
66Ga: A Novelty or a Valuable Preclinical Screening Tool for the Design of Targeted Radiopharmaceuticals?


Abstract
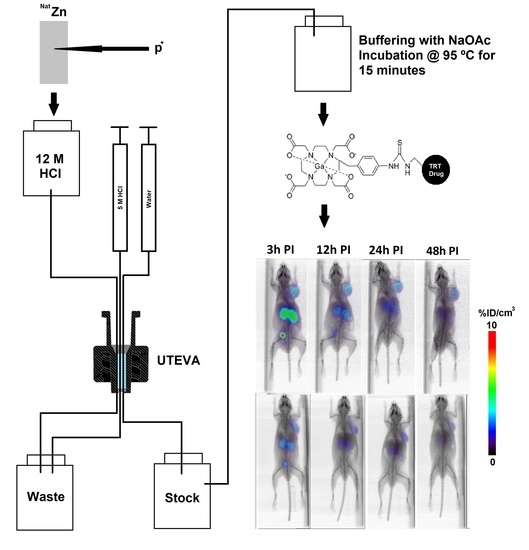
:
1. Introduction
2. Results
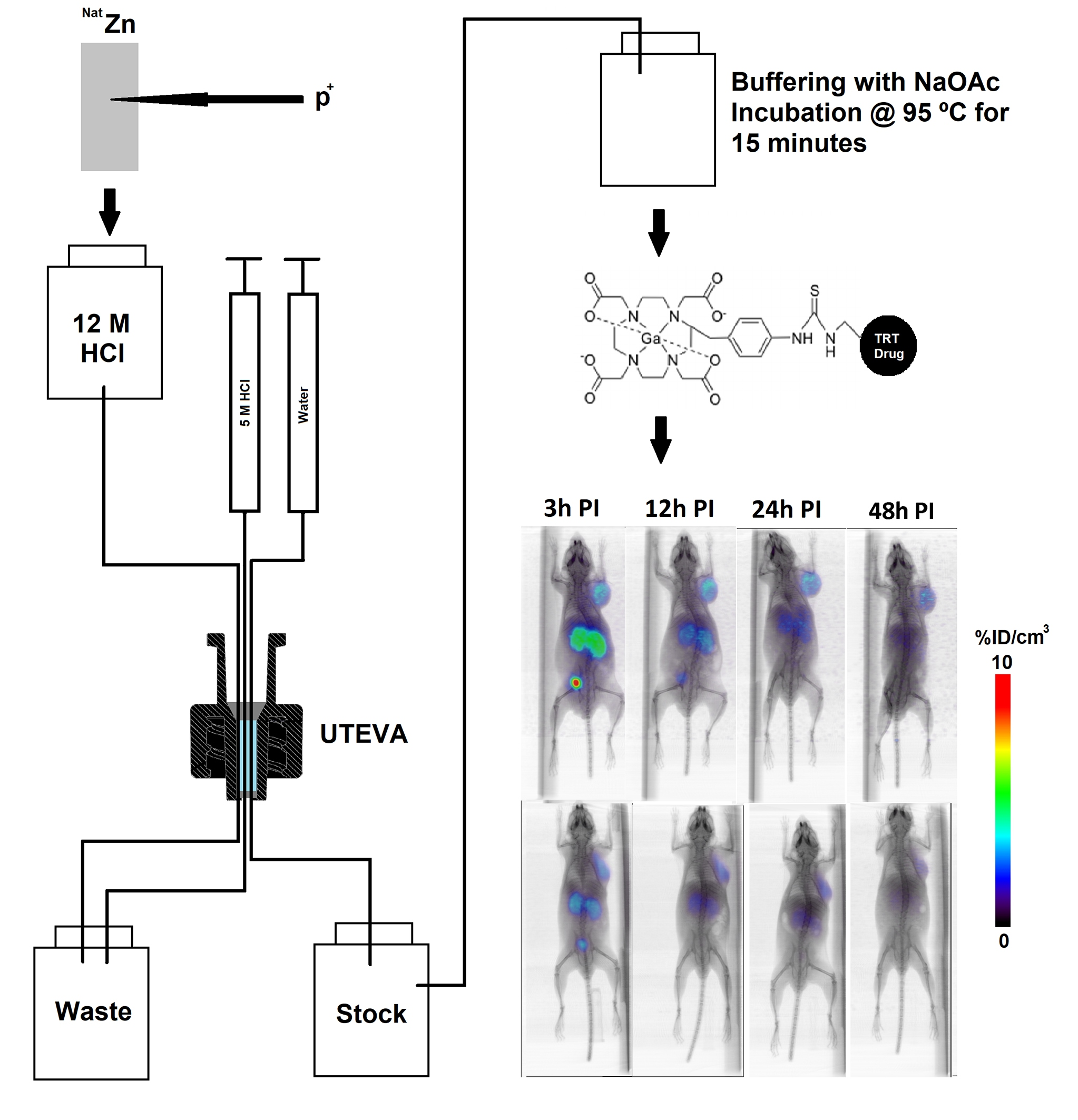
2.1. Production and Purification of 66Ga
2.2. Measurement of Metal Contaminants in the Purified 66Ga Solution
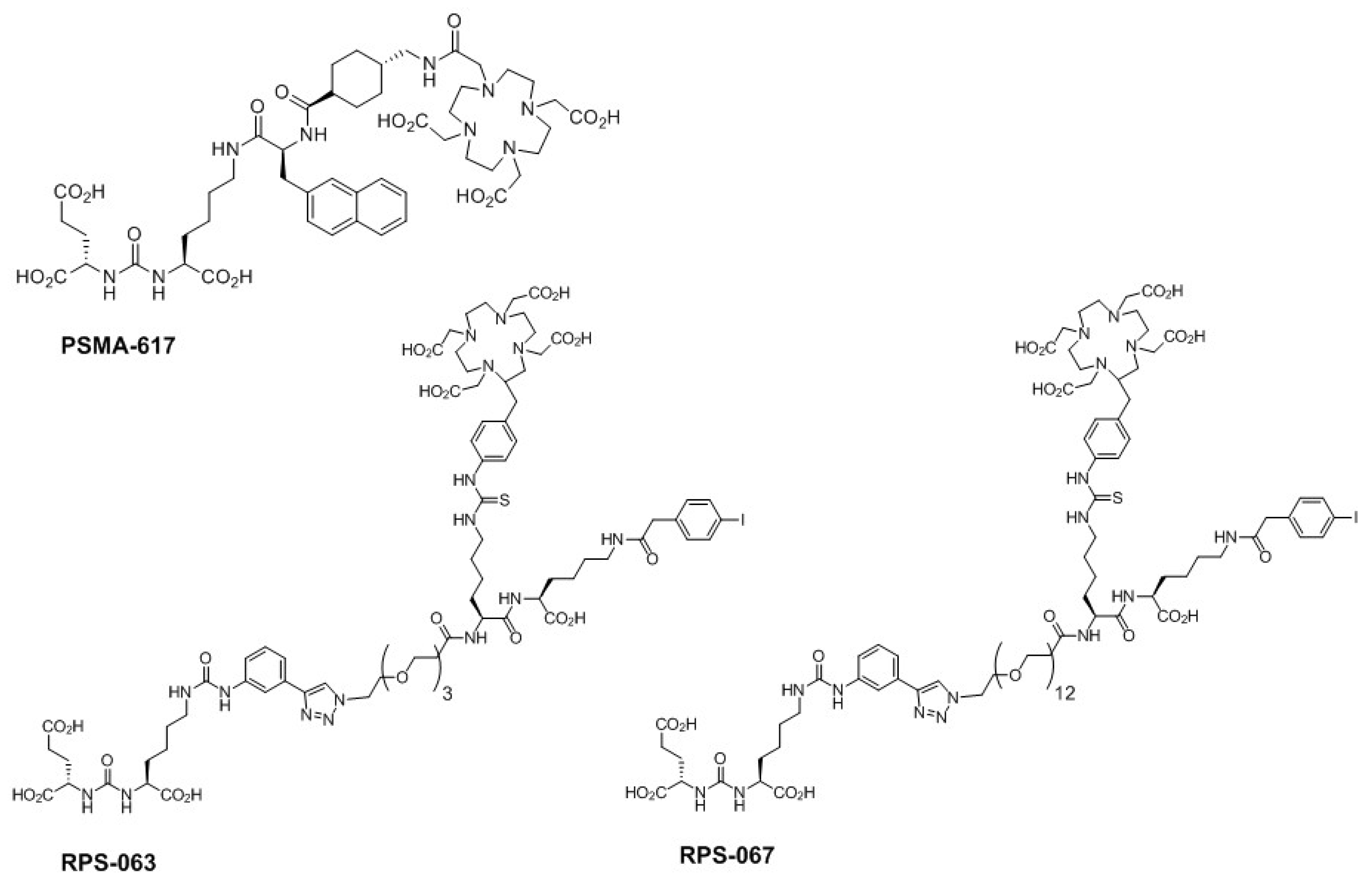
2.3. Radiolabeling of DOTA-Containing PSMA-Targeting Ligands with Purified 66Ga
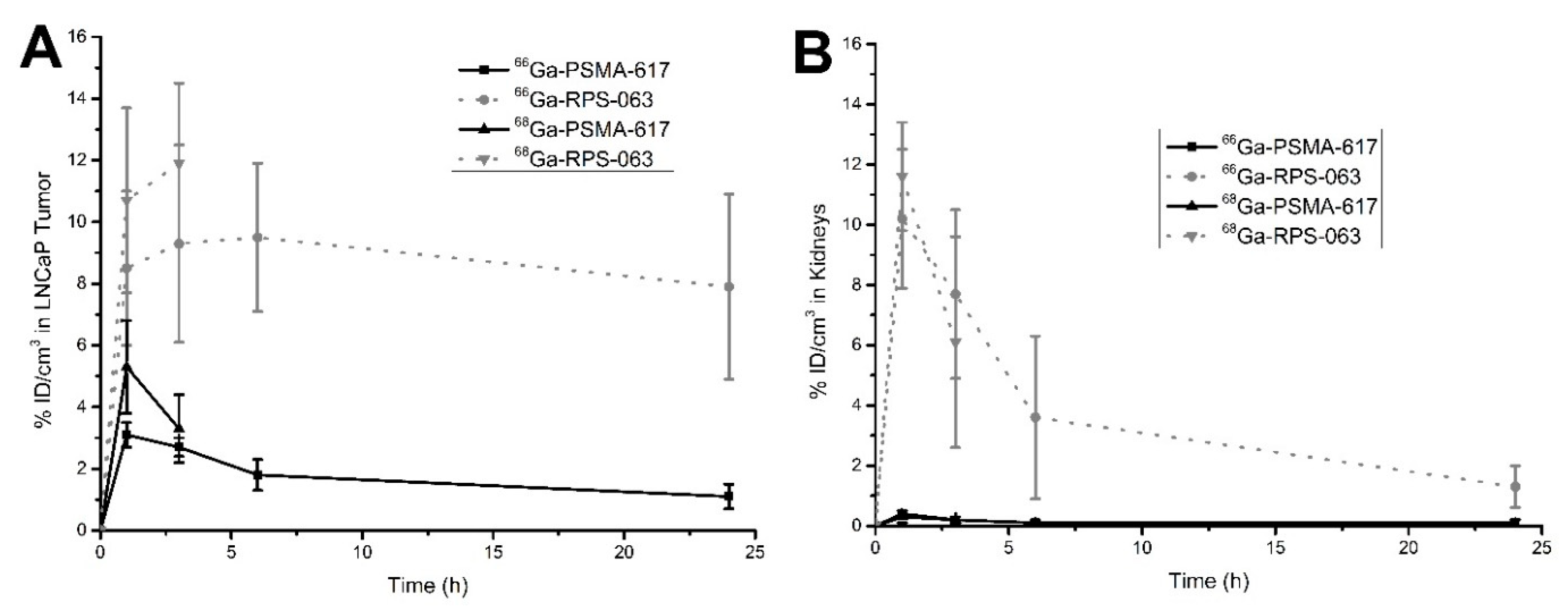
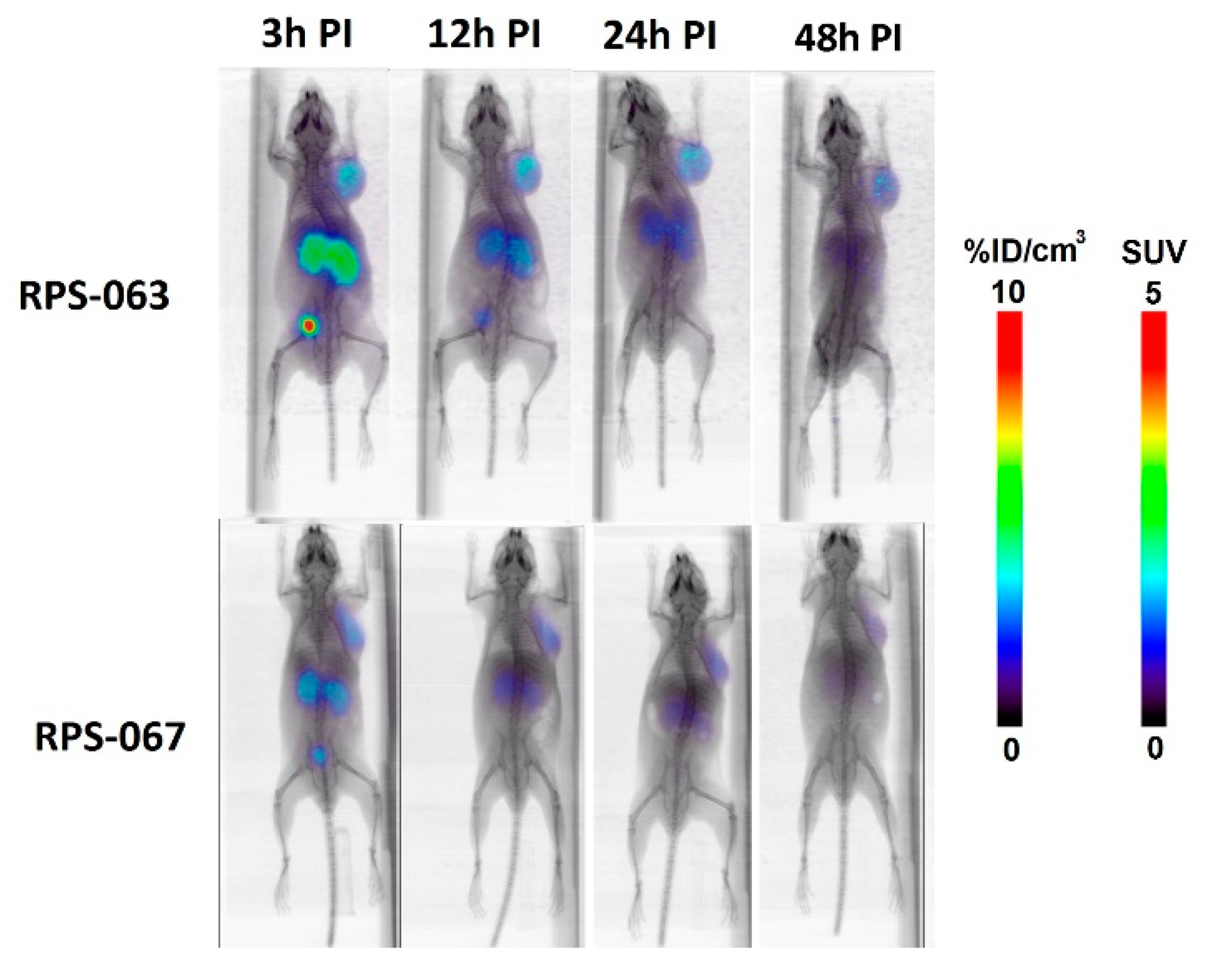
2.4. µPET/CT Imaging of [66/68Ga]RPS-063, [66Ga]RPS-067 and [66/68Ga]PSMA-617
3. Discussion
4. Materials and Methods
4.1. Synthesis of Precursors and Ligands
4.2. Radiochemistry
4.2.1. General Methods
4.2.2. Production of 66Ga
4.2.3. Radiolabeling of PSMA-617, RPS-063, and RPS-067
4.3. Sample Analysis by ICP-MS
4.4. µPET Imaging Studies in LNCaP Xenograft Tumor-Bearing Mice
4.4.1. Inoculation of Mice with Xenografts
4.4.2. PET Imaging Studies
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rudin, M.; Weissleder, R. Molecular Imaging in Drug Discovery and Development. Nat. Rev. Drug Discov. 2003, 2, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Czernin, J.; Weber, W.A.; Herschmann, H.R. Molecular Imaging in the Development of Cancer Therapeutics. Annu. Rev. Med. 2006, 57, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.R.; Pomper, M.G. Clinical Applications of Gallium-68. Appl. Radiat. Isot. 2013, 76, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Struthers, H.; Winiger, C.; Zhernosekov, K.; Schibli, R. DOTA Conjugate with an Albumin-Binding Entity Enables the First Folic Acid-Targeted 177Lu-Radionuclide Tumor Therapy in Mice. J. Nucl. Med. 2013, 54, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Choy, C.J.; Ling, X.; Geruntho, J.J.; Beyer, S.K.; Latoche, J.D.; Langton-Webster, B.; Anderson, C.J.; Berkman, C.E. 177Lu-Labeled Phosphoramidate-Based PSMA Inhibitors: The Effect of an Albumin Binder on Biodistribution and Therapeutic Efficacy in Prostate Tumor-Bearing Mice. Theranostics 2017, 7, 1928–1939. [Google Scholar] [CrossRef] [PubMed]
- Benešová, M.; Umbricht, C.A.; Schibli, R.; Müller, C. Albumin-Binding PSMA Ligands: Optimization of the Tissue Distribution Profile. Mol. Pharm. 2018, 15, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Umbricht, C.A.; Benešová, M.; Schibli, R.; Müller, C. Preclinical Development of Novel PSMA-Targeting Radioligands: Modulation of Albumin-Binding Properties to Improve Prostate Cancer Therapy. Mol. Pharm. 2018, 15, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jacobson, O.; Tian, R.; Mease, R.C.; Kiesewetter, D.O.; Niu, G.; Pomper, M.G.; Chen, X. Radioligand Therapy of Prostate Cancer with a Long-Lasting Prostate-Specific Membrane Antigen Targeting Agent 90Y-DOTA-EB-MCG. Bioconjug. Chem. 2018, 29, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.; Amor-Coarasa, A.; Ponnala, S.; Nikolopoulou, A.; Williams, C., Jr.; Schlyer, D.; Zhao, Y.; Kim, D.; Babich, J.W. Trifunctional PSMA-targeting constructs for prostate cancer with unprecedented localization to LNCaP tumors. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Börjesson, P.K.; Jauw, Y.W.; Boellaard, R.; de Bree, R.; Comans, E.F.; Roos, J.C.; Castelijns, J.A.; Vosjan, M.J.; Kummer, J.A.; Leemans, C.R.; et al. Performance of immune-positron emission tomography with zirconium-89-labeled chimeric monoclonal antibody U36 in the detection of lymph node metastases in head and neck cancer patients. Clin. Cancer Res. 2006, 12, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- Dijkers, E.C.; Oude Munnink, T.H.; Kosterink, J.G.; Brouwers, A.H.; Jager, P.L.; de Jong, J.R.; van Dongen, G.A.; Schröder, C.P.; Lub-de Hooge, M.N.; de Vries, E.G. Biodistribution of 89Zr-trastuzumab and PET imaging of HER2-positive lesions in patients with metastatic breast cancer. Clin. Pharmacol. Ther. 2010, 87, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Dehdashti, F.; Cutler, P.D.; Schwarz, S.W.; Laforest, R.; Bass, L.A.; Lewis, J.S.; McCarthy, D.W. 64Cu-TETA-octreotide as a PET imaging agent for patients with neuroendocrine tumors. J. Nucl. Med. 2001, 42, 213–221. [Google Scholar] [PubMed]
- Sevcenco, S.; Klingler, H.C.; Eredics, K.; Friedl, A.; Schneeweiss, J.; Knoll, P.; Kunit, T.; Lusuardi, L.; Mirzaei, S. Application of Cu-64 NODAGA-PSMA PET in Prostate Cancer. Adv. Ther. 2018, 35, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Kurihara, H.; Yonemori, K.; Tsuda, H.; Suzuki, J.; Kono, Y.; Honda, N.; Kodaira, M.; Yamamoto, H.; Yunokawa, M.; et al. 64Cu-DOTA-trastuzumab PET imaging in patients with HER2-positive breast cancer. J. Nucl. Med. 2013, 54, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.C.; Pinkston, K.L.; Robinson, H.; Harvey, B.R.; Wilganowski, N.; Gore, K.; Sevick-Muraca, E.M.; Azhdarinia, A. Comparison of DOTA and NODAGA as chelators for (64)Cu-labeled immunoconjugates. Nucl. Med. Biol. 2015, 42, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Kang, C.S.; Sin, I.; Ren, S.; Liu, D.; Ruthengael, V.C.; Lewis, M.R.; Chong, H.-S. Promising Bifunctional Chelators for Copper 64-PET Imaging: Practical 64Cu Radiolabeling and High In Vitro and In Vivo Complex Stability. J. Biol. Inorg. Chem. 2016, 21, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Rösch, F.; Herzog, H.; Stolz, B.; Brockmann, J.; Köhle, M.; Mühlensiepen, H.; Marbach, P.; Müller-Gärtner, H.W. Uptake kinetics of the somatostatin receptor ligand [86Y]DOTA-DPhe1-Tyr3-octreotide ([86Y]SMT487) using positron emission tomography in non-human primates and calculation of radiation doses of the 90Y-labelled analogue. Eur. J. Nucl. Med. 1999, 26, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Förster, G.J.; Engelbach, M.J.; Brockmann, J.J.; Reber, H.J.; Buchholz, H.G.; Mäcke, H.R.; Rösch, F.R.; Herzog, H.R.; Bartenstein, P.R. Preliminary Data on Biodistribution and Dosimetry for Therapy Planning of Somatostatin Receptor Positive Tumours: Comparison of (86)Y-DOTATOC and (111)In-DTPA-octreotide. Eur. J. Nucl. Med. 2001, 28, 1743–1750. [Google Scholar] [CrossRef] [PubMed]
- Biddlecombe, G.B.; Rogers, B.E.; de Visser, M.; Parry, J.J.; de Jong, M.; Erion, J.L.; Lewis, J.S. Molecular imaging of gastrin-releasing peptide receptor-positive tumors in mice using 64Cu and 86Y-DOTA-(Pro1, Tyr4)-bombesin (1-14). Bioconjug. Chem. 2007, 18, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.R.; Foss, C.A.; Pullambhatla, M.; Wang, Y.; Srinivasan, S.; Hobbs, R.F.; Baidoo, K.E.; Brechbiel, M.W.; Nimmagadda, S.; Mease, R.C.; et al. Preclinical evaluation of 86Y-labeled inhibitors of prostate-specific membrane antigen for dosimetry estimates. J. Nucl. Med. 2015, 56, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Avila-Rodriguez, M.A.; Nye, J.A.; Nickles, R.J. Production and separation of non-carrier-added 86Y from enriched 86Sr targets. Appl. Radiat. Isot. 2008, 66, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Lukić, D.; Tamburella, C.; Buchegger, F.; Beyer, G.-J.; Čomor, J.J.; Seimbille, Y. High efficiency production and purification of 86Y based on electrochemical separation. Appl. Radiat. Isot. 2009, 67, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Rösch, F.; Herzog, H.; Qaim, S.M. The Beginning and Development of the Theranostic Approach in Nuclear Medicine, as Exemplified by the Radionuclide Pair 86Y and 90Y. Pharmaceuticals (Basel) 2017, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Ehlerding, E.B.; Ferreira, C.A.; Aluicio-Sarduy, E.; Jiang, D.; Lee, H.J.; Theuer, C.P.; Engle, J.W.; Cai, W. 86/90Y-Based Theranostics Targeting Angiogenesis in a Murine Breast Cancer Model. Mol. Pharm. 2018, 15, 2606–2613. [Google Scholar] [CrossRef] [PubMed]
- Pruszyński, M.; Majkowska-Pilip, A.; Loktionova, N.S.; Eppard, E.; Roesch, F. Radiolabeling of DOTATOC with the long-lived positron emitter 44Sc. Appl. Radiat. Isot. 2012, 70, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, R.; Valdovinos, H.F.; Yang, Y.; Chakravarty, R.; Hong, H.; Barnhart, T.E.; Cai, W. 44Sc: An Attractive Isotope for Peptide-Based PET Imaging. Mol. Pharm. 2014, 11, 2954–2961. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Bunka, M.; Reber, J.; Fischer, C.; Zhernosekov, K.; Türler, A.; Schibli, R. Promises of Cyclotron-Produced 44Sc as a Diagnostic Match for Trivalent β--Emitters: In Vitro and In Vivo Study of a 44Sc-DOTA-Folate Conjugate. J. Nucl. Med. 2013, 54, 2168–2174. [Google Scholar] [CrossRef] [PubMed]
- Eppard, E.; de la Fuente, A.; Benešová, M.; Khawar, A.; Bundschuh, R.A.; Gärtner, F.C.; Kreppel, B.; Kopka, K.; Essler, M.; Rösch, F. Clinical Translation and First In-Human Use of [44Sc]Sc-PSMA-617 for PET Imaging of Metastasized Castrate-Resistant Prostate Cancer. Theranostics 2017, 7, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Zweit, J.; Sharma, H.; Downey, S. Production of Gallium-66, a Short-lived Positron Emitting Radionuclide. Appl. Radiat. Isot. 1987, 38, 499–501. [Google Scholar] [CrossRef]
- Lewis, M.R.; Reichert, D.E.; Laforest, R.; Margenau, W.H.; Shefer, R.E.; Klinkowstein, R.E.; Hughey, B.J.; Welch, M.J. Production and purification of gallium-66 for preparation of tumor-targeting radiopharmaceuticals. Nucl. Med. Biol. 2002, 29, 701–706. [Google Scholar] [CrossRef]
- Ugur, Ö.; Kothari, P.J.; Finn, R.D.; Zanzonico, P.; Ruan, S.; Guenther, I.; Maecke, H.R.; Larson, S.M. Ga-66 labeled somatostatin analogue DOTA-DPhe1-Tyr3-octreotide as a potential agent for positron emission tomography imaging and receptor mediated internal radiotherapy of somatostatin receptor positive tumors. Nucl. Med. Biol. 2002, 29, 147–157. [Google Scholar] [CrossRef]
- Engle, J.W.; Lopez-Rodriguez, V.; Gaspar-Carcamo, R.E.; Valdovinos, H.F.; Valle-Gonzalez, M.; Trejo-Ballado, F.; Severin, G.W.; Barnhart, T.E.; Nickles, R.J.; Avila-Rodriguez, M.A. Very high specific activity 66/68Ga from zinc targets for PET. Appl. Radiat. Isot. 2012, 70, 1792–1796. [Google Scholar] [CrossRef] [PubMed]
- Engle, J.W.; Hong, H.; Zhang, Y.; Valdovinos, H.F.; Myklejord, D.V.; Barnhart, T.E.; Theuer, C.P.; Nickles, R.J.; Cai, W. Positron emission tomography imaging of tumor angiogenesis with a 66Ga-labeled monoclonal antibody. Mol. Pharm. 2012, 9, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.C.; Pentlow, K.S.; Malawi, O.; Finn, R.D.; Daghighian, F.; Larson, S.M. An investigation of the physical characteristics of 66Ga as an isotope for PET imaging and quantification. Med. Phys. 1997, 24, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Sabet, M.; Rowshanfarzad, P.; Jalilian, M.R.; Rajamand, A.A. Production and quality control of 66Ga radionuclide. Nukleonika 2006, 51, 147–154. [Google Scholar]
- Amor-Coarasa, A.; Milera, A.; Carvajal, D.; Gulec, S.; McGoron, A.J. Lyophilized Kit for the Preparation of the PET Perfusion Agent [68Ga]-MAA. Int. J. Mol. Imaging 2014, 2014, 269365. [Google Scholar] [CrossRef] [PubMed]
- Benešová, M.; Schäfer, M.; Bauder-Wüst, U.; Afshar-Oromieh, A.; Kratochwil, C.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Preclinical Evaluation of a Tailor-Made DOTA-Conjugated PSMA Inhibitor with Optimized Linker Moiety for Imaging and Endoradiotherapy of Prostate Cancer. J. Nucl. Med. 2015, 56, 914–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Andrade Martins, P.; Osso, J.A., Jr. Thermal diffusion of 67Ga from irradiated Zn targets. Appl. Rad. Isot. 2013, 82, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Ometáková, J.; Rajec, P.; Csiba, V.; Leporis, M.; Štefečka, M.; Vlk, P.; Galamboš, M.; Rosskopfová, O. Automated production of 64Cu prepared by 18 MeV cyclotron. J. Radioanal. Nucl. Chem. 2012, 293, 217–222. [Google Scholar] [CrossRef]
- Wadas, T.J.; Wong, E.H.; Weisman, G.R.; Anderson, C.J. Coordinating Radiometals of Copper, Gallium, Indium, Yttrium, and Zirconium for PET and SPECT Imaging of Disease. Chem. Rev. 2010, 110, 2858–2902. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Beam Current (µA) | Irradiation Time (h) | 66Ga Activity at EOB (GBq) | % 66Ga at EOB | % 67Ga at EOB | % 68Ga at EOB | 66Ga Production Yield (MBq/µAh) |
|---|---|---|---|---|---|---|
| 20 (n = 3) | 2 | 8.6 ± 0.2 | 9.54 ± 0.22 | 0.24 ± 0.01 | 90.22 ± 0.22 | 215 ± 6 |
| 17 (n = 6) | 2 | 7.2 ± 1.1 | 8.06 ± 1.16 | 0.20 ± 0.02 | 91.74 ± 1.16 | 211 ± 33 |
| Metal | Al | Co | Cr | Cu |
|---|---|---|---|---|
| Column A | 21.95 ± 6.73 | <0.10 | 10.94 ± 1.10 | 52.92 ± 1.98 |
| Column B | 3.73 ± 4.76 | <0.10 | < 0.77 | 1.38 ± 1.77 |
| Detection Limit (ppb) | 6.67 | 0.10 | 0.77 | 0.37 |
| Metal | Fe | Mn | Ni | Zn |
| Column A | 57.70 ± 10.44 | <1.04 | 9.50 ± 0.96 | 322,300 ± 21,500 |
| Column B | 48.93 ± 14.33 | <1.04 | 6.18 ± 7.43 | 12,800 ± 6100 |
| Detection Limit (ppb) | 20.34 | 1.04 | 0.23 | 0.127 |
| Group | Isotope | Half Life (T1/2) | Max 𝛃+ Energy (MeV) | 𝛃+ Emission (%) | Target Material and Natural Abundance |
|---|---|---|---|---|---|
| A | 11C | 20.4 min | 0.960 | 99.8 | 14N (99.6%) |
| 13N | 10.0 min | 1.199 | 100 | 16O (99.76%) | |
| 15O | 2.07 min | 1.732 | 100 | 15N (0.4%) | |
| 18F | 109.4 min | 0.635 | 97.0 | 18O (0.2%) | |
| 68Ga | 68.2 min | 1.897 | 89.3 | Gen (68Ge), 68Zn (18.5%) | |
| B | 44Sc | 3.92 h | 1.470 | 94.3 | Gen (44Ti), 44Ca (2%) |
| 52Mn | 5.6 days | 0.575 | 29.6 | 52Cr (82%) | |
| 64Cu | 12.8 h | 0.656 | 17.4 | 64Ni (0.9%) | |
| 66Ga | 9.49 h | 4.153 | 56.5 | 66Zn (27.8%) | |
| 76Br | 16.2 h | 3.980 | 57.0 | 76Se (9.1%) | |
| 86Y | 14.74 h | 3.150 | 34.0 | 86Sr (9.9%) | |
| 89Zr | 3.27 d | 0.900 | 22.7 | 89Y (100%) | |
| 124I | 4.18 d | 2.130 | 25.0 | 124Te (4.8%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amor-Coarasa, A.; Kelly, J.M.; Ponnala, S.; Nikolopoulou, A.; Williams, C., Jr.; Babich, J.W. 66Ga: A Novelty or a Valuable Preclinical Screening Tool for the Design of Targeted Radiopharmaceuticals? Molecules 2018, 23, 2575. https://doi.org/10.3390/molecules23102575
Amor-Coarasa A, Kelly JM, Ponnala S, Nikolopoulou A, Williams C Jr., Babich JW. 66Ga: A Novelty or a Valuable Preclinical Screening Tool for the Design of Targeted Radiopharmaceuticals? Molecules. 2018; 23(10):2575. https://doi.org/10.3390/molecules23102575
Chicago/Turabian StyleAmor-Coarasa, Alejandro, James M. Kelly, Shashikanth Ponnala, Anastasia Nikolopoulou, Clarence Williams, Jr., and John W. Babich. 2018. "66Ga: A Novelty or a Valuable Preclinical Screening Tool for the Design of Targeted Radiopharmaceuticals?" Molecules 23, no. 10: 2575. https://doi.org/10.3390/molecules23102575