom, leading to the release of H atom in the opposite direction of the O− (Figure 4) and the side H atom (Figure 5), respectively. The charge transfers of LP(O−) → can also be observed in these two cases. Compared with the above apex attack mode in Figure 3, remarkable variations of the total kinetic energy can be observed here, suggesting that the internal energy distributions of the products are different depending on the attack modes.
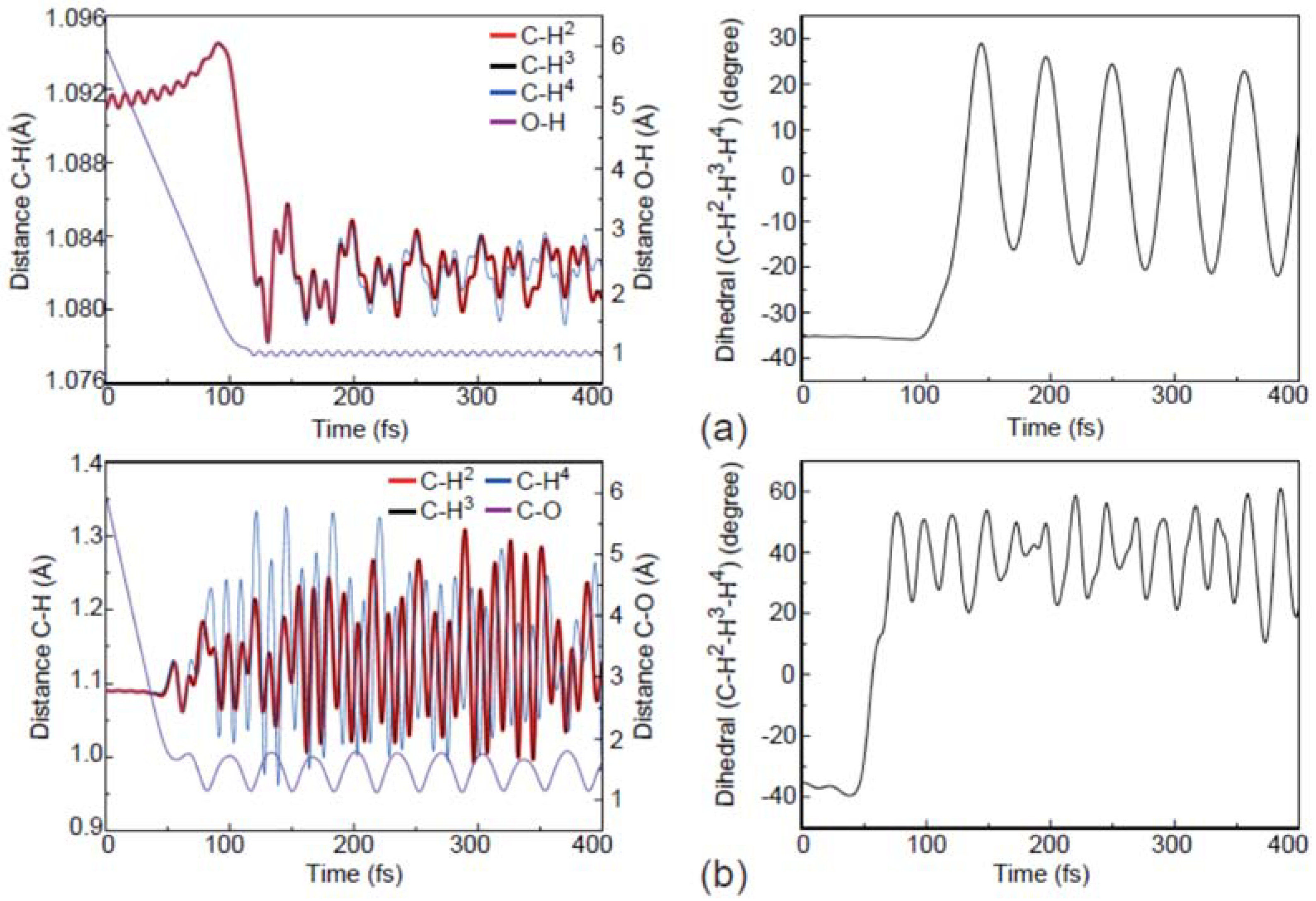
As shown in Figure 6a, the apex attack reaction results in the activation of the internal motions mostly for the umbrella vibrational mode of the methyl radical. On the other hand, as shown in Figure 6b,c, the internal motions of OCH3− are distinctly different. In particular, the stretching motion of the C-H, the umbrella motion of CH3, and the stretching motion of OH− are much more active in the reaction induced by the center-of-plane attack. As a result, as can be seen from the stereodynamics effects for the above three typical trajectories, the sites of the H atom released and the internal energy distributions of the products are highly dependent on the O− attack directions. Here, it should be noted that the present simulations are performed for the collisions along the different attractive interaction lines (i.e., the impact parameter b is zero). Meanwhile, the geometry of the target CH4 is motionless initially in the simulations.
As shown in Figure 2a, reaction (2) is a double-well reaction. In comparison with the mechanisms proposed for the double-well SN2 anion–molecule reactions [39], the apex and center-of-plane attack modes correspond to the reactions involving the formation of the frontside complex. While the center-of-edge-angle attack mode is similar to those of the SN2 reactions leading to the formation of the ion-dipole complex. Certainly, there are other mechanisms for the SN2 reactions [39,40], largely depending on the impact parameter and the internal atomic motions of the target molecule.
3.3. Reactions with Thermally Vibrating CH4
As mentioned above, the internal motion (molecular vibration) and the rotation of the target molecule can influence the collision process [11,19,20,21]. To further confirm this point, the reaction of O− with thermally vibrating CH4 has also been investigated. As shown in Figure S1 of the SI, we stochastically select 50 geometries of CH4 from thermally equilibrated simulation after 200 fs at an interval of 4 fs. At the initial steps of the reaction simulations, the atomic coordinates and velocities of oxygen and carbon atom are set up in the center-of-mass Cartesian coordinate (Tables S3 and S4 of the SI), where the O− kinetic energies are 0.5 (2.46 × 103 m/s), 1.0 (3.47 × 103 m/s), 1.5 (4.25 × 103 m/s), 2.0 (4.91 × 103 m/s), 2.5 (5.49 × 103 m/s), 3.0 (6.01 × 103 m/s), and 3.5 eV (6.50 × 103 m/s), respectively.
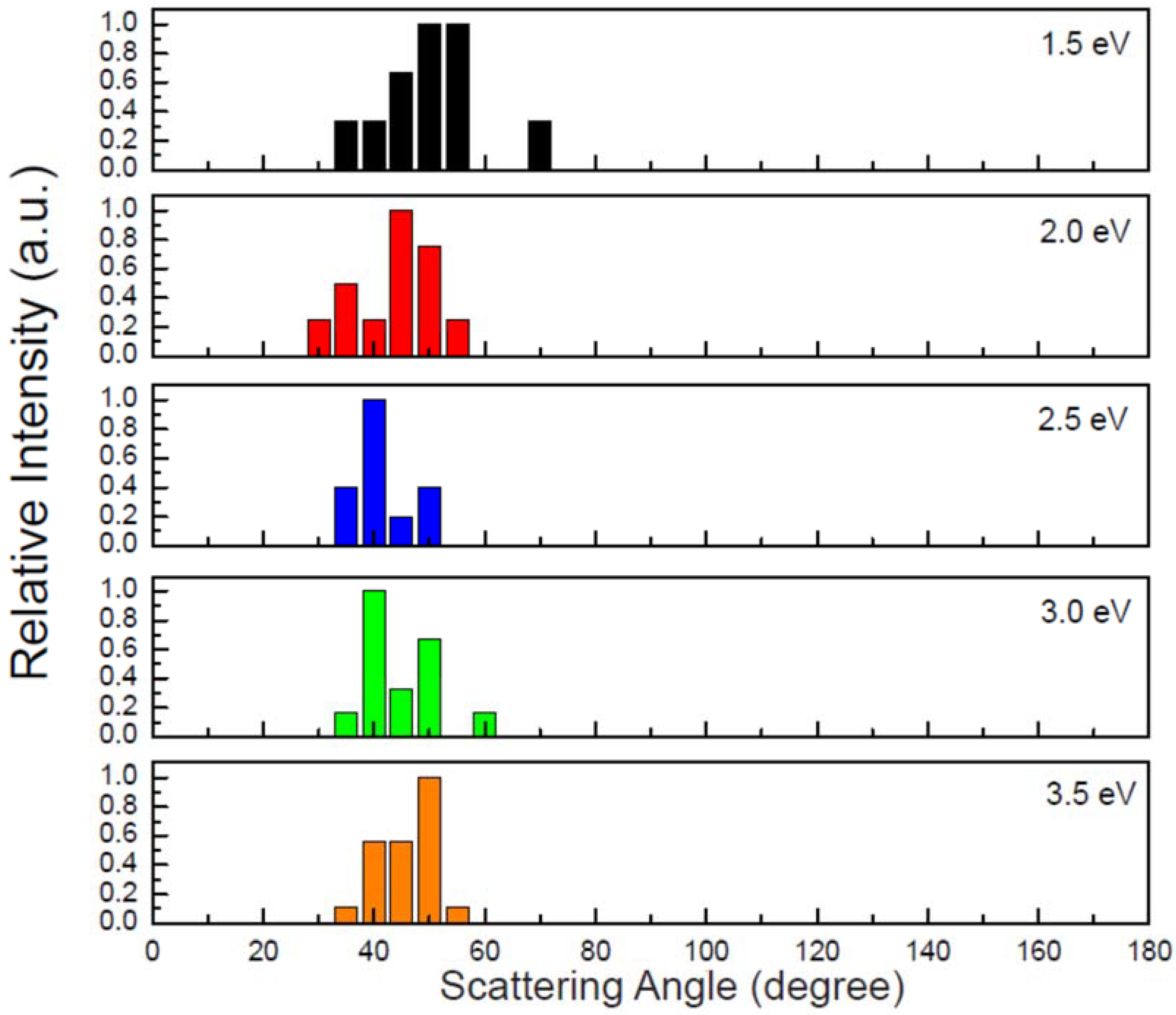
Similar to the above reaction, reaction (2) is also the predominant pathway here. As shown in Figure 7, the reaction of the O− having the collision energy of 1.0 eV leads to OH− and CH3 products via a direct H-abstraction. However, the detailed process is distinctly different from that observed in Figure 3a although the charge transfers depicted in Figure 3b and Figure S2 of the SI are similar to each other. As shown in Figure S3 of the SI, the C-H2,3,4 bond stretching motions are more significant. Moreover, the molecular rotations for both OH− and CH3 are remarkable. In view of the fact that the excess energy of the reaction can be redistributed to the internal atomic motions and molecular rotations, the translational energies of the products at this collision energy (1.0 eV) are relatively small. At the higher collision energy, the products can obtain more translational energies and the angular distributions of OH− are anisotropic. The calculated relative intensities of the scattering angular distributions (in laboratory coordinate) of OH− have been plotted in Figure 8. Obviously, the maxima are distributed between 40° and 60°, which is generally in agreement with experimental results [23].
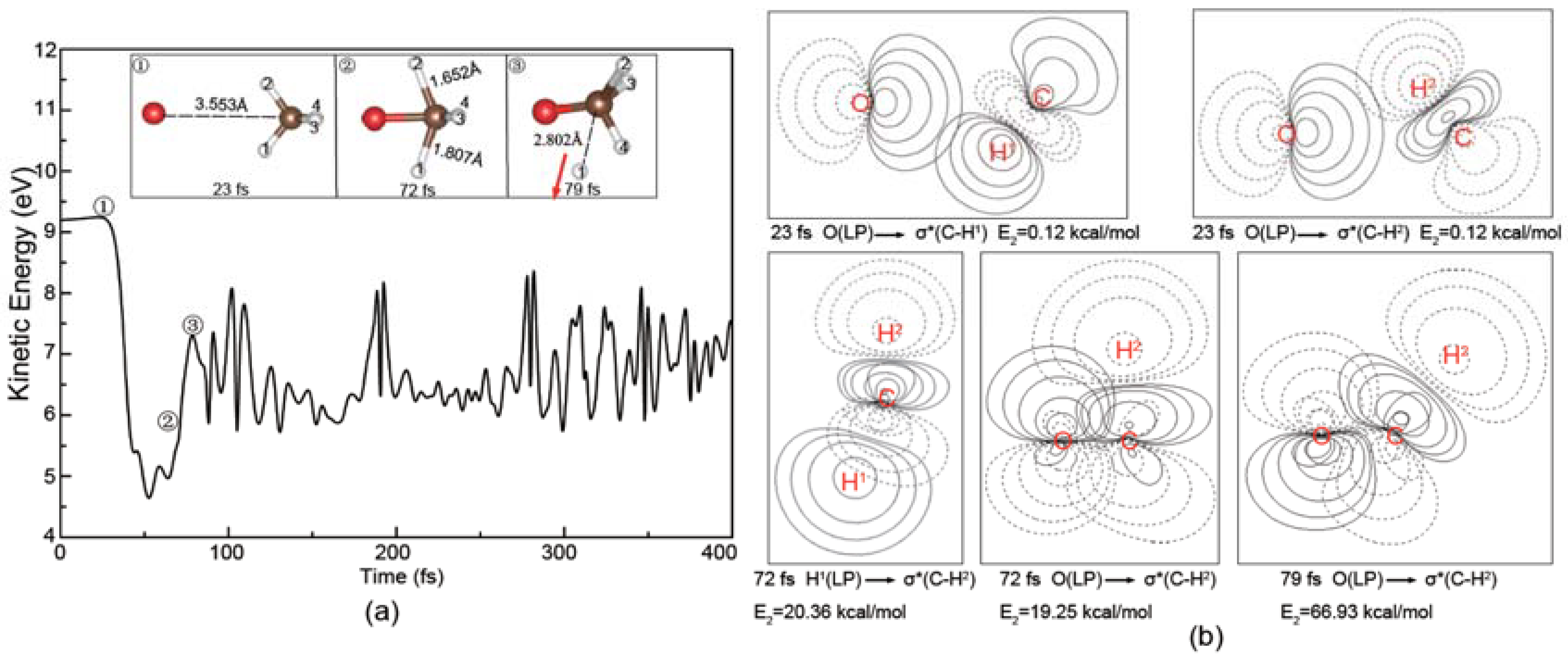
In addition, a few new trajectories leading to the products of OCH3− and H have also been observed. As shown in Figure 9, the dynamic process of reaction (3) is completely different from those observed in Figure 4a and Figure 5a. In this process, an H atom is firstly abstracted to form the slowly rotating OH−. Then, this OH− group is recombined with CH3. Finally, the H atom attached to the O atom is released and the vibrating OCH3− is formed. Therefore, the above phenomena suggest that the reaction (3) is a typical roaming process, which has also been observed in the molecular dynamics of the other systems [39,40,41,42]. Given the fact that no roaming mechanism for reactions (3) has been reported previously, further experimental and theoretical studies are required to confirm this point.
4. Conclusions
In this study, the reaction between O− and CH4 has been systematically investigated using the ab initio molecular dynamics simulations. The stereodynamics properties for the different attack modes of O− to the initially motionless CH4 have been revealed. As a result, besides the major exothermic pathway to produce the OH− and CH3, a new endothermic channel into the yields of H and OCH3− has been observed. Meanwhile, the translational and internal energies of the products are dependent on the attack modes of O−. Moreover, considering the internal atomic motions of the gas-phase target CH4, we also performed the dynamics simulations for the reaction between O− and the thermally vibrating CH4. It was shown that most of the trajectories can lead to the production of OH− and CH3, and the radical products are populated at the ro-vibrational states and in the forward distributions. Especially, it is worth noting that a typical roaming pathway to produce the H and OCH3− has also been observed, which deserves further explorations in the future.
Supplementary Materials
The following are available online, Figure S1: Variation of the kinetic energy for the equilibration process of the isolated CH4 at the vibrational ground state, Figure S2: NBO contour maps of the charge transfers in one of the trajectory of the O− (with the kinetic energy of 1.0 eV) reaction with the vibrating CH4. E2 is the hyperconjugative energy and represents the strength of orbital-orbital interaction, Figure S3: Evolution of the C-H atomic distance and dihedral angle in one of the trajectory of the O− (with the kinetic energy of 1.0 eV) reaction with the vibrating CH4, Figure S4: Atomic labels of methane molecule used in Tables S2 and S3, Table S1: The Cartesian coordinates of the transition state possessing C3v symmetry in the major pathway of the title reaction, Table S2: Charge distributions of the reactants and the products in the reactions of O− with the initially fixed-structure CH4. The charge values are obtained with natural bond orbital analysis, Table S3: Geometric parameters of methane molecule at the 10 moments of vibration, Table S4: The speeds (m/s) of individual atoms of methane molecule at the 10 moments of vibration.
Author Contributions
Data curation, W.W. (Wenliang Wang); Formal analysis, W.F.; Investigation, W.W. (Weihua Wang); Methodology, W.F. and W.W. (Wenliang Wang); Supervision, P.L.; Writing—original draft, W.W. (Weihua Wang); Writing—review & editing, P.L.
Funding
This work is supported by National Natural Science Foundation of China (No. 21577076, 21303093, 21805163), Natural Science Foundation of Shandong Province (No. ZR2018MB020), and the Doctoral Foundation of Shandong Province (No. ZR2016BB20, ZR2017BB055).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cui, X.; Li, H.; Wang, Y.; Hu, Y.; Hua, L.; Li, H.; Han, X.; Liu, Q.; Yang, F.; He, L.; et al. Room-temperature methane conversion by graphene-confined single iron atoms. Chemistry 2018, 4, 1902–1910. [Google Scholar] [CrossRef]
- Schwarz, H.; Shaik, S.; Li, J. Electronic effects on room-temperature, gas-phase C–H Bond activations by cluster oxides and metal carbides: The methane challenge. J. Am. Chem. Soc. 2017, 139, 17201–17212. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Kim, T.Y.; Kwon, G.; Yi, J.; Lee, H. Selective activation of methane on single-atom catalyst of rhodium dispersed on zirconia for direct conversion. J. Am. Chem. Soc. 2017, 139, 17694–17699. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, R.H.; Lei, A. Introduction: CH activation. Chem. Rev. 2017, 117, 8481–8482. [Google Scholar] [CrossRef] [PubMed]
- Schwach, P.; Pan, X.; Bao, X. Direct conversion of methane to value-added chemicals over heterogeneous catalysts: Challenges and prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.N.; Ding, X.L.; Li, Z.Y.; Zhao, Y.X.; He, S.G. Hydrogen atom abstraction from CH4 by nanosized vanadium oxide cluster cations. J. Phys. Chem. 2014, 118, 24062–24071. [Google Scholar] [CrossRef]
- Ding, X.L.; Wu, X.N.; Zhao, Y.X.; He, S.G. C–H bond activation by oxygen-centered radicals over atomic clusters. Acc. Chem. Res. 2011, 45, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Dietl, N.; Schlangen, M.; Schwarz, H. Thermal hydrogen-atom transfer from methane: The role of radicals and spin states in oxo-cluster chemistry. Angew. Chem. Int. Ed. 2012, 51, 5544–5555. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Grabowski, J.J. Reactions of the atomic oxygen radical anion and the synthesis of organic reactive intermediates. Chem. Rev. 1992, 92, 1611–1647. [Google Scholar] [CrossRef]
- Fehsenfeld, F.; Schmeltekopf, A.; Schiff, H.; Ferguson, E. Laboratory measurements of negative ion reactions of atmospheric interest. Plan. Space Sci. 1967, 15, 373–379. [Google Scholar] [CrossRef]
- Fehsenfeld, F.; Ferguson, E.; Schmeltekopf, A. Thermal-energy associative-detachment reactions of negative ions. J. Chem. Phys. 1966, 45, 1844–1845. [Google Scholar] [CrossRef]
- Fehsenfeld, F.C.; Howard, C.J.; Schmeltekopf, A.L. Gas phase ion chemistry of HNO3. J. Chem. Phys. 1975, 63, 2835–2841. [Google Scholar] [CrossRef]
- Guo, Y.; Grabowski, J.J. Gas phase ion chemistry of the vinylidene radical anion and the acidity of the vinyl radical. Int. J. Mass Spectrom. Ion Process. 1990, 97, 253–264. [Google Scholar] [CrossRef]
- Chou, P.K.; Kass, S.R. C4H4 negative ions: Formation of the bicyclo[1.1.0]but-1(3)-ene radical anion and an experimental determination of the heat of formation of bicyclo[1.1.0]but-1(3)-ene. J. Am. Chem. Soc. 1991, 113, 697–698. [Google Scholar] [CrossRef]
- Guo, Y.; Grabowski, J.J. Reactions of the benzyne radical anion in the gas phase, the acidity of the phenyl radical, and the heat of formation of o-benzyne. J. Am. Chem. Soc. 1991, 113, 5923–5931. [Google Scholar] [CrossRef]
- Carrascosa, E.; Meyer, J.; Wester, R. Imaging the dynamics of ion–molecule reactions. Chem. Soc. Rev. 2017, 46, 7498–7516. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.; Wester, R. Ion–Molecule Reaction Dynamics. Annu. Rev. Phys. Chem. 2017, 68, 333–353. [Google Scholar] [CrossRef] [PubMed]
- Comer, J.; Schulz, G.J. Measurements of electron-detachment cross sections from O− and S−. Phys. Rev. A 1974, 10, 2100–2106. [Google Scholar] [CrossRef]
- Lindinger, W.; Albritton, D.L.; Fehsenfeld, F.C.; Ferguson, E.E. Reactions of O− with N2, N2O, SO2, NH3, CH4, and C2H4 and C2H2− with O2 from 300 K to relative kinetic energies of ~2 eV. J. Chem. Phys. 1975, 63, 3238–3242. [Google Scholar] [CrossRef]
- Bohme, D.K.; Fehsenfeld, F.C. Thermal reactions of O− ions with saturated hydrocarbon molecules. Can. J. Chem. 1969, 47, 2717–2719. [Google Scholar] [CrossRef]
- Viggiano, A.A.; Morris, R.A.; Miller, T.M.; Friedman, J.F.; Menedez-Barreto, M.; Paulson, J.F.; Michels, H.H.; Hobbs, R.H. Reaction on the O− + CH4 potential energy surface: Dependence on translational and internal energy and on isotopic composition, 93–1313 K. J. Chem. Phys. 1997, 106, 8455–8463. [Google Scholar] [CrossRef]
- Ferguson, E.E.; Fehsenfeld, F.C.; Albritton, D.L. Ion chemistry of the earth’s atmosphere. In Gas Phase Ion Chemistry; Bowers, M.T., Ed.; Academic Press: New York, NY, USA, 1979; Chapter 2; pp. 45–82. [Google Scholar]
- Carpenter, M.A.; Farrar, J.M. Dynamics of hydrogen atom abstraction in the O− + CH4 reaction: Product energy disposal and angular distributions. J. Chem. Phys. 1997, 106, 5951–5960. [Google Scholar] [CrossRef]
- Diken, E.G.; Weddle, G.H.; Headrick, J.M.; Weber, J.M.; Johnson, M.A. Argon cluster-mediated trapping and vibrational spectroscopic characterization of an OH−·HCH2• intermediate in the O•− + CH4 reaction. J. Phys. Chem. A 2004, 108, 10116–10121. [Google Scholar] [CrossRef]
- Arai, H.; Kato, S.; Koda, S. Ab initio potential surfaces for the atomic oxygen(1D) + methane reaction. J. Phys. Chem. 1994, 98, 12–16. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Head-Gordon, T. Analytic MP2 frequencies without fifth-order storage. Theory and application to bifurcated hydrogen bonds in the water hexamer. Chem. Phys. Lett. 1994, 220, 122–128. [Google Scholar] [CrossRef]
- Sæbø, S.; Almlöf, J. Avoiding the integral storage bottleneck in LCAO calculations of electron correlation. Chem. Phys. Lett. 1989, 154, 83–89. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. A direct MP2 gradient method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. Semi-direct algorithms for the MP2 energy and gradient. Chem. Phys. Lett. 1990, 166, 281–289. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Schlegel, H.B. Accurate reaction paths using a Hessian based predictor–corrector integrator. J. Chem. Phys. 2004, 120, 9918–9924. [Google Scholar] [CrossRef] [PubMed]
- Hratchian, H.P.; Schlegel, H.B. Using hessian updating to increase the efficiency of a hessian based predictor-corrector reaction path following method. J. Chem. Theory Comput. 2005, 1, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Millam, J.M.; Bakken, V.R.; Chen, W.; Hase, W.L.; Schlegel, H.B. Ab initio classical trajectories on the Born–Oppenheimer surface: Hessian-based integrators using fifth-order polynomial and rational function fits. J. Chem. Phys. 1999, 111, 3800–3805. [Google Scholar] [CrossRef]
- Bakken, V.; Millam, J.M.; Bernhard Schlegel, H. Ab initio classical trajectories on the Born–Oppenheimer surface: Updating methods for Hessian-based integrators. J. Chem. Phys. 1999, 111, 8773–8777. [Google Scholar] [CrossRef]
- Gaussian 09, Revision D.01; Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. (Eds.) Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F.D. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef]
- Xie, J.; Otto, R.; Mikosch, J.; Zhang, J.; Wester, R.; Hase, W.L. Identification of atomic-level mechanisms for gas-phase X– + CH3Y SN2 reactions by combined experiments and simulations. Acc. Chem. Res. 2014, 47, 2960–2969. [Google Scholar] [CrossRef] [PubMed]
- Mikosch, J.; Trippel, S.; Eichhorn, C.; Otto, R.; Lourderaj, U.; Zhang, J.; Hase, W.; Weidemüller, M.; Wester, R. Imaging nucleophilic substitution dynamics. Science 2008, 319, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Hua-Gen, Y. An ab initio molecular dynamics study of the roaming mechanism of the H2 + HOC+ reaction. Phys. Scr. 2011, 84, 028104. [Google Scholar]
- Li, A.; Li, J.; Guo, H. Quantum manifestation of roaming in H + MgH → Mg + H2: The birth of roaming resonances. J. Phys. Chem. A 2013, 117, 5052–5060. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |
Figure 1.
Interaction energy profiles for the attack of O− (in red) to CH4 (H in white, C in brown) in the apex (a), center-of-plane (b) and center-of-edge-angle (c) directions.
Figure 1.
Interaction energy profiles for the attack of O− (in red) to CH4 (H in white, C in brown) in the apex (a), center-of-plane (b) and center-of-edge-angle (c) directions.

Figure 2.
The reaction profiles for the attack of O− to CH4 in the apex (a) and center-of-plane (b) attack modes.
Figure 2.
The reaction profiles for the attack of O− to CH4 in the apex (a) and center-of-plane (b) attack modes.

Figure 3.
(a) Evolution of the total kinetic energy and snapshots in the reaction beginning with the O− apex attack (1.8 eV); (b) The corresponding natural bond orbital (NBO) contour maps at the selected times and E2 is the hyperconjugative energy. The red arrow in (a) shows the translational direction of the product CH3.
Figure 3.
(a) Evolution of the total kinetic energy and snapshots in the reaction beginning with the O− apex attack (1.8 eV); (b) The corresponding natural bond orbital (NBO) contour maps at the selected times and E2 is the hyperconjugative energy. The red arrow in (a) shows the translational direction of the product CH3.

Figure 4.
(a) Evolution of the total kinetic energy and snapshots in the reaction beginning with the O− center-of-plane attack (6.6 eV); (b) The corresponding NBO contour maps at the selected times and E2 is the hyperconjugative energy. The red arrow in (a) shows the translational direction of the product H.
Figure 4.
(a) Evolution of the total kinetic energy and snapshots in the reaction beginning with the O− center-of-plane attack (6.6 eV); (b) The corresponding NBO contour maps at the selected times and E2 is the hyperconjugative energy. The red arrow in (a) shows the translational direction of the product H.

Figure 5.
(a) Evolution of the total kinetic energy and snapshots in the reaction beginning with the O− center-of-edge-angle attack (9.2 eV); (b) The corresponding NBO contour maps at the selected times and E2 is the hyperconjugative energy. The red arrow in (a) shows the translational direction of the product H.
Figure 5.
(a) Evolution of the total kinetic energy and snapshots in the reaction beginning with the O− center-of-edge-angle attack (9.2 eV); (b) The corresponding NBO contour maps at the selected times and E2 is the hyperconjugative energy. The red arrow in (a) shows the translational direction of the product H.

Figure 6.
Evolution of the atomic distances and dihedral angles for the apex (a), center-of-plane (b), and center-of-edge-angle (c) attack modes.
Figure 6.
Evolution of the atomic distances and dihedral angles for the apex (a), center-of-plane (b), and center-of-edge-angle (c) attack modes.


Figure 7.
Evolution of the total kinetic energy and snapshots in the reaction of the vibrating CH4 with the O− with the kinetic energy of 1.0 eV. The circle and the arrow represent the molecular rotating and moving directions.
Figure 7.
Evolution of the total kinetic energy and snapshots in the reaction of the vibrating CH4 with the O− with the kinetic energy of 1.0 eV. The circle and the arrow represent the molecular rotating and moving directions.

Figure 8.
Angular distributions of the OH− product. The relative intensities are normalized with the respective maximum values.
Figure 8.
Angular distributions of the OH− product. The relative intensities are normalized with the respective maximum values.

Figure 9.
Evolution of the total kinetic energy and snapshots in the roaming reaction with the vibrating CH4 at the O− kinetic energy of 2.5 eV. The arrows represent the molecular moving directions.
Figure 9.
Evolution of the total kinetic energy and snapshots in the roaming reaction with the vibrating CH4 at the O− kinetic energy of 2.5 eV. The arrows represent the molecular moving directions.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, W.; Feng, W.; Wang, W.; Li, P. Ab Initio Molecular Dynamics Simulation Study on the Stereo Reactions between Atomic Oxygen Anion and Methane. Molecules 2018, 23, 2495. https://doi.org/10.3390/molecules23102495
AMA Style
Wang W, Feng W, Wang W, Li P. Ab Initio Molecular Dynamics Simulation Study on the Stereo Reactions between Atomic Oxygen Anion and Methane. Molecules. 2018; 23(10):2495. https://doi.org/10.3390/molecules23102495
Chicago/Turabian StyleWang, Weihua, Wenling Feng, Wenliang Wang, and Ping Li. 2018. "Ab Initio Molecular Dynamics Simulation Study on the Stereo Reactions between Atomic Oxygen Anion and Methane" Molecules 23, no. 10: 2495. https://doi.org/10.3390/molecules23102495