Theoretical Investigations on the Reactivity of Methylidyne Radical toward 2,3,7,8-Tetrachlorodibenzo-p-Dioxin: A DFT and Molecular Dynamics Study

Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
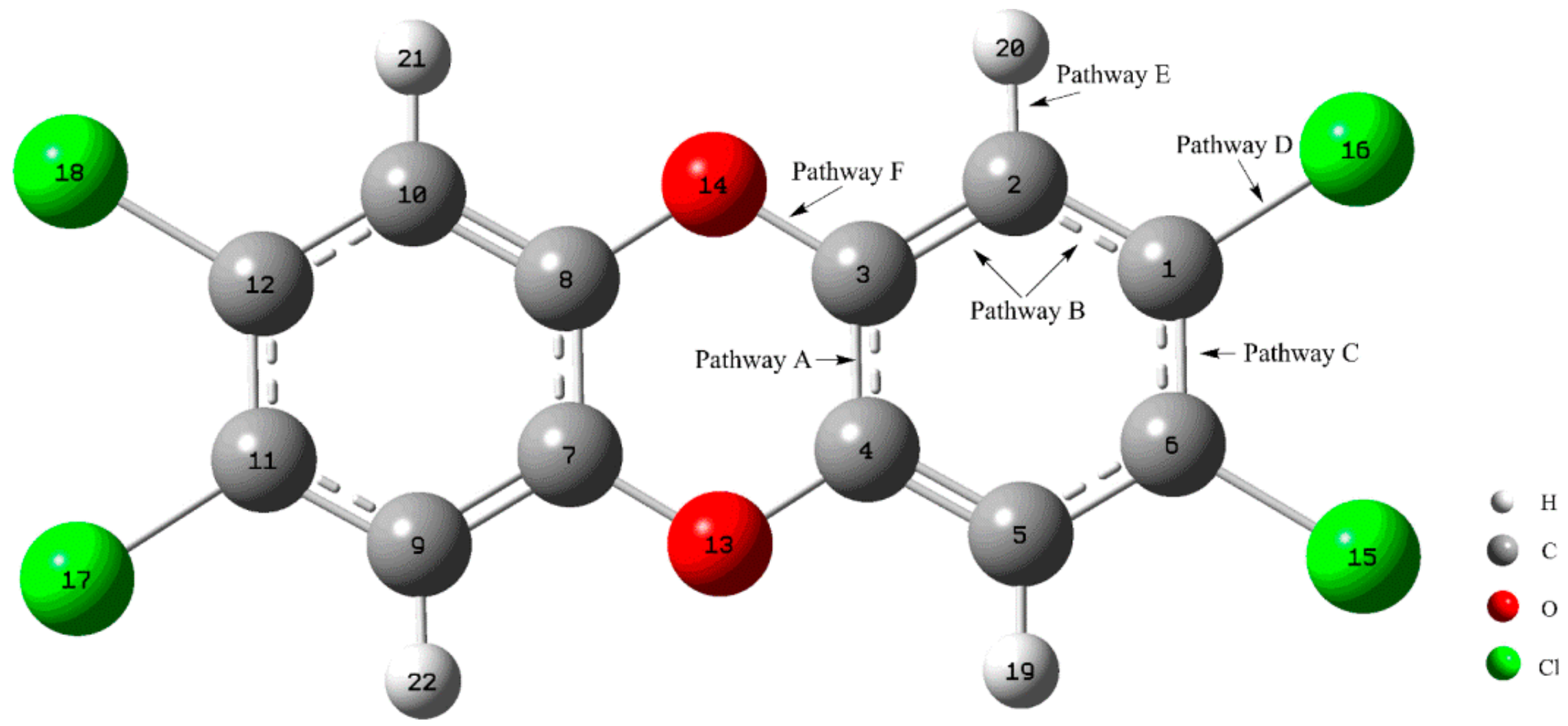
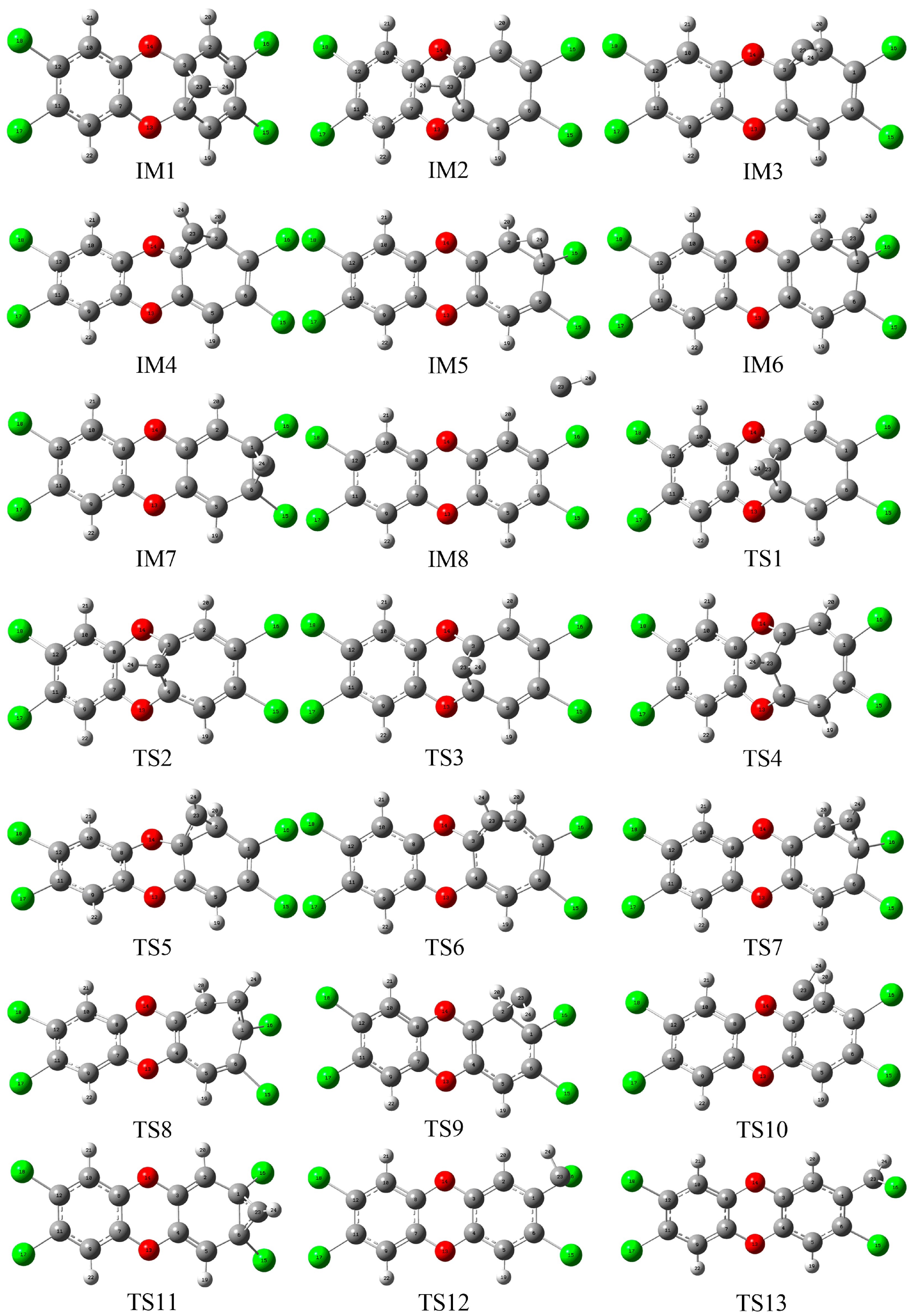
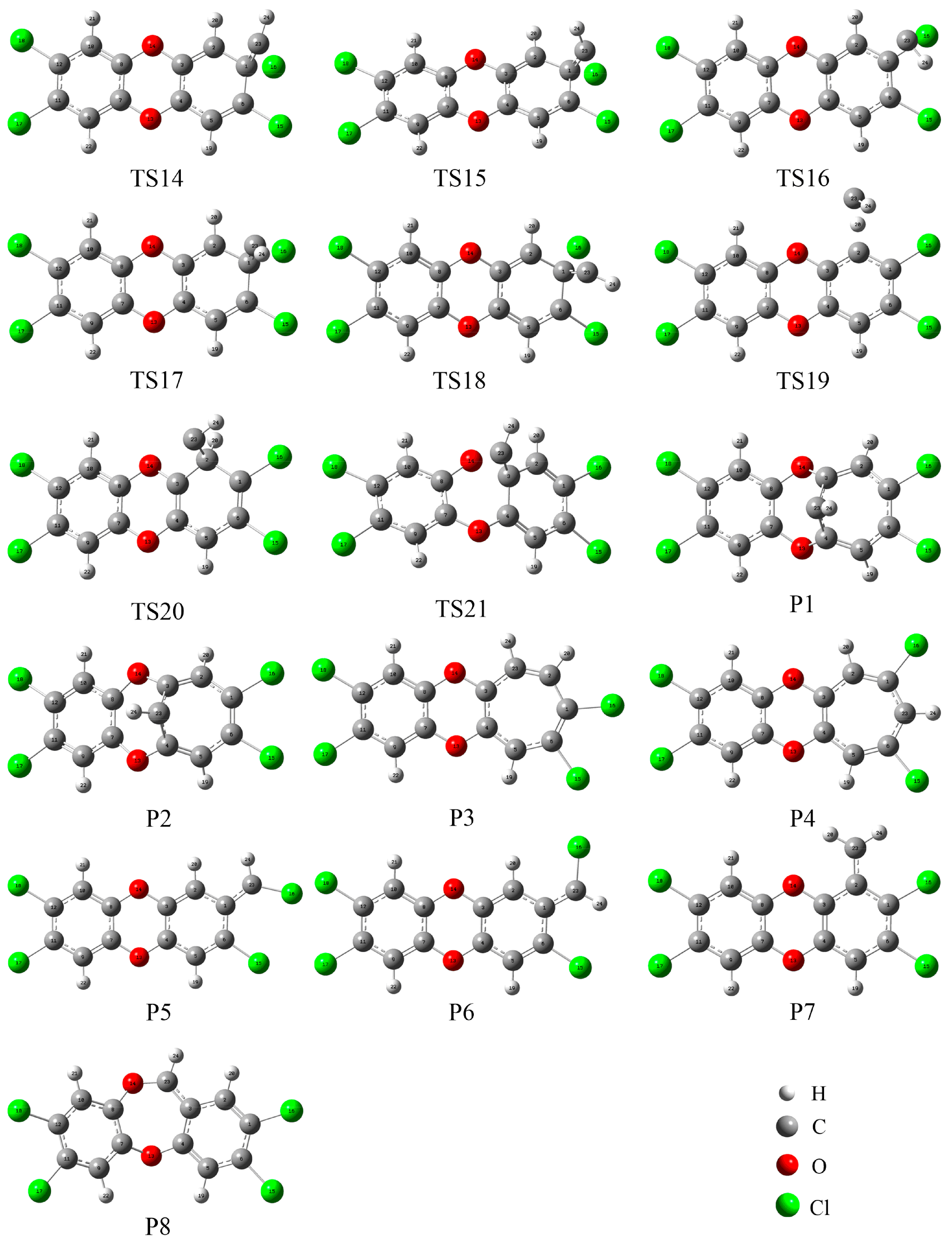
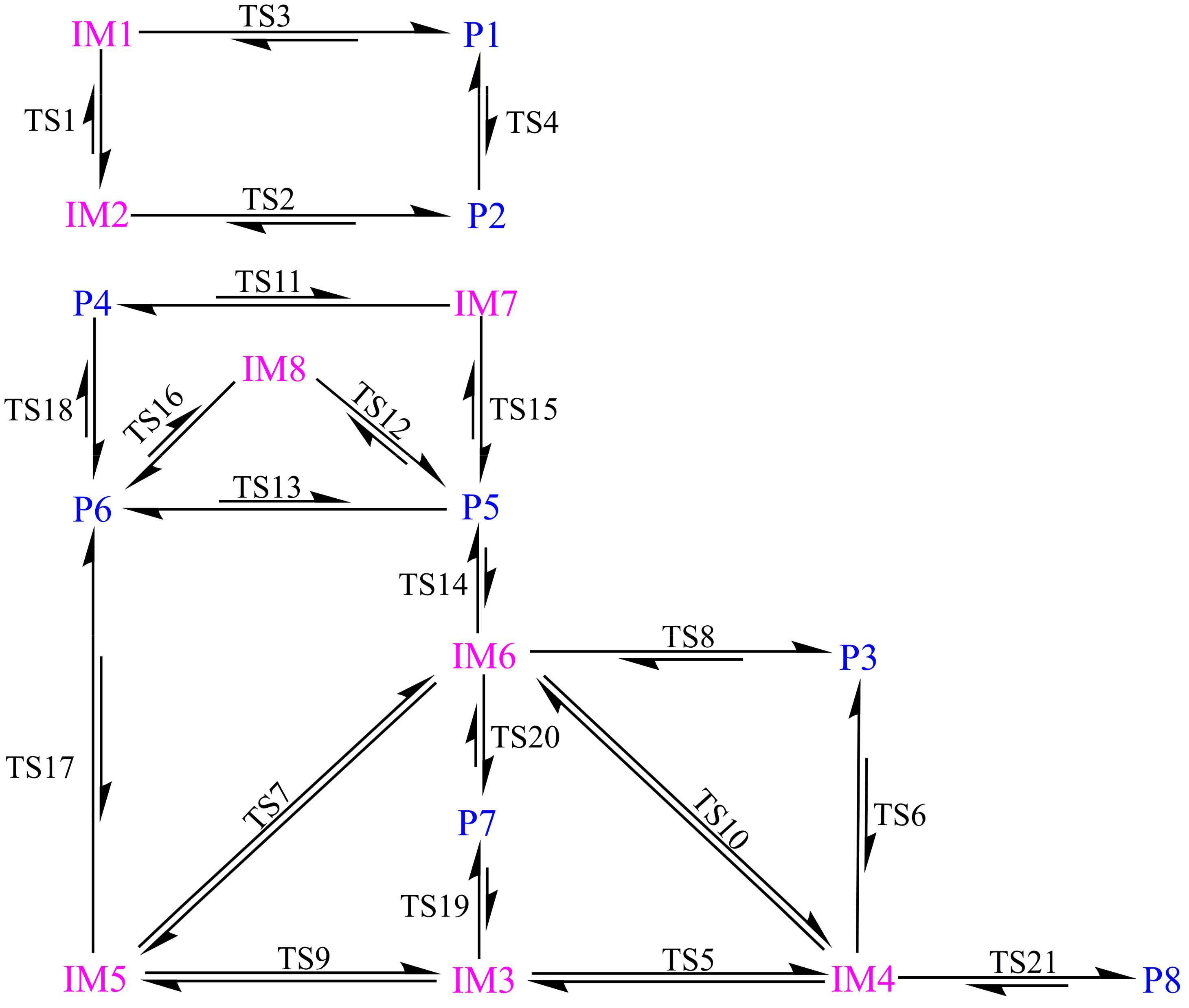
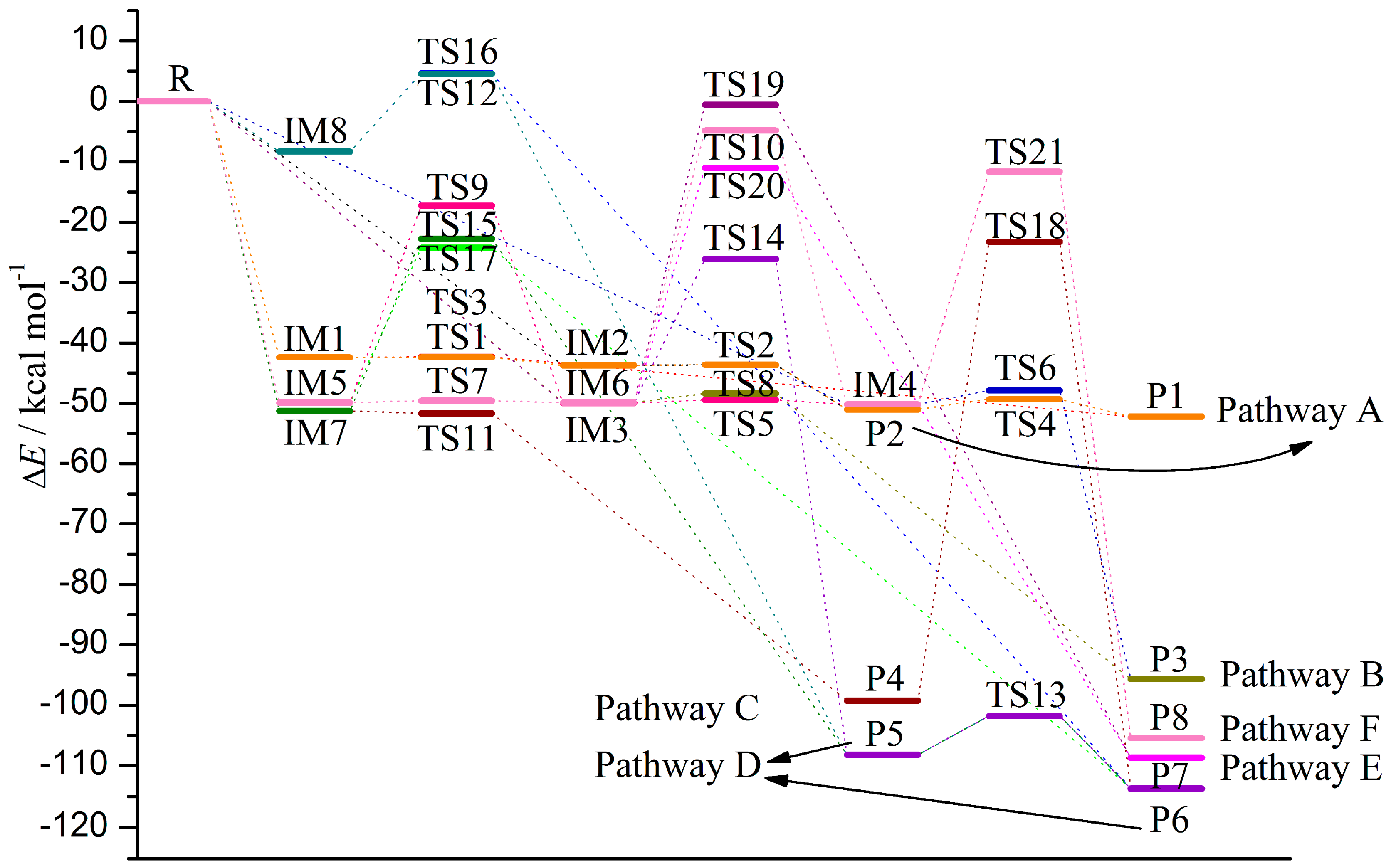
3.1. Formation of Initial Intermediates
3.2. Reaction Pathways
3.2.1. Pathway A
3.2.2. Pathway B
3.2.3. Pathway C
3.2.4. Pathway D
3.2.5. Pathway E
3.2.6. Pathway F
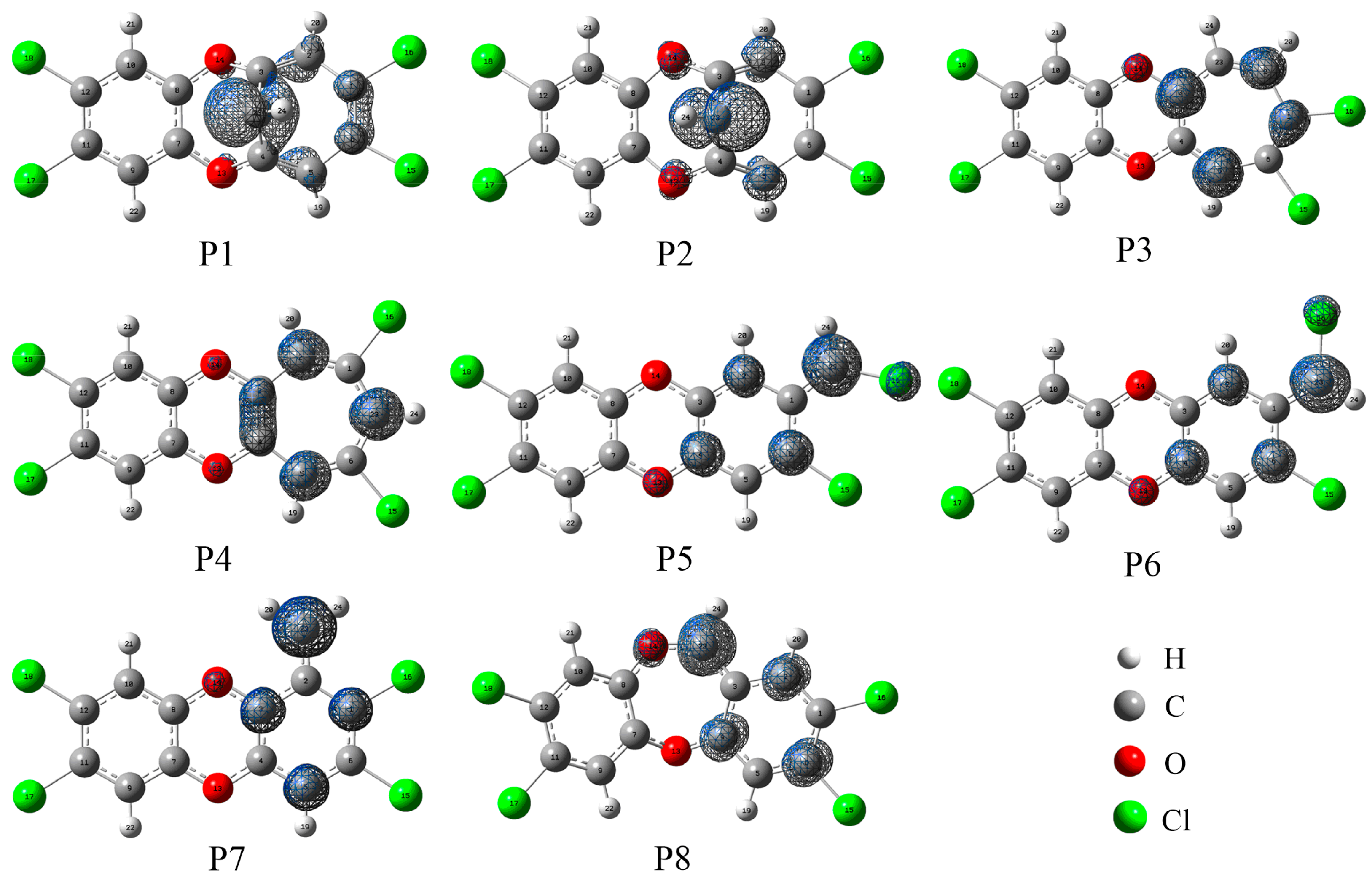
3.3. Relative Stability and Interconversion for Products
3.3.1. Relative Stability
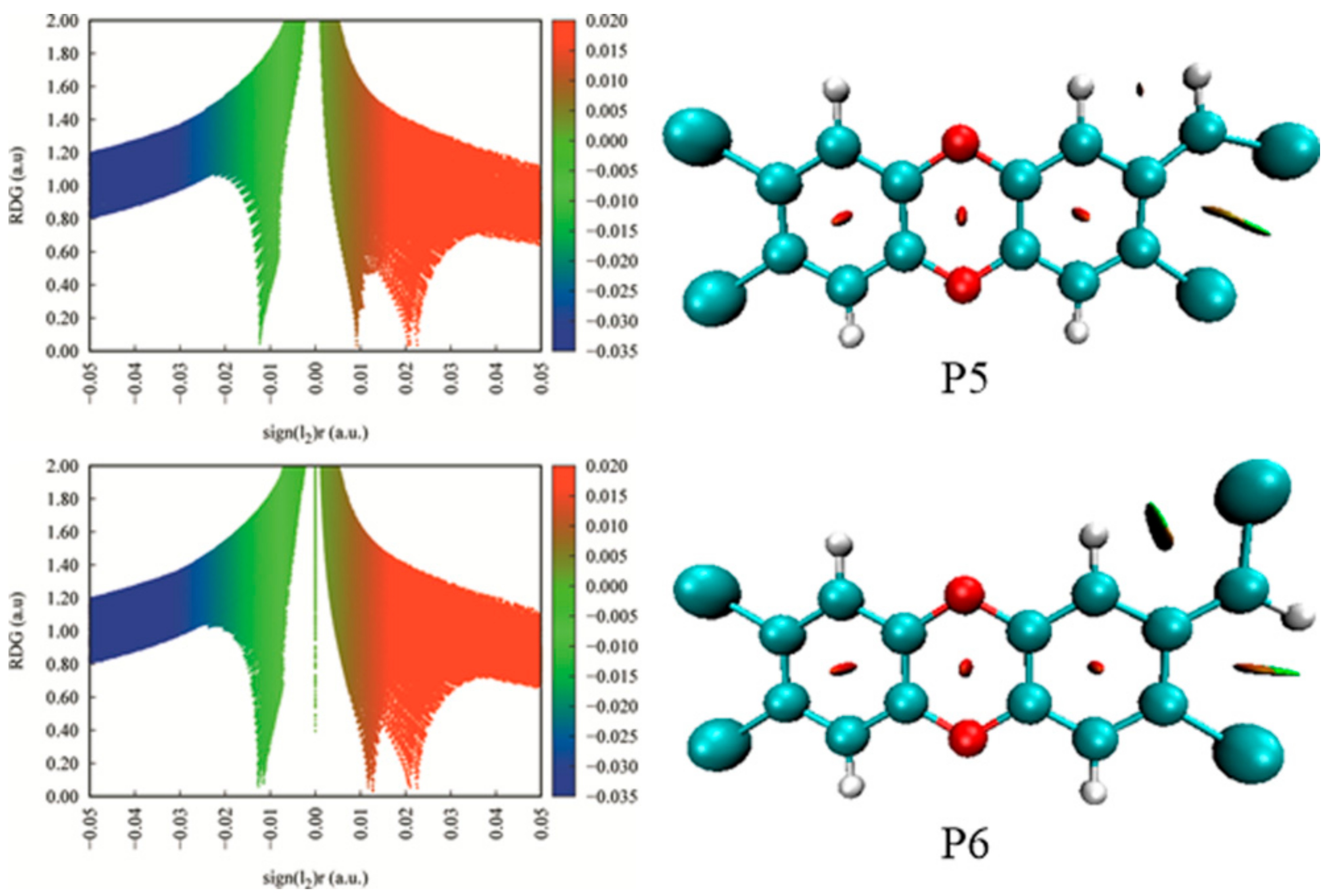
3.3.2. Interconversion among Products
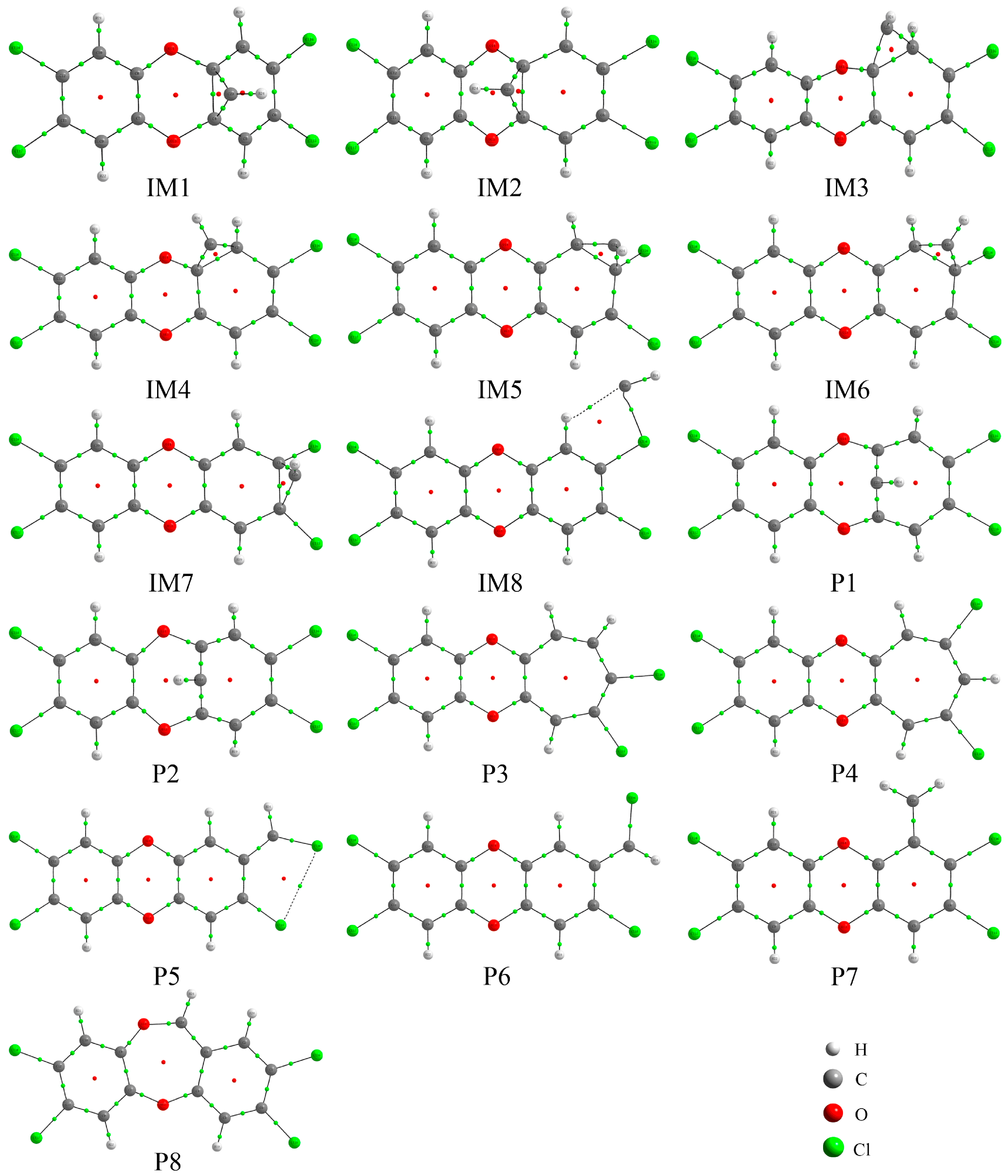
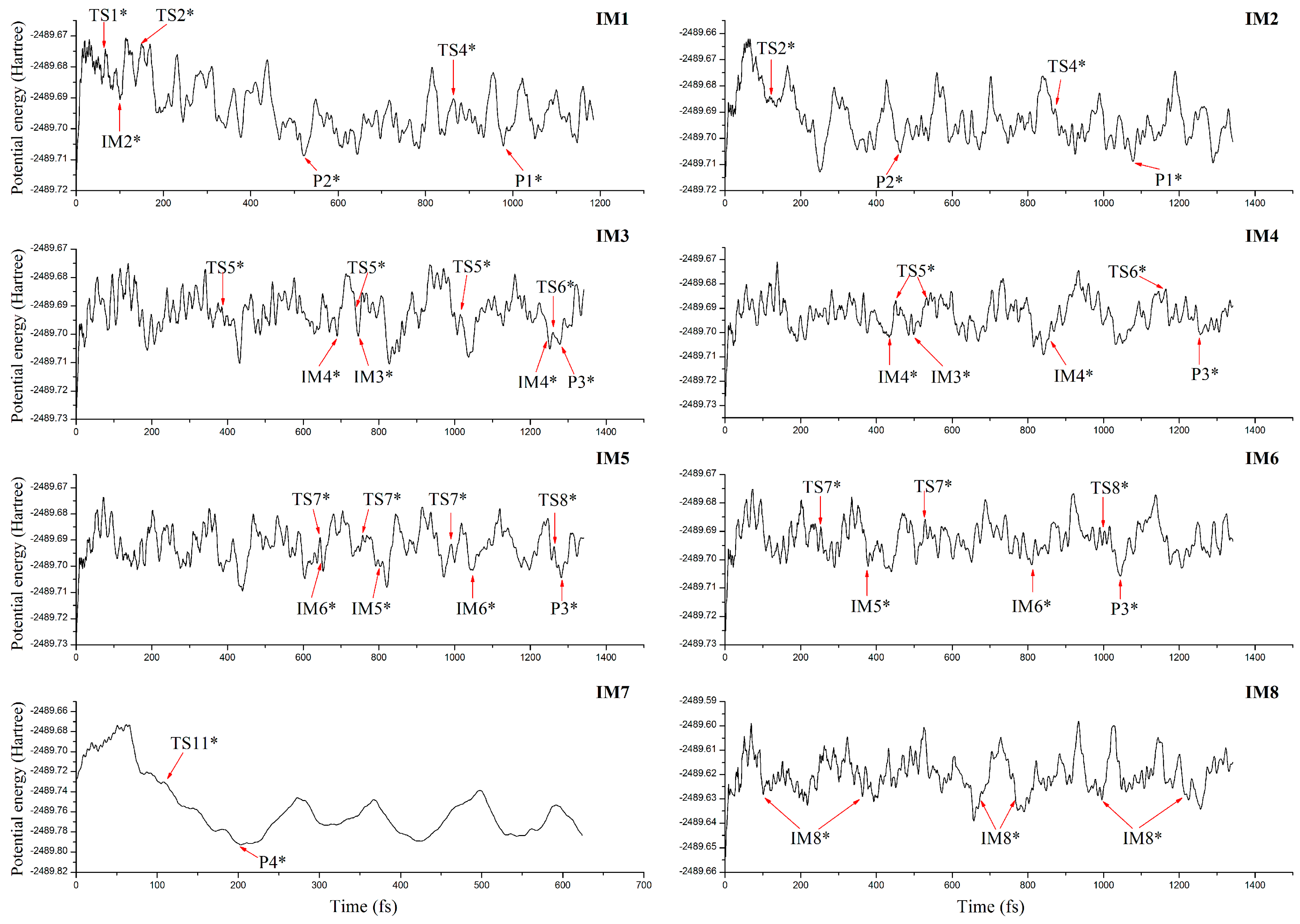
3.4. Molecular Dynamics Simulations
3.5. Analyses of IR Spectra and Hyperfine Coupling Constants
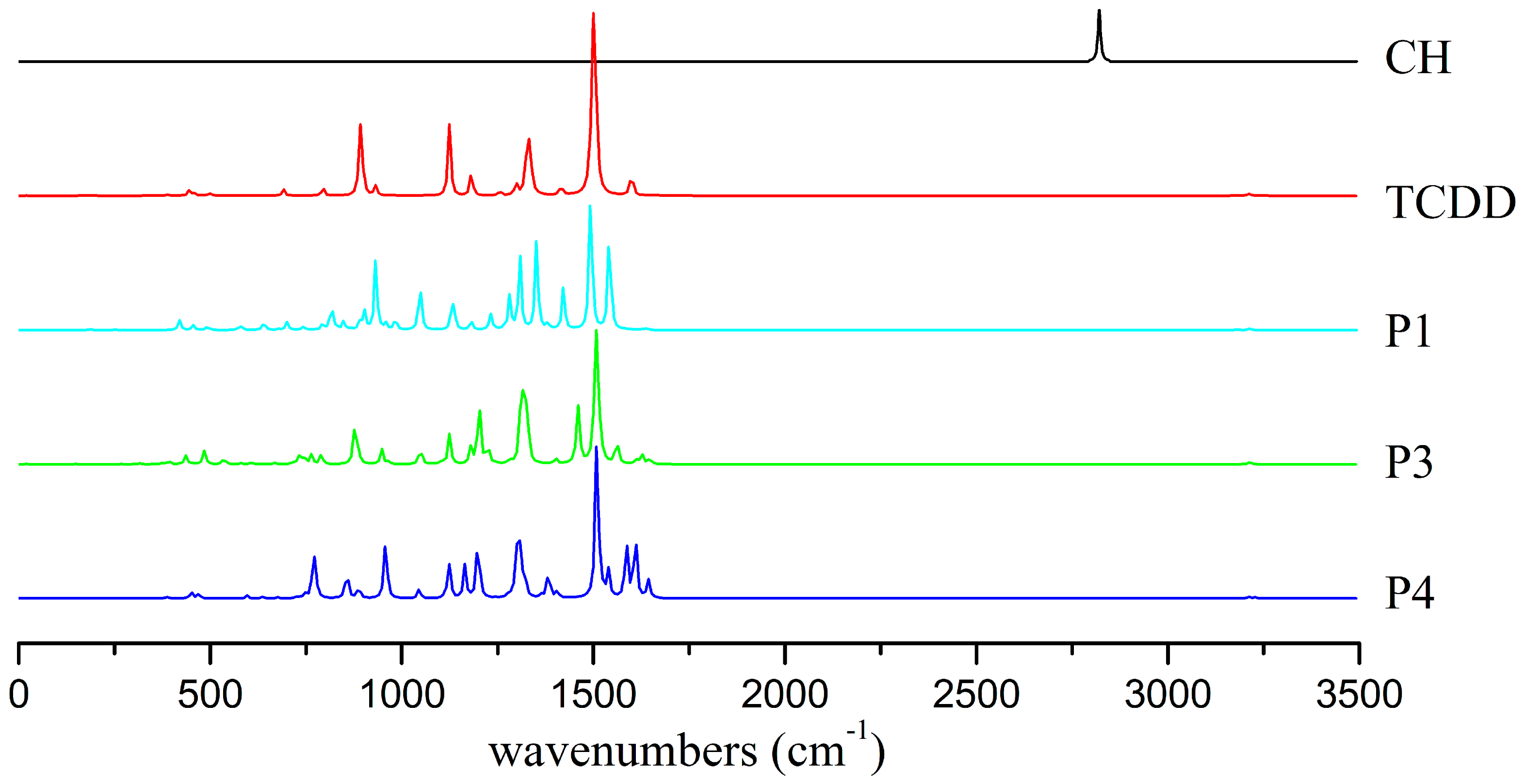
3.5.1. IR Spectra
3.5.2. Hyperfine Coupling Constants
3.6. Substitution Effects
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zou, L.L.; Ni, Y.W.; Gao, Y.; Tang, F.M.; Jin, J.; Chen, J.P. Spatial variation of PCDD/F and PCB emissions and their composition profiles in stack flue gas from the typical cement plants in China. Chemosphere 2018, 195, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Sappington, E.N.; Balasubramani, A.; Rifai, H.S. Polychlorinated dibenzo-p-dioxins and polychlorinated dibenzofurans (PCDD/Fs) in municipal and industrial effluents. Chemosphere 2015, 133, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Ni, Y.W.; Zhang, H.J.; Zhang, X.P.; Chen, J.P. Formation and emission of PCDD/Fs in Chinese non-wood pulp and paper mills. Environ. Sci. Technol. 2012, 46, 12234–12240. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Zhang, H.J.; Ni, Y.W.; Du, Q.Q.; Zhang, X.P.; Chen, J.P. Kinetics of PCDD/Fs formation from non-wood pulp bleaching with chlorine. Environ. Sci. Technol. 2014, 48, 4361–4367. [Google Scholar] [CrossRef] [PubMed]
- Salamanca, M.; Chandia, C.; Hernandez, A. Impact of forest fires on the concentrations of polychlorinated dibenzo-p-dioxin and dibenzofurans in coastal waters of central Chile. Sci. Total Environ. 2016, 573, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, H.; Themelis, N.J. Inventory of US 2012 dioxin emissions to atmosphere. Waste Manag. 2015, 46, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Heeb, N.V.; Rey, M.D.; Zennegg, M.; Haag, R.; Wichser, A.; Schmid, P.; Seiler, C.; Honegger, P.; Zeyer, K.; Mohn, J.; et al. Biofuel-Promoted polychlorinated dibenzodioxin/furan formation in an iron-catalyzed diesel particle filter. Environ. Sci. Technol. 2015, 49, 9273–9279. [Google Scholar] [CrossRef] [PubMed]
- Heeb, N.V.; Zennegg, M.; Haag, R.; Wichser, A.; Schmid, P.; Seiler, C.; Ulrich, A.; Honegger, P.; Zeyer, K.; Emmenegger, L.; et al. PCDD/F formation in an iron/potassium-catalyzed diesel particle filter. Environ. Sci. Technol. 2013, 47, 6510–6517. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Furuichi, T.; Tanikawa, N.; Kuboshima, M. Estimation of the biodegradation rate of 2,3,7,8-tetrachlorodibenzo-p-dioxin by using dioxin-degrading fungus, Pseudallescheria boydii. J. Hazard. Mater. 2009, 162, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, T.; Munetsuna, E. Enzyme systems for biodegradation of polychlorinated dibenzo-p-dioxins. Appl. Microbiol. Biot. 2010, 88, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Hieke, A.S.C.; Brinkmeyer, R.; Yeager, K.M.; Schindler, K.; Zhang, S.J.; Xu, C.; Louchouarn, P.; Santschi, P.H. Widespread distribution of Dehalococcoides mccartyi in the Houston Ship Channel and Galveston Bay, Texas, sediments and the potential for reductive dechlorination of PCDD/F in an estuarine environment. Mar. Biotechnol. 2016, 18, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Chang-Chien, G.P.; Lee, W.S. Photodegradation of tetra- and hexachlorodibenzo-p-dioxins. J. Hazard. Mater. 2005, 120, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Finocchio, E.; Busca, G.; Notaro, M. A review of catalytic processes for the destruction of PCDD and PCDF from waste gases. Appl. Catal. B 2006, 62, 12–20. [Google Scholar] [CrossRef]
- Yu, M.F.; Lin, X.Q.; Li, X.D.; Chen, T.; Yan, J.H. Catalytic decomposition of PCDD/Fs over Nano-TiO2 based V2O5/CeO2 catalyst at low temperature. Aerosol. Air. Qual. Res. 2016, 16, 2011–2022. [Google Scholar] [CrossRef]
- Hung, P.C.; Chang, S.H.; Ou-Yang, C.C.; Chang, M.B. Simultaneous removal of PCDD/Fs, pentachlorophenol and mercury from contaminated soil. Chemosphere 2016, 144, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Govindan, M.; Moon, I.S. Expeditious removal of PCDD/Fs from industrial waste incinerator fly ash using electrogenerated homogeneous Ag(II) ions. Chem. Eng. J. 2015, 272, 145–150. [Google Scholar] [CrossRef]
- Palanisami, N.; Chung, S.J.; Moon, I.S. Cerium(IV)-mediated electrochemical oxidation process for removal of polychlorinated dibenzo-p-dioxins and dibenzofurans. J. Ind. Eng. Chem. 2015, 28, 28–31. [Google Scholar] [CrossRef]
- Boyd, S.A.; Sallach, J.B.; Zhang, Y.J.; Crawford, R.; Li, H.; Johnston, C.T.; Teppen, B.J.; Kaminski, N.E. Sequestration of 2,3,7,8-tetrachlorodibenzo-p-dioxin by activated carbon eliminates bioavailability and the suppression of immune function in mice. Environ. Toxicol. Chem. 2017, 36, 2671–2678. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.C.; Wang, Z.H.; Xu, J.R.; Liu, Y.; Cen, K.F. Quantum chemistry study on the destruction mechanism of 2,3,7,8-TCDD by OH and O3 radicals. Chemosphere 2013, 92, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.X.; Sun, T.L.; Sun, X.M. Mechanism for OH-initiated degradation of 2,3,7,8-tetrachlorinated dibenzo-p-dioxins in the presence of O2 and NO/H2O. Environ. Sci. Technol. 2011, 45, 4756–4762. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.M.; Tang, A.L. Atmospheric oxidation mechanisms of polychlorinated dibenzo-p-dioxins are different from those of benzene and dibenzofuran: A theoretical prediction. Chemosphere 2011, 82, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.X.; Sun, X.M.; Xu, Y.S.; Qi, C.S.; Zhang, J.H. Atmospheric chemistry of 2,3,7,8-TCDD/F: Mechanism and kinetics study. J. Environ. Chem. Eng. 2014, 2, 1098–1103. [Google Scholar] [CrossRef]
- Wang, Z.H.; Wen, Z.C.; Xu, J.R.; Zhou, J.H.; Cen, K.F. Theoretical study on destruction mechanism of 2,3,7,8-TCDD by O3 and NO3. Chin. J. Chem. Phys. 2010, 23, 303–309. [Google Scholar] [CrossRef]
- Bai, J.; Sun, X.M.; Zhang, C.X.; Gong, C.; Hu, J.T.; Zhang, J.H. Mechanism and kinetics study on the ozonolysis reaction of 2,3,7,8-TCDD in the atmosphere. J. Environ. Sci. (China) 2014, 26, 181–188. [Google Scholar] [CrossRef]
- Lu, G.N.; Dang, Z.; Fennell, D.E.; Huang, W.L.; Li, Z.; Liu, C.Q. Rules of thumb for assessing reductive dechlorination pathways of PCDDs in specific systems. J. Hazard. Mater. 2010, 177, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.X.; Qi, Y.Y.; Wang, R.X.; Han, Z.; Zhang, D.J.; Zhan, J.H. Adsorption of TCDD with 1-butyl-3-methylimidazolium dicyanamide ionic liquid: A combined molecular dynamics simulation and quantum chemistry study. Chemosphere 2013, 91, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.L.; Hu, W.H.; You, Z.M.; Ge, F.; Tian, K.X. Molecular dynamics simulation of TCDD adsorption on organo-montmorillonite. J. Colloid Interface Sci. 2012, 377, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Love, N.; Parthasarathy, R.N.; Gollahalli, S.R. Concentration measurements of CH and OH radicals in laminar biofuel flames. Int. J. Green Energy 2011, 8, 113–120. [Google Scholar] [CrossRef]
- Tinaut, F.V.; Reyes, M.; Gimenez, B.; Pastor, J.V. Measurements of OH* and CH* chemiluminescence in premixed flames in a constant volume combustion bomb under autoignition conditions. Energy Fuels 2011, 25, 119–129. [Google Scholar] [CrossRef]
- Mann, A.P.C.; Williams, D.A. List of interstellar molecules. Nature 1980, 283, 721–725. [Google Scholar] [CrossRef]
- Kaiser, R.I. Experimental investigation on the formation of carbon-bearing molecules in the interstellar medium via neutral-neutral reactions. Chem. Rev. 2002, 102, 1309–1358. [Google Scholar] [CrossRef] [PubMed]
- Krasnopolsky, V.A. A photochemical model of Titan’s atmosphere and ionosphere. Icarus 2009, 201, 226–256. [Google Scholar] [CrossRef]
- Goulay, F.; Trevitt, A.J.; Meloni, G.; Selby, T.M.; Osborn, D.L.; Taatjes, C.A.; Vereecken, L.; Leone, S.R. Cyclic versus linear isomers produced by reaction of the methylidyne radical (CH) with small unsaturated hydrocarbons. J. Am. Chem. Soc. 2009, 131, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- McKee, K.; Blitz, M.A.; Hughes, K.J.; Pilling, M.J.; Qian, H.B.; Taylor, A.; Seakins, P.W. H atom branching ratios from the reactions of CH with C2H2, C2H4, C2H6, and neo-C5H12 at room temperature and 25 Torr. J. Phys. Chem. A 2003, 107, 5710–5716. [Google Scholar] [CrossRef]
- Fleurat-Lessard, P.; Rayez, J.C.; Bergeat, A.; Loison, J.C. Reaction of methylidyne CH(X2Π) radical with CH4 and H2S: Overall rate constant and absolute atomic hydrogen production. Chem. Phys. 2002, 279, 87–99. [Google Scholar] [CrossRef]
- Ribeiro, J.M.; Mebel, A.M. Reaction mechanism and product branching ratios of the CH + C3H8 reaction: A theoretical study. J. Phys. Chem. A 2014, 118, 9080–9086. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.M.; Mebel, A.M. Reaction mechanism and rate constants of the CH + CH4 reaction: A theoretical study. Mol. Phys. 2015, 113, 1865–1872. [Google Scholar] [CrossRef]
- Ribeiro, J.M.; Mebel, A.M. Reaction mechanism and product branching ratios of the CH + C3H6 reaction: A theoretical study. J. Phys. Chem. A 2016, 120, 1800–1812. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.M.; Mebel, A.M. Reaction mechanism and product branching ratios of the CH + C3H4 reaction: A theoretical study. Phys. Chem. Chem. Phys. 2017, 19, 14543–14554. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, H.L.; Zhou, Z.J.; Huang, X.R.; Sun, C.C. Reaction mechanism of CH + C3H6: A theoretical study. J. Phys. Chem. A 2010, 114, 9496–9506. [Google Scholar] [CrossRef] [PubMed]
- Trevitt, A.J.; Prendergast, M.B.; Goulay, F.; Savee, J.D.; Osborn, D.L.; Taatjes, C.A.; Leone, S.R. Product branching fractions of the CH + propene reaction from synchrotron photoionization mass spectrometry. J. Phys. Chem. A 2013, 117, 6450–6457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Maksyutenko, P.; Kaiser, R.I. Chemical dynamics of the CH(X2Π) + C2H4(X1A1g), CH(X2Π) + C2D4(X1A1g), and CD(X2Π) + C2H4(X1A1g) reactions studied under single collision conditions. Phys. Chem. Chem. Phys. 2012, 14, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Loison, J.C.; Bergeat, A. Rate constants and the H atom branching ratio of the reactions of the methylidyne CH(X2Π) radical with C2H2, C2H4, C3H4 (methylacetylene and allene), C3H6 (propene) and C4H8 (trans-butene). Phys. Chem. Chem. Phys. 2009, 11, 655–664. [Google Scholar] [CrossRef] [PubMed]
- McCunn, L.R.; FitzPatrick, B.L.; Krisch, M.J.; Butler, L.J.; Liang, C.W.; Lin, J.J. Unimolecular dissociation of the propargyl radical intermediate of the CH + C2H2 and C + C2H3 reactions. J. Chem. Phys. 2006, 125, 133306. [Google Scholar] [CrossRef] [PubMed]
- Trevitt, A.J.; Goulay, F. Insights into gas-phase reaction mechanisms of small carbon radicals using isomer-resolved product detection. Phys. Chem. Chem. Phys. 2016, 18, 5867–5882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soorkia, S.; Taatjes, C.A.; Osborn, D.L.; Selby, T.M.; Trevitt, A.J.; Wilson, K.R.; Leone, S.R. Direct detection of pyridine formation by the reaction of CH (CD) with pyrrole: A ring expansion reaction. Phys. Chem. Chem. Phys. 2010, 12, 8750–8758. [Google Scholar] [CrossRef] [PubMed]
- Lockyear, J.F.; Welz, O.; Savee, J.D.; Goulay, F.; Trevitt, A.J.; Taatjes, C.A.; Osborn, D.L.; Leone, S.R. Isomer specific product detection in the reaction of CH with acrolein. J. Phys. Chem. A 2013, 117, 11013–11026. [Google Scholar] [CrossRef] [PubMed]
- Goulay, F.; Rebrion-Rowe, C.; Biennier, L.; Le Picard, S.D.; Canosa, A.; Rowe, B.R. Reaction of anthracene with CH radicals: An experimental study of the kinetics between 58 and 470 K. J. Phys. Chem. A 2006, 110, 3132–3137. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.G.; Blitz, M.A.; Seakins, P.W. The reaction of methylidene (CH) with methanol isotopomers. Phys. Chem. Chem. Phys. 2000, 2, 2549–2553. [Google Scholar] [CrossRef]
- Thiesemann, H.; MacNamara, J.; Taatjes, C.A. Deuterium kinetic isotope effect and temperature dependence in the reactions of CH2Π with methane and acetylene. J. Phys. Chem. A 1997, 101, 1881–1886. [Google Scholar] [CrossRef]
- Wei, W.J.; Wang, W.H.; Xu, K.N.; Feng, W.L.; Li, X.P.; Li, P. Theoretical insights into the reaction mechanisms between 2,3,7,8-tetrachlorodibenzofuran and the methylidyne radical. RSC Adv. 2018, 8, 21150–21163. [Google Scholar] [CrossRef]
- Mazarei, E.; Mousavipour, S.H. A theoretical study on the dynamics of the reaction of CH radicals with water molecules. J. Phys. Chem. A 2017, 121, 8033–8047. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, W.H.; Feng, W.L.; Wang, W.L.; Li, P. Theoretical insights into the interaction mechanisms between nitric acid and nitrous oxide initiated by an excess electron. J. Phys. Chem. A 2018, 122, 7312–7319. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Feng, W.L.; Wang, W.L.; Li, P. Theoretical insights into the electron capture behavior of H2SO4···N2O complex: A DFT and molecular dynamics study. Molecules 2018, 23, 2349. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wang, W.H.; Feng, W.L.; Li, P. Insights into the one-electron reduction behavior of tetrachloro-o-benzoquinone: A DFT and molecular dynamics study. RSC Adv. 2017, 7, 12775–12782. [Google Scholar] [CrossRef]
- Xu, K.N.; Wang, W.H.; Wei, W.J.; Feng, W.L.; Sun, Q.; Li, P. Insights into the reaction mechanism of Criegee intermediate CH2OO with methane and implications for the formation of methanol. J. Phys. Chem. A 2017, 121, 7236–7245. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Guo, C.; Feng, W.L.; Sun, Q.; Li, P. Theoretical insights into the reaction mechanism between tetrachloro-o-benzoquinone and N-methyl benzohydroxamic acid. RSC Adv. 2017, 7, 32419–32426. [Google Scholar] [CrossRef]
- Feng, W.L.; Ren, C.; Wang, W.H.; Guo, C.; Sun, Q.; Li, P. An identification of the C–C bonding spin adduct in the spin trapping of N-methyl benzohydroxamic acid radical by 5,5-dimethyl-1-pyrroline-N-oxide. Theor. Chem. Acc. 2016, 135, 190. [Google Scholar] [CrossRef]
- Feng, W.L.; Ren, C.; Wang, W.H.; Guo, C.; Sun, Q.; Li, P. Theoretical studies on the spin trapping of the 2-chloro-5-hydroxy-1,4-benzoquinone radical by 5,5-dimethyl-1-pyrroline N-oxide (DMPO): The identification of the C–O bonding spin adduct. RSC Adv. 2016, 6, 48099–48108. [Google Scholar] [CrossRef]
- Wang, W.H.; Zhang, X.X.; Li, P.; Sun, Q.; Li, Z.; Ren, C.; Guo, C. CO2 capture and separation from N2/CH4 mixtures by Co@B8/Co@B8¯ and M@B9/M@B9¯ (M = Ir, Rh, Ru) clusters: A theoretical study. J. Phys. Chem. A 2015, 119, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, W.H.; Sun, Q.; Li, Z.; Du, A.J.; Bi, S.W.; Zhao, Y. Insights into the mechanism of the reaction between tetrachloro-p-benzoquinone and hydrogen peroxide and their implications in the catalytic role of water molecules in producing the hydroxyl radical. ChemPhysChem 2013, 14, 2737–2743. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1998, 37, 785–789. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Morisánchez, P.; Contrerasgarcía, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, H.B.; Millam, J.M.; Iyengar, S.S.; Voth, G.A.; Daniels, A.D.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. J. Chem. Phys. 2001, 114, 9758–9763. [Google Scholar] [CrossRef]
- Iyengar, S.S.; Schlegel, H.B.; Millam, J.M.; Voth, G.A.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. II. Generalizations based on mass-weighting, idempotency, energy conservation and choice of initial conditions. J. Chem. Phys. 2001, 115, 10291–10302. [Google Scholar] [CrossRef]
- Schlegel, H.B.; Iyengar, S.S.; Li, X.; Millam, J.M.; Voth, G.A.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. III. Comparison with Born-Oppenheimer dynamics. J. Chem. Phys. 2002, 117, 8694–8704. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Creve, S.; Vanquickenborne, L.G. Efficient calculation of isotropic hyperfine constants of phosphorus radicals using Density Functional Theory. J. Phys. Chem. A 1997, 101, 3174–3181. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intermediates | ΔErela | ΔH | ΔG |
|---|---|---|---|
| IM1 | −42.41 (−44.30/−42.17) [−41.37] | −43.70 (−45.77/−43.48) [−42.67] | −34.07 (−35.32/−33.95) [−33.03] |
| IM2 | −43.72 (−45.54/−43.49) [−42.72] | −45.06 (−46.93/−44.85) [−44.06] | −35.16 (−36.63/−34.87) [−34.16] |
| IM3 | −49.98 | −51.37 | −41.21 |
| IM4 | −50.16 (−48.74/−50.43) | −51.54 (−50.13/−51.82) | −41.44 (−39.76/−41.64) |
| IM5 | −49.88 | −51.15 | −41.52 |
| IM6 | −49.90 (−49.87/−51.03) | −51.15 (−51.17/−52.28) | −41.59 (−41.31/−42.68) |
| IM7 | −51.26 (−50.22/−53.89) | −52.36 (−51.40/−54.81) | −43.06 (−41.76/−46.16) |
| IM8 | −8.30 | −8.83 | −1.03 |
| Reactions | TSs | ΔErela | ΔEfor* | ΔErev* | ΔH | ΔG |
|---|---|---|---|---|---|---|
| IM1 ⇌ IM2 | TS1 | −42.41 | −0.01 (0.69) | 1.31 | −1.36 | −1.09 |
| IM2 ⇌ P2 | TS2 | −43.63 | 0.09 | 7.39 | −7.40 | −6.96 |
| −45.36/−43.42 | 0.18/ 0.07 | 6.82/7.77 | −6.77/−7.79 | −6.36/−7.39 | ||
| IM1 ⇌ P1 | TS3 | −42.33 | 0.08 | 9.86 | −10.00 | −9.00 |
| −42.08/−41.99 | 2.22/0.19 | 13.15/10.01 | −11.06/−10.02 | −10.49/−8.88 | ||
| P2 ⇌ P1 | TS4 | −49.30 | 1.71 | 2.89 | −1.24 | −0.95 |
| −50.28/−49.51 | 1.90/1.68 | 4.95/2.49 | −3.13/−0.86 | −2.82/−0.56 | ||
| IM3 ⇌ IM4 | TS5 | −49.44 | 0.55 | 0.72 | −0.17 | −0.23 |
| IM4 ⇌ P3 | TS6 | −47.89 | 2.28 | 47.80 | −45.12 | −46.89 |
| −47.04/−48.30 | 1.70/2.14 | 51.71/46.37 | −49.49/−43.91 | −51.77/−45.60 | ||
| IM5 ⇌ IM6 | TS7 | −49.55 | 0.33 | 0.35 | −0.01 | −0.08 |
| IM6 ⇌P3 | TS8 | −48.39 | 1.51 | 47.29 | −45.50 | −46.73 |
| −48.70/−49.55 | 1.17/1.48 | 50.04/45.12 | −48.45/−43.44 | −50.22/−44.55 | ||
| IM5 ⇌ IM3 | TS9 | −17.27 | 32.62 | 32.72 | −0.22 | 0.31 |
| IM6 ⇌ IM4 | TS10 | −4.82 | 45.08 | 45.34 | −0.38 | 0.15 |
| IM7 ⇌ P4 | TS11 | −51.67 | −0.41 (0.21) | 47.56 | −47.96 | −48.41 |
| −50.57/−54.25 | −0.35 (0.35)/−0.36 (0.00) | 54.19/44.89 | −54.50/−45.36 | −55.27/−45.62 | ||
| IM8 ⇌ P5 | TS12 | 4.58 | 12.88 | 112.79 | −100.58 | −99.14 |
| P5 ⇌ P6 | TS13 | −101.79 | 6.42 | 11.94 | −5.57 | −5.35 |
| IM6 ⇌ P5 | TS14 | −26.14 | 23.76 | 82.06 | −58.26 | −58.58 |
| IM7 ⇌ P5 | TS15 | −22.76 | 28.50 | 85.44 | −57.06 | −57.11 |
| IM8 ⇌ P6 | TS16 | 4.64 | 12.94 | 118.36 | −106.15 | −104.49 |
| IM5 ⇌ P6 | TS17 | −24.27 | 25.61 | 89.46 | −63.83 | −64.00 |
| P4 ⇌ P6 | TS18 | −23.31 | 75.91 | 90.41 | −14.66 | −14.06 |
| IM3 ⇌ P7 | TS19 | −0.58 | 49.41 | 108.12 | −58.56 | −59.03 |
| IM6 ⇌ P7 | TS20 | −11.03 | 38.87 | 97.67 | −58.77 | −58.64 |
| IM4⇌ P8 | TS21 | −11.67 | 38.50 | 93.79 | −55.26 | −55.53 |
| Atomic Labels | Atoms | P1 | P3 | P4 |
|---|---|---|---|---|
| 1 | 13C | 0.1 | 2.4 | −11.0 |
| 2 | 13C | 6.2 | −8.4 | 6.4 |
| 3 | 13C | −8.1 | 12.5 | −2.7 |
| 4 | 13C | −8.1 | −12.7 | −2.5 |
| 5 | 13C | 6.2 | 10.0 | 6.2 |
| 6 | 13C | 0.1 | −7.0 | −10.9 |
| 7 | 13C | −0.1 | 0.4 | −0.3 |
| 8 | 13C | −0.1 | −0.7 | −0.3 |
| 9 | 13C | 0.2 | −0.5 | −0.1 |
| 10 | 13C | 0.2 | 0.2 | −0.1 |
| 11 | 13C | 0.2 | 0.7 | 0.0 |
| 12 | 13C | 0.2 | −0.7 | 0.0 |
| 13 | 17O | −1.0 | 0.2 | −0.1 |
| 14 | 17O | −1.0 | −0.4 | −0.1 |
| 15 | 35Cl | 0.2 | −0.1 | −0.1 |
| 16 | 35Cl | 0.2 | 0.1 | −0.1 |
| 17 | 35Cl | 0.0 | 0.0 | 0.0 |
| 18 | 35Cl | 0.0 | 0.0 | 0.0 |
| 19 | 1H | −0.9 | −9.8 | −7.7 |
| 20 | 1H | −0.9 | 1.1 | −7.8 |
| 21 | 1H | 0.1 | −0.3 | 0.0 |
| 22 | 1H | 0.1 | 0.2 | 0.0 |
| 23 | 13C | 60.5 | 4.2 | 10.9 |
| 24 | 1H | −12.0 | −6.3 | −10.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Feng, W.; Wang, W.; Li, P. Theoretical Investigations on the Reactivity of Methylidyne Radical toward 2,3,7,8-Tetrachlorodibenzo-p-Dioxin: A DFT and Molecular Dynamics Study. Molecules 2018, 23, 2685. https://doi.org/10.3390/molecules23102685
Wang W, Feng W, Wang W, Li P. Theoretical Investigations on the Reactivity of Methylidyne Radical toward 2,3,7,8-Tetrachlorodibenzo-p-Dioxin: A DFT and Molecular Dynamics Study. Molecules. 2018; 23(10):2685. https://doi.org/10.3390/molecules23102685
Chicago/Turabian StyleWang, Weihua, Wenling Feng, Wenliang Wang, and Ping Li. 2018. "Theoretical Investigations on the Reactivity of Methylidyne Radical toward 2,3,7,8-Tetrachlorodibenzo-p-Dioxin: A DFT and Molecular Dynamics Study" Molecules 23, no. 10: 2685. https://doi.org/10.3390/molecules23102685
APA StyleWang, W., Feng, W., Wang, W., & Li, P. (2018). Theoretical Investigations on the Reactivity of Methylidyne Radical toward 2,3,7,8-Tetrachlorodibenzo-p-Dioxin: A DFT and Molecular Dynamics Study. Molecules, 23(10), 2685. https://doi.org/10.3390/molecules23102685