3. Experimental Section
3.1. General
Vinblastine, vincristine, and catharanthine were derived from the corresponding sulfate salts; the bases were released directly before using them in the reactions. m-Chloroperoxybenzoic acid (m-CPBA) of 77% assay was purchased from Sigma-Aldrich (Budapest, Hungary) and was used as received. Melting points were measured on a VEB Analytik Dresden PHMK-77/1328 apparatus (Dresden, Germany) and are uncorrected. IR spectra were recorded on the Zeiss IR 75 and 80 instruments (Thornwood, NY, USA). NMR measurements were performed on a Varian VNMRS 800 MHz NMR spectrometer equipped with a 1H{13C/15N} Triple Resonance 13C Enhanced Salt Tolerant Cold Probe, a Varian VNMRS 500 MHz NMR spectrometer equipped with a 1H {13C/15N} 5 mm PFG Triple Resonance 13C Enhanced Cold Probe (Varian, Inc., Palo Alto, CA, USA), and a Bruker Avance III HDX 500 MHz NMR spectrometer equipped with a 1H {13C/15N} 5 mm TCI CryoProbe (Bruker Corporation, Billerica, MA, USA). 1H And 13C chemical shifts are given on the delta scale as parts per million (ppm) with tetramethylsilane (TMS) (1H, 13C) or dimethylsulfoxide-d6 (13C) as the internal standard (0.00 ppm and 39.4 ppm, respectively). 1H-1H, direct 1H-13C, and long-range 1H-13C scalar spin-spin connectivites were established from 2D COSY, TOCSY, HSQC, and HMBC experiments. 1H-1H spatial proximities were determined using either two-dimensional NOESY/ROESY experiments or their selective 1D counterparts. All pulse sequences were applied by using the standard spectrometer software package. All experiments were performed at 298 K. NMR spectra were processed using VnmrJ 2.2 Revision C (Varian, Inc. Palo Alto, CA, USA), Bruker TopSpin 3.5 pl 6 (Bruker Corporation, Billerica, MA, USA) and ACD/Spectrus Processor version 2017.1.3 (Advanced Chemistry Development, Inc., Toronto, ON, Canada). HRMS and MS analyses were performed on an LTQ FT Ultra as well as an LTQ XL (Thermo Fisher Scientific, Bremen, Germany) system. The ionization method was ESI operated in the positive ion mode. For the CID (collision-induced dissociation) experiment, helium was used as the collision gas, and the normalized collision energy (expressed in percentage), which is a measure of the amplitude of the resonance excitation RF voltage applied to the endcaps of the linear ion trap, was used to induce fragmentation. The protonated molecular ion peaks were fragmented by CID at a normalized collision energy of 35–55%. The samples were dissolved in methanol. Data acquisition and analysis were accomplished with Xcalibur software version 2.0 (Thermo Fisher Scientific, Bremen, Germany). TLC was carried out using Kieselgel 60F254 (Merck, Budapest, Hungary) glass plates.
3.2. Bromocyclopropanation of Vindoline (4)
Vindoline (4) (228 mg, 0.50 mmol) was dissolved in dichloromethane (5 mL) and under Ar with 1.28 mL (1.28 mmol in 1 M hexane solution) of diethylzinc and then 88 μL (1.00 mmol) bromoform was injected to the solution. After stirring for 2 h at room temperature the reaction mixture was filtered and the filtrate was diluted with dichloromethane (50 mL) and washed with water (100 mL). The aqueous phase was extracted with dichloromethane (4 × 50 mL) and the combined organic phase was dried over magnesium sulfate, filtered, and the filtrate was evaporated to dryness. The crude product was separated by preparative TLC (dichloromethane-methanol 19:1) and 86 mg (31%) of product (8) was isolated. Mp 118–119 °C.
TLC (dichloromethane-methanol 20:1); Rf = 0.71.
IR (KBr) 2963, 1744, 1617, 1502, 1230, 1039 cm−1.
1H NMR (499.9 MHz, CDCl3) δ (ppm) 0.68 (t, J = 7.3 Hz, 3H, H3-18), 0.97 (dd, J = 9.7, 3.8 Hz, 1H, H-15), 1.00 (dq, J = 14.4, 7.3 Hz, 1H, Hx-19), 1.62 (dddd J = 9.7, 3.8, 3.5, 0.7 Hz, 1H, H-14), 1.91 (dq, J = 14.4, 7.3 Hz, 1H, Hy-19), 2.21 (s, 3H, C(17)-OC(O)CH3), 2.22–2.40 (m, 5H, H2-6, H-21, Hx-3, Hx-5), 2.63 (s, 3H, N(1)-CH3), 3.23–3.33 (br m, 1H, Hy-5), 3.44 (br d, J = 11.4 Hz, 1H, Hy-3), 3.52 (t, J = 3.8 Hz, 1H, H-22), 3.57 (s, 1H, H-2), 3.78 (s, 3H, C(11)-OCH3), 3.81 (s, 3H, C(16)-COOCH3), 5.53 (s, 1H, H-17), 6.07 (d, J = 2.2 Hz, 1H, H-12), 6.30 (dd, J = 8.2, 2.2 Hz, 1H, H-10), 6.84 (d, J = 8.2 Hz, 1H, H-9), 7.96 (br s, 1H, C(16)-OH).
13C NMR (125.7 MHz, CDCl3) δ (ppm) 8.2 (C-18), 21.6 (C(17)-OC(O)CH3), 23.3 (C-14), 23.5 (C-22), 28.0 (C-15), 34.4 (C-19), 38.7 (N(1)-CH3), 41.8 (C-20), 44.6 (C-6), 51.9 (C-3), 52.2 (C-7), 52.7 (C(16)-COOCH3), 53.5 (C-5), 55.6 (C(11)-OCH3), 69.6 (C-21), 76.3 (C-17), 78.9 (C-16), 83.7 (C-2), 95.9 (C-12), 105.1 (C-10), 122.7 (C-9), 125.6 (C-8), 153.7 (C-13), 161.2 (C-11), 171.2 (C(17)-OC(O)CH3), 171.9 (C(16)-COOCH3).
HRMS: M + H = 549.15965 (C26H34O6N2Br, Δ = 0.3 ppm). HR-ESI-MS-MS (CID = 35%, rel. int. %): 489(100); 457(1); 429(1); 314(1).
3.3. Bromocyclopropanation of 10-Bromovindoline (7)
The compound 10-Bromovindoline (7) (268 mg, 0.50 mmol) was dissolved in dichloromethane (5 mL) and under Ar with 1.28 mL (1.28 mmol in 1 M hexane solution) of diethylzinc and then 88 μL (1.00 mmol), the bromoform was injected to the solution. After stirring for 3 h at room temperature the reaction mixture was diluted with dichloromethane (40 mL) and washed with water (100 mL). The aqueous phase was extracted with dichloromethane (2 × 50 mL) and the combined organic phase was dried over magnesium sulfate, filtered and the filtrate was evaporated to dryness. The crude product was purified by preparative TLC (dichloromethane-methanol 30:1) and 126 mg (40%) of product (9) was isolated. Mp 144–146 °C.
TLC (dichloromethane-methanol 30:1); Rf = 0.54.
IR (KBr) 2965, 1743, 1604, 1498, 1232, 1041 cm−1.
1H NMR (799.7 MHz, CDCl3) δ (ppm) 0.73 (t, J = 7.3 Hz, 3H, H3-18), 0.96–1.01 (m, 2H, H-15, Hx-19), 1.66 (dt, J = 9.7, 3.5 Hz, 1H, H-14), 1.93 (dq, J = 14.4, 7.3 Hz, 1H, Hy-19), 2.22–2.33 (m, 7H, C(17)-OC(O)CH3, H-21, H2-6, Hx-3), 2.35–2.40 (m, 1H, Hx-5), 2.66 (s, 3H, N(1)-CH3), 3.26–3.38 (br m, 1H, Hy-5), 3.44–3.50 (br m, 1H, Hy-3), 3.51 (br t, J = 3.5 Hz, 1H, H-22), 3.59 (s, 1H, H-2), 3.83 (s, 3H, C(16)-COOCH3), 3.90 (s, 3H, C(11)-OCH3), 5.53 (s, 1H, H-17), 6.10 (s, 1H, H-12), 7.07 (s, 1H, H-9), 7.98 (br, 1H, C(16)-OH).
13C NMR (201.1 MHz, CDCl3) δ (ppm) 8.1 (C-18), 21.7 (C(17)-OC(O)CH3), 23.2 br (C-14), 23.3 br (C-22), 27.9 (C-15), 34.5 (C-19), 38.7 (N(1)-CH3), 41.8 (C-20), 44.4 (C-6), 51.9 (C-3), 52.1 (C-7), 52.5 (C(16)-COOCH3), 53.4 (C-5), 56.3 (C(11)-OCH3), 69.5 (C-21), 76.1 (C-17), 78.7 (C-16), 83.6 (C-2), 94.5 (C-12), 100.2 (C-10), 126.1 (C-9), 126.5 br (C-8), 152.9 (C-13), 156.8 (C-11), 171.1 (C(17)-OC(O)CH3), 171.7 (C(16)-COOCH3).
HRMS: M + H = 627.07035 (C26H33O6N2Br2, Δ = 0.6 ppm), HR-ESI-MS-MS (CID = 35%; rel. int. %): 567(100); 507(2).
3.4. Iodocyclopropanation of Vindoline (4)
Vindoline (4) (228 mg, 0.50 mmol) was dissolved in dichloromethane (20 mL) and under an Ar at 0 °C 1.28 mL (1.28 mmol in 1 M hexane solution) of diethylzinc, it was injected to the solution. Then 394 mg (1.00 mmol) of iodoform was added and the reaction mixture was stirred for 30 min at 0 °C and then for 6 h at room temperature. After allowing the solution to stand overnight, the addition of diethylzinc (1.28 mL) and iodoform (394 mg) was repeated at 0 °C. After stirring for 7 h at room temperature, the reaction mixture was filtered and the filtrate was diluted with dichloromethane (30 mL) and washed with water (100 mL). The aqueous phase was extracted with dichloromethane (5 × 60 mL) and the combined organic phase was dried over magnesium sulfate, filtered, and the filtrate was evaporated to dryness. The crude product was separated by preparative TLC (dichloromethane-methanol 19:1) and 28 mg (9%) of product (10) was isolated. Mp 116−117 °C.
TLC (dichloromethane-methanol 20:1); Rf = 0.75.
IR (KBr) 2963, 1742, 1615, 1502, 1231, 1039 cm−1.
1H NMR (499,9 MHz, CDCl3) δ (ppm) 0.68 (t, J = 7.3 Hz, 3H, H3-18), 0.93 (dd, J = 9.2, 4.3 Hz, 1H, H-15), 0.99 (dq, J = 14.3, 7.3 Hz, 1H, Hx-19), 1.52–1.57 (m, 1H, H-14), 1.89 (dq, J = 14.3, 7.3 Hz, 1H, Hy-19), 2.21–2.31 (m, 7H, Hx-3, H2-6, C(17)-OC(O)CH3, H-21), 2.32–2.41 (m, 1H, Hx-5), 2.63 (s, 3H, N(1)-CH3), 3.13 (dd, J = 4.3, 3.7 Hz, 1H, H-22), 3.23–3.35 (m, 1H, Hy-5), 3.42–3.50 (m, 1H, Hy-3), 3.56 (s, 1H, H-2), 3.78 (s, 3H, C(11)-OCH3), 3.81 (s, 3H, C(16)-COOCH3), 5.53 (s, 1H, H-17), 6.07 (d, J = 2.3 Hz, 1H, H-12), 6.30 (dd, J = 8.2, 2.3 Hz, 1H, H-10), 6.83 (d, J = 8.2 Hz, 1H, H-9), 8.03 (br, 1H, C(16)-OH).
13C NMR (125.7 MHz, CDCl3) δ (ppm) -9.5 (C-22), 8.1 (C-18), 22.4 (C(17)-OC(O)CH3), 24.3 (C-14), 29.0 (C-15), 34.5 (C-19), 38.6 (N(1)-CH3), 42.1 (C-20), 44.6 (C-6), 52.2 (C-3), 52.3 (C-7), 52.4 (C(16)-COOCH3), 53.3 (C-5), 55.4 (C(11)-OCH3), 69.7 (C-21), 76.5 (C-17), 78.9 (C-16), 83.8 (C-2), 95.9 (C-12), 105.0 (C-10), 122.5 (C-9), 125.5 (C-8), 153.6 (C-13), 161.2 (C-11), 171.2 (C(17)-OC(O)CH3), 171.9 (C(16)-COOCH3).
HRMS: M + H = 597.14664 (C26H34O6N2I, Δ = 1.7 ppm). HR-ESI-MS-MS (CID = 35%) (rel. int. %): 537(100); 505(1); 477(2); 441(1); 381(3); 362(2); 188(2).
3.5. Iodocyclopropanation of 10-Bromovindoline (7)
The compound 10-Bromovindoline (7) (268 mg, 0.50 mmol) was dissolved in dichloromethane (20 mL) and under Ar at 0 °C with 1.28 mL (1.28 mmol in 1 M hexane solution) of diethylzinc being injected into the solution. Then 394 mg (1.00 mmol) of iodoform was added and the reaction mixture was stirred for 30 min at 0 °C and then for 8 h at room temperature. After allowing the solution to stand overnight, the addition of diethylzinc (1.28 mL) and iodoform (394 mg) was repeated at 0 °C. After stirring for 6 h at room temperature, the reaction mixture was filtered and the filtrate was diluted with dichloromethane (30 mL) and washed with water (100 mL). The aqueous phase was extracted with dichloromethane (5 × 60 mL) and the combined organic phase was dried over magnesium sulfate, filtered and the filtrate was evaporated to dryness. The crude product was separated by preparative TLC (dichloromethane-methanol 19:1) and 74 mg (22%) of product (11) was isolated. Mp > 350 °C.
TLC (dichloromethane-methanol 20:1); Rf = 0.86.
IR (KBr) 3444, 2927, 1740, 1228, 742 cm−1.
1H NMR (499,9 MHz, CDCl3) δ (ppm) 0.71 (t, 3H, J = 7.3 Hz, H3-18), 0.93 (dd, J = 9.2, 4.3 Hz, 1H, H-15), 0.93–1.01 (m, 1H, Hx-19), 1.54–1.58 (m, 1H, H-14), 1.89 (dq, J = 14.6, 7.3 Hz, 1H, Hy-19), 2.22–2.27 (m, 4H, Hx-3, H2-6, H-21), 2.27 (s, 3H, C(17)-OC(O)CH3), 2.32–2.39 (m, 1H, Hx-5), 2.64 (s, 3H, N(1)-CH3), 3.10 (t, J = 4.3 Hz, 1H, H-22), 3.24–3.30 (m, 1H, Hy-5), 3.43–3.47 (m, 1H, Hy-3), 3.57 (s, 1H, H-2), 3.81 (s, 3H, C(16)-COOCH3), 3.88 (s, 3H, C(11)-OCH3), 5.51 (s, 1H, H-17), 6.09 (s, 1H, H-12), 7.05 (s, 1H, H-9), 8.01 (br, 1H, C(16)-OH).
13C NMR (125.7 MHz, CDCl3) δ (ppm) -9.7 (C-22), 8.1 (C-18), 22.4 (C(17)-OC(O)CH3), 24.2 (C-14), 28.9 (C-15), 34.7 (C-19), 38.7 (N(1)-CH3), 42.1 (C-20), 44.5 (C-6), 52.17 (C-3), 52.22 (C-7), 52.5 (C(16)-COOCH3), 53.1 (C-5), 56.3 (C(11)-OCH3), 69.6 (C-21), 76.3 (C-17), 78.7 (C-16), 83.8 (C-2), 94.5 (C-12), 100.1 (C-10), 126.2 (C-9), 126.4 (C-8), 152.8 (C-13), 156.7 (C-11), 171.2 (C(17)-OC(O)CH3), 171.7 (C(16)-COOCH3).
HRMS: M + H = 675.05411 (C26H33O6N2BrI, Δ = –2.9 ppm). ESI-MS-MS (CID = 35%) (rel. int. %): 615(100); 555(2); 459(1).
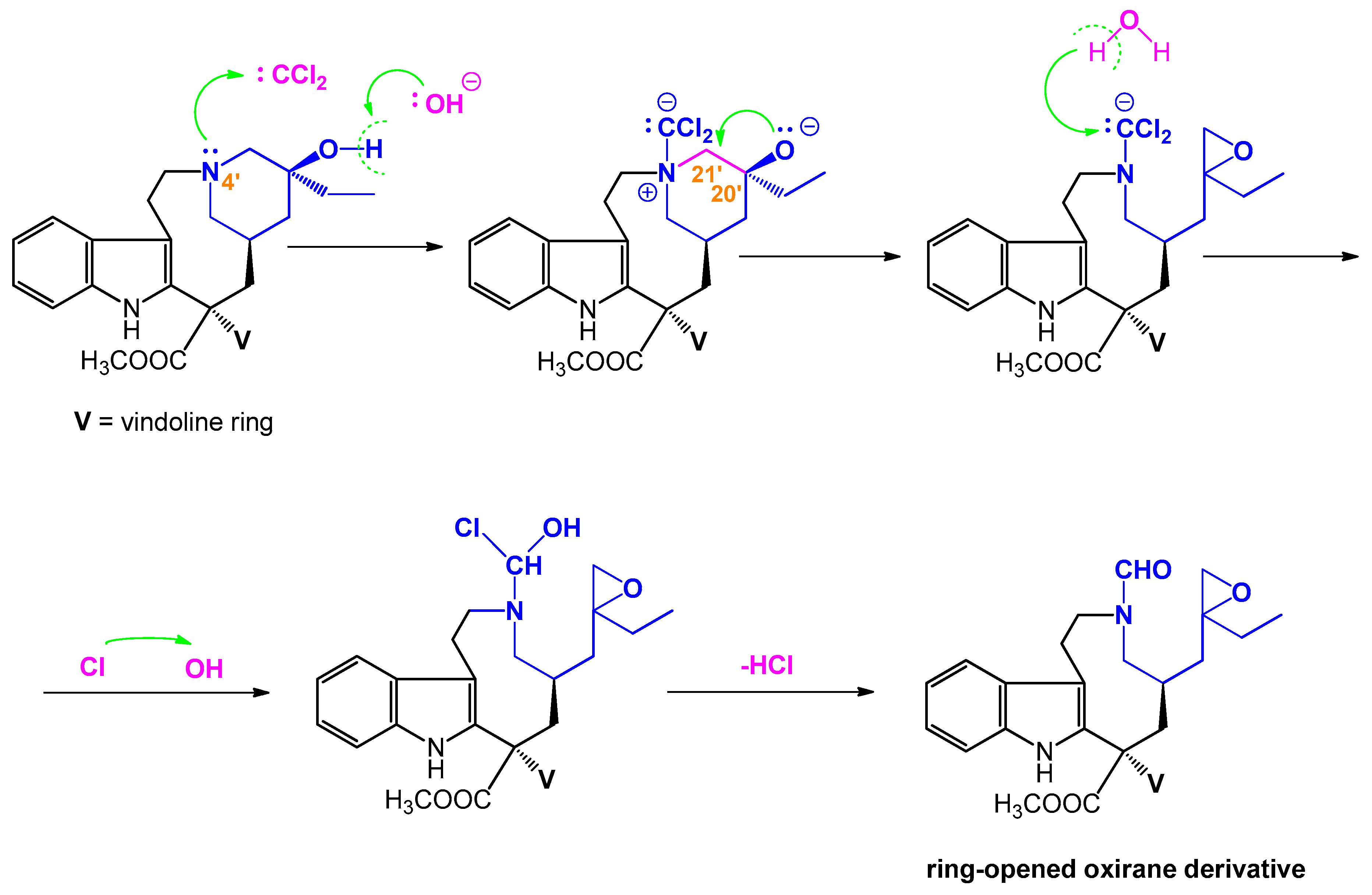
3.6. Attempted Dichlorocyclopropanation of Vindoline (4)
Vindoline (4) (256 mg, 0.56 mmol) and TEBAC (9 mg, 0.04 mmol) were dissolved in chloroform (1.4 mL), then a solution of sodium hydroxide (0.6 g, 15.00 mmol) in water (0.6 mL) was added dropwise to the reaction mixture, and stirred at room temperature for 2 h. The pH of the reaction mixture was adjusted to 7 using 1 M hydrochloric acid. Water was added and the mixture was extracted with chloroform. The combined organic phase was washed with water, dried over magnesium sulfate, and evaporated under reduced pressure. The residue was purified by preparative TLC (dichloromethane-methanol 20:1) and 24 mg (9%) of product (13) [20−22] was obtained.
3.7. Attempted Dichlorocyclopropanation of Vinblastine (1)
Vinblastine (1) (120 mg, 0.15 mmol) was dissolved in chloroform (0.4 mL), TEBAC (3 mg, 0.013 mmol) was added, and 200 mg (5.00 mmol) of NaOH in water (0.2 mL) was added dropwise. After stirring at room temperature for 2 h, the pH of the reaction mixture was neutralized using 1 M hydrochloric acid. Water (20 mL) was added and the mixture was extracted with chloroform (2 × 10 mL). The combined organic phase was dried over magnesium sulfate and evaporated under reduced pressure. The residue was purified by preparative TLC (dichloromethane-methanol 10:1) and 20 mg (16%) of product (15) was obtained. Mp 205−207 °C.
TLC (dichloromethane-methanol 10:1); Rf = 0.60.
IR (KBr) 3470, 2964, 1740, 1669, 1614, 1501, 1461, 1371, 1227, 1040, 742 cm−1.
1H NMR (799.7 MHz, CDCl3) δ (ppm) 0.39 (t, J = 7.5 Hz, 3H, H3-18’), 0.82 (t, J = 7.4 Hz, 3H, H3-18), 0.91 (dd, J = 13.8, 12.3 Hz, 1H, Hx-15’), 1.06 (dq, J = 14.7, 7.5 Hz, 1H, Hx-19’), 1.36 (dq, J = 14.4, 7.4 Hz, 1H, Hx-19), 1.46 (dd, J = 13.8, 3.4 Hz, 1H, Hy-15’), 1.66 (dq, J = 14.7, 7.5 Hz, 1H, Hy-19’), 1.83 (dq, J = 14.4, 7.4 Hz, 1H, Hy-19), 1.99–2.05 (m, 1H, H-14’), 2.11 (s, 3H, C(17)-OC(O)CH3), 2.14 (ddd, J = 14.0, 8.7, 7.4 Hz, 1H, Hx-6), 2.30 (br d, J = 16.5 Hz, 1H, Hx-17’), 2.43 (d, J = 4.9 Hz, 1H, Hx-21’), 2.44–2.47 (m, 1H, Hx-5), 2.48 (dd, J = 13.2, 11.2 Hz, 1H, Hx-3’), 2.55–2.56 (m, 1H, Hy-21’), 2.62–2.66 (m, 2H, H-21, Hy-6), 2.66 (s, 3H, N(1)-CH3), 2.79–2.83 (m, 1H, Hx-3), 3.01–3.09 (br m, 1H, Hy-17’), 3.15–3.20 (m, 1H, Hx-6’), 3.31–3.35 (m, 1H, Hy-5), 3.37–3.42 (m, 2H, Hx-5’, Hy-3), 3.50–3.61 (m, 2H, Hy-6’, Hy-5’) overlapped with 3.56 (br s, 3H, C(16’)-COOCH3), 3.73 (s, 1H, H-2), 3.80 (s, 3H, C(16)-COOCH3), 3.81 (s, 3H, C(11)-OCH3), 3.84–3.87 (m, 1H, Hy-3’), 5.28–5.31 (m, 1H, H-15), 5.52 (s, 1H, H-17), 5.87 (ddd, J = 10.4, 4.8, 0.8 Hz, 1H, H-14), 6.11 (s, 1H, H-12), 6.65 (s, 1H, H-9), 7.10–7.14 (m, 2H, H-10’, H-12’), 7.16–7.19 (m, 1H, H-11’), 7.35 (br s, 1H, N(4’)-CHO), 7.51–7.53 (m, 1H, H-9’), 7.95 (br, 1H, NH-1’), 9.85 (br, 1H, C(16)-OH).
13C NMR (201.1 MHz, CDCl3) δ (ppm) 8.4 (C-18’), 8.5 (C-18), 21.2 (C(17)-OC(O)CH3), 24.9 (C-19’), 25.1 (C-6’), 28.8 (br, C-14’), 30.9 (C-19), 38.5 (N(1)-CH3), 39.1 (C-17’), 42.6 (C-15’), 42.7 (C-20), 43.6 (C-6), 49.5 (C-5’), 50.5 (C-3), 51.37 (C-5), 51.40 (C-3’), 52.2 (C(16)-COOCH3), 52.4 (C(16’)-COOCH3), 53.3 (C-7), 54.5 (C-21’), 55.7 (C(11)-OCH3), 56.4 (br, C-16’), 58.7 (C-20’), 66.4 (C-21), 76.6 (C-17), 79.7 (C-16), 84.0 (C-2), 94.0 (br, C-12), 110.8 (C-12’), 111.1 (br, C-7’), 117.6 (C-9’), 119.3 (C-10’), 120.0 (br, C-10), 122.5 (C-11’), 123.8 (br, C-8), 124.6 (C-14), 125.3 (C-9), 128.3 (C-8’), 129.9 (C-15), 133.0 (br, C-2’), 135.3 (br, C-13’), 153.3 (C-13), 157.9 (C-11), 163.4 (N(4’)-CHO), 171.0 (C(17)-OC(O)CH3), 171.8 (C(16)-COOCH3), 174.4 (br, C(16’)-COOCH3).
HRMS: M + Na = 839.41764 (C45H60O10N4Na, Δ = –3.0 ppm). ESI-MS-MS (CID = 35%, rel. int. %): 821(12), 779(100), 761(4), 747(4), 677(7), 570(45).
3.8. Attempt to Dichlorocyclopropanation of Vincristine (2)
Vincristine (2) (130 mg, 0.16 mmol) was dissolved in chloroform (1 mL) and TEBAC (7 mg, 0.031 mmol) was added. Then 230 mg (5.75 mmol) of NaOH in water (0.23 mL) was added dropwise. After stirring at room temperature for 2 h, the pH of the reaction mixture was neutralized with 1 M hydrochloric acid. Water (10 mL) was added and the mixture was extracted with dichloromethane (2 × 10 mL). The combined organic phase was dried over magnesium sulfate and evaporated under reduced pressure. The residue was purified by preparative TLC (dichloromethane-methanol 10:1) and 49 mg (36%) of product (17) was obtained. Mp 233–235 °C (decomp.).
TLC (dichloromethane-methanol 10:1); Rf = 0.53.
IR (KBr) 3469, 1738, 1668, 1460, 1232, 744 cm−1.
NMR: two signal sets in a ratio of ca. 2:1 (conformational isomers due to hindered rotation of N-formyl group on the vindoline subunit of 2).
Major signal set:
1H NMR (499.9 MHz, CDCl3) δ (ppm) 0.35–0.41 (m, 3H, H3-18’), 0.77 (br t, J = 7.3 Hz, 3H, H3-18), 0.97–1.06 (m, 2H, Hx-19’, Hx-15’), 1.26–1.43 (m, 3H, Hx-19, Hy-19’, Hy-15’), 1.59–1.69 (m, 1H, Hy-19), 1.88–1.98 (br m, 1H, H-14’), 2.01–2.08 (br m, 1H, Hx-6) overlapped with 2.07 (br s, 3H, C(17)-OC(O)CH3), 2.33–2.44 (m, 2H, Hx-17’, Hx-21’), 2.45–2.54 (m, 2H, Hy-21’, Hx-3’), 2.54–2.70 (m, 2H, Hy-6, Hx-5), 2.84–2.93 (m, 2H, Hx-3, H-21), 3.00–3.10 (br m, 1H, Hy-17’), 3.17–3.22 (m, 1H, Hx-6’), 3.35–3.43 (m, 3H, Hy-3, Hy-5, Hx-5’), 3.46–3.53 (m, 1H, Hy-6’), 3.53–3.60 (m, 1H, Hy-5’), 3.62 (s, 3H, C(16’)-COOCH3), 3.73 (s, 3H, C(16)-COOCH3), 3.81 (br d, J = 13.7 Hz, 1H, Hy-3’), 3.89 (br s, 3H, C(11)-OCH3), 4.76 (br s, 1H, H-2), 5.25 (br s, 1H, H-17), 5.42–5.46 (m, 1H, H-15), 5.91–5.95 (m, 1H, H-14), 6.80 (br s, 1H, H-12), 6.93 (br s, 1H, H-9), 7.09–7.15 (m, 1H, H-10’), 7.15–7.22 (m, 2H, H-12’, H-11’), 7.36 (br s, 1H, N(4’)-CHO), 7.51–7.55 (m, 1H, H-9’), 7.94 (br, 1H, NH-1’), 8.74 (s, 1H, N(1)-CHO), 9.23 (br, 1H, C(16)-OH).
13C NMR (125.7 MHz, CDCl3) δ (ppm) 8.1 (C-18, C-18’), 21.1 (C(17)-OC(O)CH3), 24.9 (C-19’), 25.0 (C-6’), 28.7 (br, C-14’), 30.7 (C-19), 38.6 (br, C-17’), 40.3 (C-6), 42.3 (C-20), 42.5 (C-15’), 49.5 (C-5’), 49.7 (C-5), 49.9 (C-3), 51.6 (C-3’), 52.6 (C(16’)-COOCH3), 52.9 (C(16)-COOCH3), 53.5 (C-7), 53.8 (C-21’), 56.1 (C(11)-OCH3), 56.7 (br, C-16’), 58.4 (C-20’), 65.0 (C-21), 72.6 (C-2), 77.0 (C-17), 79.8 (C-16), 94.8 (br, C-12), 111.1 (C-12’), 111.9 (C-7’), 117.7 (C-9’), 119.6 (C-10’), 122.8 (C-11’), 124.6 (C-8), 125.0 (C-14), 126.6 (br, C-10), 126.9 (C-9), 128.3 (C-8’), 129.6 (C-15), 132.1 (C-2’), 135.6 (C-13’), 141.7 (C-13), 157.7 (C-11), 160.1 (N(1)-CHO), 163.4 (N(4’)-CHO), 170.5 (C(17)-OC(O)CH3), 170.7 (C(16)-COOCH3), 173.6 (br, C(16’)-COOCH3).
Minor signal set:
1H NMR (499.9 MHz, CDCl3) δ (ppm) 0.35–0.41 (m, 3H, H3-18’), 0.64–0.70 (m, 3H, H3-18), 0.97–1.06 (m, 2H, Hx-19’, Hx-15’), 1.26–1.43 (m, 3H, Hx-19, Hy-19’, Hy-15’), 1.59–1.69 (m, 1H, Hy-19), 1.88–1.98 (br m, 1H, H-14’), 2.01–2.08 (br m, 1H, Hx-6), 2.10 (br s, 3H, C(17)-OC(O)CH3), 2.33–2.44 (m, 2H, Hx-17’, Hx-21’), 2.45–2.54 (m, 2H, Hy-21’, Hx-3’), 2.54–2.70 (m, 2H, Hy-6, Hx-5), 2.84–2.90 (m, 1H, Hx-3), 2.95 (br s, 1H, H-21), 3.00–3.10 (br m, 1H, Hy-17’), 3.17–3.22 (m, 1H, Hx-6’), 3.35–3.43 (m, 3H, Hy-3, Hy-5, Hx-5’), 3.46–3.53 (m, 1H, Hy-6’), 3.53–3.60 (m, 1H, Hy-5’), 3.62 (s, 3H, C(16’)-COOCH3), 3.78 (s, 3H, C(16)-COOCH3), 3.81 (br d, J = 13.7 Hz, 1H, Hy-3’), 3.90 (br s, 3H, C(11)-OCH3), 4.52 (br s, 1H, H-2), 5.29 (br s, 1H, H-17), 5.42–5.46 (m, 1H, H-15), 5.92–5.96 (m, 1H, H-14), 6.86 (br s, 1H, H-9), 7.09–7.15 (m, 1H, H-10’), 7.15–7.22 (m, 2H, H-12’, H-11’), 7.36 (br s, 1H, N(4’)-CHO), 7.51–7.55 (m, 1H, H-9’), 7.80 (br s, 1H, H-12), 7.96 (br, 1H, NH-1’), 8.21 (s, 1H, N(1)-CHO), 9.23 (br, 1H, C(16)-OH).
13C NMR (125.7 MHz, CDCl3) δ (ppm) 7.9 (C-18), 8.1 (C-18’), 21.1 (C(17)-OC(O)CH3), 25.0 (C-19’, C-6’), 28.7 (br, C-14’), 30.7 (C-19), 38.6 (br, C-17’), 40.0 (C-6), 42.3 (C-20), 42.4 (C-15’), 49.6 (C-5, C-5’), 50.0 (C-3), 51.7 (C-3’), 52.6 (C(16)-COOCH3), 52.9 (C(16’)-COOCH3), 53.1 (C-7), 53.8 (C-21’), 56.1 (C(11)-OCH3), 56.7 (br, C-16’), 58.4 (C-20’), 64.3 (C-21), 74.4 (C-2), 76.0 (C-17), 81.5 (C-16), 101.7 (br, C-12), 111.1 (C-12’), 111.7 (C-7’), 117.6 (C-9’), 119.5 (C-10’), 122.7 (C-11’), 124.3 (C-8), 125.1 (C-14), 125.7 (C-9), 126.6 (br, C-10), 128.2 (C-8’), 129.6 (C-15), 132.5 (C-2’), 135.6 (C-13’), 141.6 (C-13), 157.2 (C-11), 160.5 (N(1)-CHO), 163.4 (N(4’)-CHO), 170.3 (C(16)-COOCH3), 170.4 (C(17)-OC(O)CH3), 173.7 (C(16’)-COOCH3).
HRMS: M + Na = 853.39557 (C45H58O11N4Na, Δ = –4.5 ppm). ESI-MS-MS (CID = 35%, rel. int. %): 835(19), 811(7), 793(100), 775(4), 733(18), 705(3).
3.9. Epoxidation of 10-Bromovindoline (7)
To a solution of 10-bromovindoline (7) (300 mg, 0.56 mmol) in methanol (5 mL) and 72% perchloric acid (0.17 mL), m-CPBA (306 mg, 1.37 mmol) in methanol (2 mL) was added dropwise at 0 °C, and the reaction mixture was stirred at reflux for 5 h. Methanol was evaporated, 10% aqueous sodium carbonate solution (20 mL) was added to the residue, and the mixture was extracted with dichloromethane (3 × 20 mL). The combined organic phase was dried over magnesium sulfate and evaporated under reduced pressure. The residue was purified by preparative TLC (dichloromethane-methanol 15:1) and 70 mg (27%) of product (19) was obtained. Mp. 365 °C (decomp.).
TLC (dichloromethane-methanol 20:1); Rf = 0.30.
IR (KBr) 3445, 1745, 1677, 1589, 1414, 1254 cm−1.
1H NMR (499.9 MHz, CDCl3) δ (ppm) 0.71 (t, J = 7.4 Hz, 3H, H3-18), 1.47 (dq, J = 14.8, 7.4 Hz, 1H, Hx-19), 1.75 (dq, J = 14.8, 7.4 Hz, 1H, Hy-19), 2.08 (s, 3H, C(17)-OC(O)CH3), 2.22 (ddd, J = 13.6, 11.2, 5.0 Hz, 1H, Hx-6), 2.46 (ddd, J = 13.6, 9.0, 6.2 Hz, 1H, Hy-6), 2.74 (ddd, J = 11.2, 9.8, 6.2 Hz, 1H, Hx-5), 2.92–2.98 (m, 5H, H-21, N(1)-CH3, Hx-3), 3.48–3.54 (m, 2H, Hy-5, Hy-3), 3.82 (s, 3H, C(16)-COOCH3), 4.18 (s, 1H, H-2), 5.16 (s, 1H, H-17), 5.35 (br d, J = 10.2 Hz, 1H, H-15), 5.51 (s, 1H, H-12), 5.96 (ddd, J = 10.2, 5,1, 1.3 Hz, 1H, H-14), 6.38 (s, 1H, H-9).
13C NMR (125.7 MHz, CDCl3) δ (ppm) 7.5 (C-18), 20.8 (C(17)-OC(O)CH3), 31.1 (C-19), 35.3 (N(1)-CH3), 42.2 (C-6), 42.8 (C-20), 50.3 (C-3), 50.7 (C-5), 51.5 (C-7), 52.9 (C(16)-COOCH3), 66.8 (C-21), 74.7 (C-17), 79.2 (C-16), 82.1 (C-2), 94.7 (C-12), 123.8 (C-9), 124.7 (C-14), 129.4 (C-15), 157.6 (C-8), 157.9 (C-13), 170.1 (C(17)-OC(O)CH3), 170.6 (C(16)-COOCH3), 174.6 (C-11), 180.7 (C-10).
HRMS: M + H = 457.19636 (C24H29O7N2, Δ = –1.2 ppm). ESI-MS-MS (CID = 45%, rel. int. %): 439(23), 429(34), 415(34), 411(3), 397(100), 379(10), 369(9), 368(10), 347(6), 337(27), 295(15), 290(7), 190(7).
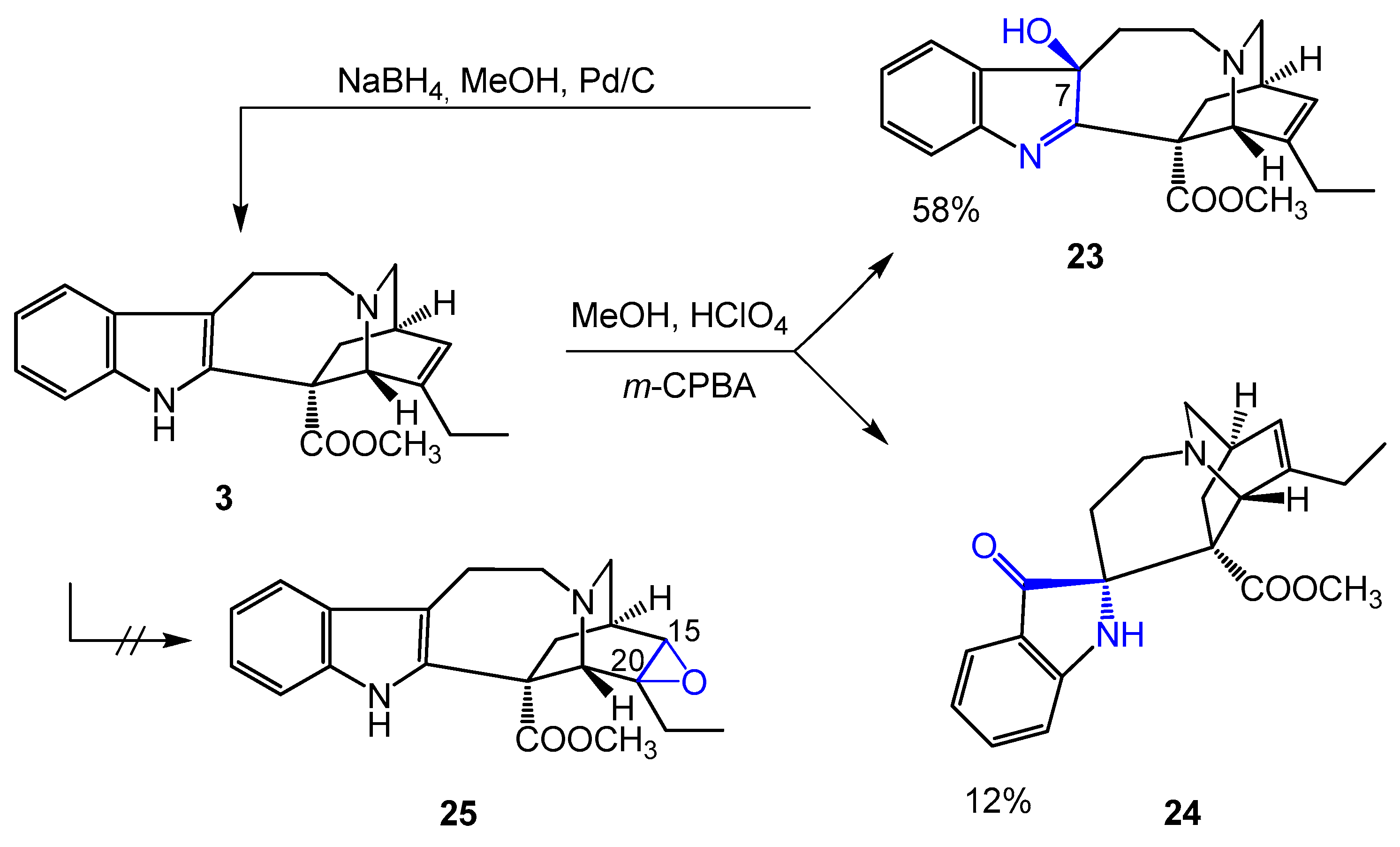
3.10. Epoxidation of Catharanthine (3)
Method A. Without perchloric acid. To a solution of catharanthine (3) (160 mg, 0.48 mmol) in dry methanol (10 mL), m-CPBA (184 mg, 0.82 mmol) was added, and the reaction mixture was stirred at room temperature for 1 h. The reaction mixture was diluted with 10% aqueous sodium carbonate (10 mL) and extracted with dichloromethane (3 × 10 mL). The combined organic phase was dried over magnesium sulfate and evaporated under reduced pressure. The residue was purified by preparative TLC (dichloromethane-methanol 4:1) and products 20 (57 mg, 34%) and 21 (62 mg, 35%) were isolated. N-oxide 20 is prone to rearrangement and transforms into isoxazolidine 22, which is a stable compound.
20 and 22; mp 116–117 °C. TLC (dichloromethane-methanol 4:1); Rf (N-oxide form (20)) = 0.76 and Rf (neutral form (22)) = 0.90.
IR (KBr) 3378, 3185, 2960, 1735, 1460, 1435, 1237, 744 cm−1.
NMR: (chemical shifts might vary slightly with concentration, pH and the exact ratio of 20 and 22)
N-oxide 20:
1H NMR (499.9 MHz, CDCl3) δ (ppm) 1.04 (t, J = 7.4 Hz, 3H, H3-18), 1.58–1.62 (m, 1H, Hx-17), 2.12–2.19 (m, 1H, Hx-19), 2.44–2.50 (m, 1H, Hy-19), 2.83–2.88 (m, 1H, Hy-17), 2.92–2.96 (m, 1H, H-14), 2.96–3.02 (m, 1H, Hx-6), 3.40–3.48 (m, 2H, Hx-3, Hy-6), 3.67 (s, 3H, C(16)-COOCH3), 3.74–3.79 (m, 1H, Hy-3), 3.92 (ddd, J = 13.2, 8.3, 1.6 Hz, 1H, Hx-5), 4.27–4.34 (m, 1H, Hy-5), 4.72–4.74 (m, 1H, H-21), 6.07–6.10 (m, 1H, H-15), 7.06–7.09 (m, 1H, H-10), 7.11–7.15 (m, 1H, H-11), 7.21–7.24 (m, 1H, H-12), 7.41–7.43 (m, 1H, H-9), 7.88 (br s, 1H, NH-1).
13C NMR (125.7 MHz, CDCl3) δ (ppm) 10.4 (C-18), 19.8 (C-6), 28.3 (C-19), 30.0 (C-14), 32.3 (C-17), 51.0 (C-16), 53.2 (C(16)-COOCH3), 73.5 (C-3), 74.4 (C-21), 76.9 (C-5), 111.1 (C-12), 111.7 (C-7), 118.2 (C-9), 120.3 (C-10), 122.8 (C-11), 124.3 (C-15), 127.4 (C-8), 133.7 (C-2), 134.9 (C-13), 145.9 (C-20), 171.5 (C(16)-COOCH3).
Isoxazolidine 22:
1H NMR (499.9 MHz, CDCl3) δ (ppm) 1.14 (t, J = 7.5 Hz, 3H, H3-18), 1.98 (dd, J = 14.2, 6.1 Hz, 1H, Hx-17), 2.18–2.26 (m, 1H, Hx-19), 2.26–2.30 (m, 1H, Hy-17), 2.36–2.45 (m, 1H, Hy-19), 2.58 (dd, J = 10.6, 5.2 Hz, 1H, Hx-3), 2.75–2.80 (m, 1H, Hx-6), 2.94–3.01 (m, 1H, Hx-5), 3.13–3.20 (m, 1H, H-14), 3.29–3.37 (m, 3H, Hy-3, Hy-5, Hy-6), 3.77 (s, 3H, C(16)-COOCH3), 4.53 (d, J = 10.0 Hz, 1H, H-15), 6.19–6.20 (m, 1H, H-21), 7.00–7.04 (m, 1H, H-10), 7.10–7.13 (m, 1H, H-11), 7.24–7.26 (m, 1H, H-12), 7.39–7.42 (m, 1H, H-9), 8.67 (br s, 1H, NH-1).
13C NMR (125.7 MHz, CDCl3) δ (ppm) 11.3 (C-18), 23.7 (C-6), 27.4 (C-19), 32.5 (C-17), 39.9 (C-14), 46.7 (C-16), 53.1 (C(16)-COOCH3), 55.5 (C-3), 55.8 (C-5), 76.6 (C-15), 110.8 (C-12), 113.6 (C-7), 118.4 (C-9), 119.5 (C-10), 121.9 (C-21), 122.7 (C-11), 127.9 (C-2), 128.0 (C-8), 135.2 (C-13), 141.5 (C-20), 174.9 (C(16)-COOCH3).
HRMS:
N-oxide 20:
M+H = 353.18559 (C21H25O3N2, Δ = –1.1 ppm). ESI-MS-MS (CID = 45%, rel. int. %): 336(100), 321(7), 303(6), 294(4), 293(3), 275(2), 266(6), 229(2), 189(2), 171(2), 144(12).
Isoxazolidine 22:
M+H = 353.18567 (C21H25O3N2, Δ = –0.9 ppm). ESI-MS-MS (CID = 45%, rel. int. %): 335(100), 321(36), 303(36), 294(22), 293(15), 275(11), 266(38), 171(5), 144(4).
21; mp 146–147 °C. TLC (dichloromethane-methanol 4:1); Rf = 0.52.
IR (KBr) 3581, 3365, 2959, 1744, 1570, 1242, 774 cm−1.
1H NMR (499.9 MHz, DMSO-d6:CDCl3 = 1:1 + 5 v/v% CF3COOH) δ (ppm) 1.07 (t, J = 7.4 Hz, 3H, H3-18), 1.99–2.11 (m, 2H, Hx-17, Hx-19), 2.16–2.25 (m, 1H, Hx-6), 2.25–2.34 (m, 1H, Hy-19), 2.49–2.56 (m, 1H, Hy-6), 2.87–2.93 (m, 1H, Hy-17), 3.11–3.15 (m, 1H, H-14), 3.34–3.40 (m, 1H, Hx-3), 3.67 (s, 3H, C(16)-COOCH3), 3.90–3.95 (m, 1H, Hy-3), 4.14–4.29 (m, 1H, Hx-5), 4.54–4.63 (m, 1H, Hy-5), 5.59 (d, J = 1.4 Hz, 1H, H-21), 6.24–6.27 (m, 1H, H-15), 7.28–7.32 (m, 1H, H-10), 7.36–7.40 (m, 1H, H-11), 7.44–7.46 (m, 1H, H-12), 7.47–7.49 (m, 1H, H-9).
13C NMR (125.7 MHz, DMSO-d6:CDCl3 = 1:1 + 5 v/v% CF3COOH) δ (ppm) 10.2 (C-18), 26.9 (C-19), 29.0 (C-14), 30.0 (C-6), 34.6 (C-17), 52.3 (C-16), 53.3 (C(16)-COOCH3), 64.3 (br, C-3), 65.2 (br, C-5), 70.8 (C-21), 82.8 (C-7), 120.6 (C-12), 122.5 (C-9), 125.9 (C-15), 126.9 (C-10), 129.7 (C-11), 140.2 (br, C-8), 141.4 (C-20), 152.0 (C-13), 168.1 (C(16)-COOCH3), 184.0 (C-2).
HRMS: M + H = 369.18063 (C21H25O4N2, Δ = –0.7 ppm). ESI-MS-MS (CID = 45%, rel. int. %): 351(16), 335(9), 324(29), 307(100), 292(5), 264(14), 205(3), 187(4), 160(6).
Method B. Using perchloric acid. To a solution of catharanthine (3) (247 mg, 0.73 mmol) in methanol (10 mL) and 72% perchloric acid (0.10 mL), m-CPBA (184 mg, 0.82 mmol) was added at 0 °C, and the reaction mixture was stirred at room temperature for 18 h. The reaction mixture was diluted with 10% aqueous sodium carbonate (10 mL) and extracted with dichloromethane (3x10 mL). The combined organic phase was dried over magnesium sulfate and evaporated under reduced pressure. The residue was purified by preparative TLC (dichloromethane-methanol 10:1) and products 23 (149 mg, 58%) and 24 (30 mg, 12%) were isolated.
Compound 23; mp 87–89 °C. TLC (dichloromethane-methanol 10:1); Rf = 0.55.
IR (KBr) 3436, 2960, 1741, 1459, 1222, 1078, 757 cm−1.
1H NMR (499.9 MHz, DMSO-d6) δ (ppm) 0.96 (t, J = 7.4 Hz, 3H, H3-18), 1.73 (dd, J = 13.2, 2.3 Hz, 1H, Hx-17), 1.75–1.83 (m, 1H, Hx-6), 1.87–1.96 (m, 2H, Hy-6, Hx-19), 2.07 (dqd, J = 16.1, 7.4, 2.0 Hz, Hy-19), 2.52 (dt, J = 8.3, 2.6 Hz, 1H, Hx-3), 2.55–2.58 (br m, 1H, H-14), 2.59 (br d, J = 8.3 Hz, 1H, Hy-3), 2.73–2.78 (m, 1H, Hy-17), 2.79–2.84 (m, 1H, Hx-5), 3.50 (s, 3H, C(16)-COOCH3), 3.61 (ddd, J = 14.3, 12.4, 2.3 Hz, 1H, Hy-5), 4.57 (~d, J = 1.1 Hz, 1H, H-21), 5.75–5.77 (m, 1H, H-15), 6.01 (d, J = 0.9 Hz, 1H, C(7)-OH), 7.18–7.22 (m, 1H, H-10), 7.28–7.32 (m, 1H, H-11), 7.33–7.36 (m, 1H, H-12), 7.36–7,39 (m, 1H, H-9).
13C NMR (125.7 MHz, DMSO-d6) δ (ppm) 11.2 (C-18), 25.9 (C-19), 30.7 (C-14), 32.9 (C-6), 39.2 (C-17), 46.7 (C-5), 47.6 (C-3), 52.0 (C(16)-COOCH3), 57.4 (C-16), 57.8 (C-21), 87.1 (C-7), 119.9 (C-12), 121.9 (C-9), 122.4 (C-15), 126.1 (C-10), 128.8 (C-11), 143.0 (C-8), 148.1 (C-20), 152.4 (C-13), 171.6 (C(16)-COOCH3), 190.3 (C-2).
HRMS: M + H = 353.18553 (C21H25O3N2, Δ = –1.2 ppm). ESI-MS-MS (CID = 35%, rel. int. %): 335(80), 321(100), 303(3), 189(4), 171(8).
Compound 24; mp 99–101 °C (decomp.). TLC (dichloromethane-methanol 10:1); Rf = 0.13.
IR (KBr) 1730, 1693, 1618, 755 cm−1.
1H NMR (499.9 MHz, DMSO-d6:CDCl3 = 2:1 (v/v)) δ (ppm) 1.03 (t, J = 7.4 Hz, 3H, H-18), 1.71 (br d, J = 14.8 Hz, 1H, Hx-6), 1.95 (br d, J = 13.3 Hz, 1H, Hx-17), 2.06 (dqd, J = 16.4, 7.4, 1.6 Hz, Hx-19), 2.30–2.43 (m, 3H, Hy-19, Hy-6, Hy-17), 2.83 (br d, 1H, J = 10.8 Hz, Hx-3), 2.90–2.94 (m, 1H, H-14), 3.03–3.08 (m, 2H, Hy-3, Hx-5), 3.24 (s, 3H, C(16)-COOCH3), 4.10 (td, J = 12.9, 3.2 Hz, 1H, Hy-5), 4.92 (br s, 1H, H-21), 6.17 (br d, J = 6.4 Hz, 1H, H-15), 6.55–6.64 (br, 1H, NH-1), 6.72–6.77 (m, 1H, H-10), 6.79–6.82 (m, 1H, H-12), 7.37–7.42 (m, 1H, H-11), 7.49–7.52 (m, 1H, H-9).
13C NMR (125.7 MHz, DMSO-d6:CDCl3 = 2:1 (v/v)) δ (ppm) 10.7 (C-18), 23.4 (C-6), 26.5 (C-19), 28.9 (C-14), 30.3 (C-17), 44.7 (C-5), 50.3 (C-3), 51.9 (C-16), 52.0 (C(16)-COOCH3), 54.0 (C-21), 65.0 (C-2), 112.1 (C-12), 118.5 (C-10), 119.4 (C-8), 124.2 (C-9), 128.0 (C-15), 137.3 (C-11), 144.2 (C-20), 159.9 (C-13), 171.2 (C(16)-COOCH3), 202.2 (C-7).
HRMS: M + H = 353.18512 (C21H25O3N2, Δ = –2.4 ppm). ESI-MS-MS (CID = 45%, rel. int. %): 335(3), 321(2), 276(2), 189(100), 172(6), 160(38), 146(23).
3.11. Reduction of the 7-Hydroxy Derivative of Catharanthine (23)
To a solution of 7-hydroxyindolenine catharanthine (23) (300 mg, 0.85 mmol) in methanol (20 mL), 700 mg of 10% Pd/C and NaBH4 (483 mg, 12.77 mmol) was added at 10 °C. The reaction mixture was stirred under an argon atmosphere for 30 min. After filtering the catalyst, a few drops of acetic acid was added, the filtrate was diluted with dichloromethane (50 mL) and washed with 10% aqueous sodium carbonate. The aqueous phase was extracted with dichloromethane (2 × 20 mL), the combined organic phase was washed with water (50 mL), dried over magnesium sulfate and evaporated under reduced pressure. The residue was purified by preparative TLC (dichloromethane-methanol 9:1) and catharanthine (3) was obtained (111 mg, 39%).
3.12. Coupling of the Catharanthine Derivative (24) with Vindoline (4)
Compound 24 (210 mg, 0.60 mmol) and vindoline (271 mg, 0.59 mmol) (4) were added to a mixture that consisted of water (21.3 mL), 1 M hydrochloric acid (1.1 mL) and 2,2,2-trifluoroethanol (2.2 mL). Under an argon atmosphere, FeCl3·6 H2O (802 mg, 2.97 mmol) was added. The reaction mixture was stirred at room temperature for 2 h. At 0 °C, sodium borohydride (24 mg, 0.63 mmol) in water (1.9 mL) was added dropwise. After 30 min of stirring, the pH was adjusted to 8 with cc. ammonium hydroxide. The reaction mixture was extracted with dichloromethane (2 × 60 mL), the combined organic phase was washed with water (100 mL), dried over magnesium sulfate and evaporated under reduced pressure. The residue was purified by preparative TLC (dichloromethane-methanol 6:1). A cationic vindoline trimer (27) (18 mg, 7%), a vindoline trimer ketone (28) (26 mg, 10%), and the N-methyl-spiro derivative of catharanthine (29) (24 mg, 11%) were obtained.
Compound 27:
TLC (dichloromethane-methanol 7:1), Rf = 0.27.
NMR: two signal sets in a ratio of ca. 3:2 (conformational isomers). Chemical shifts vary slightly with pH, temperature, and the composition of the NMR solvent. The three vindoline subunits of 27 are denoted as A, B, and B’. The two subunits that have the same constitution (vindoline-10-yl) are called B and B’.
Major signal set:
1H NMR (799.7 MHz, CD3OD:CD3CN:D2O = 1:1:1 (v/v)) δ (ppm) 0.35 (t, J = 7.4 Hz, 3H, H3-18B), 0.52 (t, J = 7.4 Hz, 3H, H3-18B’), 0.57 (t, J = 7.4 Hz, 3H, H3-18A), 1.15–1.20 (m, 1H, Hx-19B’), 1.25–1.30 (m, 1H, Hx-19B), 1.47–1.53 (m, 1H, Hx-19A), 1.53–1.58 (m, 1H, Hy-19A), 1.55–1.61 (m, 1H, Hy-19B’), 1.58–1.63 (m, 1H, Hy-19B), 1.62–1.67 (m, 1H, Hx-6A), 2.02 (s, 6H, C(17B)-OC(O)CH3, C(17B’)-OC(O)CH3), 2.06 (s, 3H, C(17A)-OC(O)CH3), 2.15–2.20 (m, 1H, Hx-6B’), 2.20–2.24 (m, 2H, Hy-6A, Hy-6B’), 2.26–2.30 (m, 2H, Hx-6B, H-21B), 2.32–2.35 (m, 1H, Hx-5B), 2.37–2.41 (m, 1H, Hy-6B), 2.54–2.58 (m, 1H, Hx-5B’), 2.64 (s, 3H, N(1B)-CH3), 2.65 (s, 1H, H-21B’), 2.67–2.71 (m, 1H, Hx-5A), 2.71 (s, 3H, N(1B’)-CH3), 2.73–2.77 (m, 1H, Hx-3B), 2.85–2.89 (m, 1H, Hx-3B’), 2.94–2.98 (m, 1H, Hx-3A), 3.12 (s, 1H, H-21A), 3.28 (s, 3H, N(1A)-CH3), 3.30–3.34 (m, 1H, Hy-5A), 3.37–3.43 (m, 3H, Hy-5B’, Hy-5B, Hy-3A), 3.45–3.53 (m, 2H, Hy-3B’, Hy-3B), 3.66 (s, 1H, H-2B), 3.69 (s, 1H, H-2B’), 3.73 (s, 3H, C(11B’)-OCH3), 3.74 (s, 3H, C(11B)-OCH3), 3.78 (s, 3H, C(16B’)-COOCH3), 3.79 (s, 3H, C(16B)-COOCH3), 3.83 (s, 3H, C(16A)-COOCH3), 3.97 (s, 3H, C(11A)-OCH3), 4.39 (s, 1H, H-2A), 5.08 (s, 1H, H-17A), 5.08–5.11 (m, 1H, H-15B), 5.19–5.22 (m, 1H, H-15B’), 5.32 (s, 1H, H-17B’), 5.38–5.41 (s, 1H, H-15A), 5.43 (s, 1H, H-17B), 5.79–5.82 (m, 1H, H-14B), 5.87–5.90 (m, 1H, H-14B’), 5.94–5.97 (m, 1H, H-14A), 6.27 (s, 1H, H-12B), 6.36 (s, 1H, H-12B’), 6.39 (s, 1H, H-12A), 6.42 (s, 1H, H-9B’), 6.63 (s, 1H, H-9B), 7.29 (s, 1H, H-9A).
13C NMR (201.1 MHz, CD3OD:CD3CN:D2O = 1:1:1 (v/v)) δ (ppm) 7.6 (C-18A), 9.2 (C-18B), 9.5 (C-18B’), 21.0 (C(17B)-OC(O)CH3, C(17A)-OC(O)CH3, C(17B’)-OC(O)CH3), 30.8 (C-19A), 31.9 (C-19B’), 32.7 (C-19B), 37.3 (N(1A)-CH3), 38.3 (N(1B’)-CH3), 38.7 (N(1B)-CH3), 43.2 (C-6A), 43.3 (C-20A), 44.0 (C-6B, C-6B’), 44.1 (C-20B’), 44.4 (C-20B), 49.2 (C-5A), 50.6 (C-3A), 51.6 (C-3B’), 51.9 (C-3B, C-7A), 52.1 (C-5B’), 53.3 (C(16B’)-COOCH3), 53.4 (C(16B)-COOCH3), 53.7 (C-7B), 53.8 (C-5B), 54.0 (C(16A)-COOCH3), 54.1 (C-7B’), 56.5 (C(11B’)-OCH3), 57.2 (C(11B)-OCH3), 59.3 (C(11A)-OCH3), 59.4 (C-10A), 63.4 (C-21A), 67.8 (C-21B’), 69.2 (C-21B), 76.8 (C-17A), 76.9 (C-17B’), 77.0 (C-17B), 80.1 (C-16B, C-16A), 80.5 (C-16B’), 83.8 (C-2B’), 84.0 (C-2B), 84.6 (C-2A), 91.3 (C-12A), 95.1 (C-12B’), 95.5 (C-12B), 115.9 (C-10B), 116.9 (C-10B’), 124.0 (C-9B’), 126.0 (C-14B), 126.1 (C-9B), 126.2 (C-14B’), 126.4 (C-14A), 126.5 (C-8B’), 126.6 (C-8B), 130.2 (C-15A), 130.6 (C-15B, C-15B’), 132.3 (C-8A), 145.9 (C-9A), 155.3 (C-13B), 155.7 (C-13B’), 159.9 (C-11B), 160.9 (C-11B’), 169.9 (C-13A), 172.0 (C(16A)-COOCH3), 172.2 (C(17A)-OC(O)CH3), 172.8 (C(17B’)-OC(O)CH3), 173.0 (C(17B)-OC(O)CH3), 173.4 (C(16B’)-COOCH3), 173.7 (C(16B)-COOCH3), 190.6 (C-11A).
Minor signal set:
1H NMR (799.7 MHz, CD3OD:CD3CN:D2O = 1:1:1 (v/v)) δ (ppm) 0.29 (t, J = 7.4 Hz, 3H, H3-18A), 0.46 (t, J = 7.4 Hz, 3H, H3-18B’), 0.61 (t, J = 7.4 Hz, 3H, H3-18B), 0.85–0.89 (m, 1H, Hx-19A), 1.09–1.14 (m, 2H, Hx-19B, Hx-19B’), 1.41–1.45 (m, 1H, Hy-19A), 1.50–1.54 (m, 1H, Hy-19B’), 1.57–1.61 (m, 1H, Hy-19B), 2.00 (s, 3H, C(17A)-OC(O)CH3), 2.01 (s, 3H, C(17B)-OC(O)CH3), 2.02 (s, 3H, C(17B’)-OC(O)CH3), 2.07–2.11 (m, 2H, Hx-6B’, Hx-6B), 2.19–2.26 (m, 3H, Hy-6B, Hx-6A, Hy-6B’), 2.43–2.47 (m, 1H, Hx-5B’), 2.47–2.51 (m, 1H, Hy-6A), 2.57 (s, 1H, H-21B’), 2.63–2.66 (m, 1H, Hx-5B), 2.68 (s, 3H, N(1B’)-CH3), 2.72 (s, 3H, N(1B)-CH3), 2.77–2.80 (m, 1H, Hx-5A), 2.79–2.83 (m, 1H, Hx-3B’), 2.87 (s, 1H, H-21B), 2.90–2.96 (m, 2H, Hx-3A, Hx-3B), 2.98 (s, 1H, H-21A), 3.23 (s, 3H, N(1A)-CH3), 3.37–3.42 (m, 2H, Hy-5B’, Hy-5B), 3.42–3.45 (m, 1H, Hy-5A), 3.44–3.49 (m, 3H, Hy-3A, Hy-3B, Hy-3B’), 3.62 (s, 3H, C(11B’)-OCH3), 3.65 (s, 1H, H-2B’), 3.67 (s, 1H, H-2B), 3.73 (s, 3H, C(11B)-OCH3), 3.75 (s, 3H, C(16B)-COOCH3), 3.76 (s, 3H, C(16B’)-COOCH3), 3.80 (s, 3H, C(16A)-COOCH3), 4.01 (s, 3H, C(11A)-OCH3), 4.50 (s, 1H, H-2A), 4.95 (s, 1H, H-17A), 5.19 (s, 1H, H-17B), 5.22–5.26 (m, 2H, H-15B’, H-15A), 5.24 (s, 1H, H-17B’), 5.35–5.38 (m, 1H, H-15B), 5.87–5.90 (m, 1H, H-14B’), 5.90–5.93 (m, 2H, H-14A, H-14B), 6.20 (s, 1H, H-12A), 6.23 (s, 2H, H-12B, H-12B’), 6.52 (s, 1H, H-9B), 6.55 (s, 1H, H-9B’), 7.39 (br s, 1H, H-9A).
13C NMR (201.1 MHz, CD3OD:CD3CN:D2O = 1:1:1 (v/v)) δ (ppm) 7.6 (C-18A), 8.1 (C-18B), 8.7 (C-18B’), 20.8 (C(17A)-OC(O)CH3), 21.0 (C(17B)-OC(O)CH3), 21.1 (C(17B’)-OC(O)CH3), 31.4 (C-19B), 31.6 (C-19B’), 32.1 (C-19A), 37.2 (N(1A)-CH3), 38.2 (N(1B)-CH3), 38.3 (N(1B’)-CH3), 42.7 (C-6A), 43.4 (C-20A), 43.8 (C-20B), 43.9 (C-20B’), 44.3 (C-6B’), 44.9 (C-6B), 50.1 (C-5A), 50.7 (C-5B), 50.9 (C-3A), 51.2 (C-3B), 51.4 (C-7A), 51.6 (C-3B’), 51.9 (C-5B’), 53.2 (C(16B)-COOCH3), 53.3 (C(16B’)-COOCH3), 53.9 (C(16A)-COOCH3), 54.1 (C-7B’), 54.4 (C-7B), 56.5 (C(11B)-OCH3), 56.7 (C(11B’)-OCH3), 58.0 (C-10A), 59.5 (C(11A)-OCH3), 65.2 (C-21A), 66.1 (C-21B), 67.1 (C-21B’), 76.6 (C-17A), 76.9 (C-17B), 77.2 (C-17B’), 79.6 (C-16A), 80.8 (C-16B’), 81.2 (C-16B), 83.3 (C-2B), 83.4 (C-2B’), 84.9 (C-2A), 89.4 (C-12A), 94.7 (C-12B), 95.6 (C-12B’), 115.9 (C-10B), 117.3 (C-10B’), 124.3 (C-9B), 125.2 (C-8B), 125.3 (C-9B’), 125.4 (C-8B’), 125.7 (C-14B), 125.9 (C-14B’), 126.1 (C-14A), 130.1 (C-15A), 130.8 (C-15B’), 131.0 (C-15B), 132.5 (C-8A), 147.2 (br, C-9A), 154.6 (C-13B), 154.9 (C-13B’), 159.8 (C-11B), 160.7 (C-11B’), 169.9 (C-13A), 171.7 (C(17A)-OC(O)CH3), 172.1 (C(16A)-COOCH3), 172.5 (C(17B)-OC(O)CH3), 172.9 (C(17B’)-OC(O)CH3), 173.0 (C(16B)-COOCH3), 173.3 (C(16B’)-COOCH3), 191.8 (C-11A).
HRMS: M + H = 1365.65951 (C75H93O18N6, Δ = 3.97 ppm). ESI-MS-MS (CID = 35%, rel. int. %): 1347(17), 1305(73), 1245(9), 1203(10), 1096(100), 1036(17).
Compound 28:
TLC (dichloromethane-methanol 10:1), Rf = 0.35.
IR (KBr) 3433, 1749, 1619, 1289, 1207, 1151, 1025, 821 cm−1.
NMR: The three vindoline subunits of 28 are denoted as A, B, and B’. The two subunits that have the same constitution (vindoline-10-yl) are called B and B’. A minor signal set (ca. 10%) can also be detected due to conformational isomerism. The assignment of the major signal set is given below.
1H NMR (799.7 MHz, DMSO-d6:CD3CN:D2O = 3:1:1 (v/v)) δ (ppm) 0.30 (t, J = 7.3 Hz, 3H, H3-18A), 0.43 (t, J = 7.3 Hz, 3H, H3-18B), 0.50 (t, J = 7.3 Hz, 3H, H3-18B’), 0.82–0.86 (m, 1H, Hx-19B’), 0.92–0.96 (m, 1H, Hx-19A), 1.21 (dq, J = 15.2, 7.3 Hz, 1H, Hx-19B), 1.39–1.43 (m, 1H, Hy-19A), 1.43–1.50 (m, 2H, Hy-19B’ Hy-19B), 1.91 (s, 3H, C(17A)-OC(O)CH3), 1.94 (s, 3H, C(17B’)-OC(O)CH3), 1.93–1.97 (m, 1H, Hx-6B), 1.96 (s, 3H, C(17B)-OC(O)CH3), 2.08–2.16 (m, 3H, Hy-6B, Hx-6B’, Hy-6B’), 2.16–2.20 (m, 2H, Hx-5B, Hx-6A), 2.31 (s, 1H, H-21B), 2.34–2.38 (m, 1H, Hy-6A), 2.55–2.59 (m, 2H, Hx-5A, Hx-5B’), 2.58 (s, 3H, N(1B)-CH3), 2.61–2.65 (m, 8H, N(1B’)-CH3, Hx-3B, N(1A)-CH3, H-21A), 2.73 (br s, 1H, H-21B’), 2.79 (br d, J = 16.4 Hz, 1H, Hx-3A), 2.85 (br d, J = 16.7 Hz, 1H, Hx-3B’), 3.23–3.27 (m, 1H, Hy-5B), 3.29–3.35 (m, 2H, Hy-5A, Hy-5B’), 3.37–3.42 (m, 3H, Hy-3B’, Hy-3B, Hy-3A), 3.49 (s, 1H, H-2B), 3.53 (s, 3H, C(11B)-OCH3), 3.55 (s, 1H, H-2B’), 3.58 (s, 3H, C(11B’)-OCH3), 3.65 (s, 3H, C(16A)-COOCH3), 3.67 (s, 3H, C(16B’)-COOCH3), 3.68 (s, 3H, C(16B)-COOCH3), 3.91 (s, 1H, H-2A), 4.97 (s, 1H, H-17A), 5.09 (s, 1H, H-12A), 5.10–5.12 (m, 2H, H-15A, H-17B’), 5.16 (br d, J = 9.6 Hz, 1H, H-15B), 5.20–5.23 (m, 2H, H-17B, H-15B’), 5.81–5.83 (m, 2H, H-14B, H-14A), 5.83–5.86 (m, 1H, H-14B’), 6.12 (s, 1H, H-12B), 6.18 (s, 1H, H-9B), 6.20 (s, 1H, H-12B’), 6.80 (s, 1H, H-9B’), 7.17 (s, 1H, H-9A).
13C NMR (201.1 MHz, DMSO-d6:CD3CN:D2O = 3:1:1 (v/v)) δ (ppm) 7.2 (C-18B’), 7.7 (C-18A), 8.1 (C-18B), 20.6 (C(17A)-OC(O)CH3), 20.77 (C(17B’)-OC(O)CH3), 20.81 (C(17B)-OC(O)CH3), 30.7 (C-19B’), 30.9 (C-19B), 31.2 (C-19A), 34.4 (N(1A)-CH3), 38.2 (N(1B)-CH3), 38.8 (N(1B’)-CH3), 42.6 (C-20A), 42.7 (C-20B’), 42.8 (C-6A), 43.0 (C-20B), 43.2 (C-6B), 44.4 (C-6B’), 50.3 (C-7A), 50.4 (C-3A), 50.5 (C-5B’), 50.6 (C-3B’), 50.9 (C-5A), 51.0 (C-3B), 51.8 (C-5B), 52.1 (C(16B’)-COOCH3), 52.3 (C(16B)-COOCH3), 52.5 (C(16A)-COOCH3), 52.9 (C-7B), 53.1 (C-7B’), 55.3 (C(11B)-OCH3), 55.4 (C(11B’)-OCH3), 59.6 (C-10A), 65.6 (C-21B’), 67.1 (C-21B), 67.2 (C-21A), 75.7 (C-17A), 76.1 (C-17B’), 76.2 (C-17B), 78.7 (C-16A), 79.5 (C-16B), 79.7 (C-16B’), 81.7 (C-2A), 82.6 (C-2B), 83.4 (C-2B’), 92.1 (C-12A), 93.8 (C-12B), 94.3 (C-12B’), 118.5 (C-10B’), 123.0 (C-10B), 123.36 (C-8B), 123.42 (C-8B’), 124.56 (C-14A), 124.67 (C-14B’), 124.73 (C-9B’), 124.88(C-9B), 124.89 (C-14B), 129.9 (C-15A), 130.3 (C-15B), 130.6 (C-15B’), 133.4 (C-8A), 140,0 (C-9A), 152.4 (C-13B), 152.6 (C-13B’), 158.3 (C-11B), 159.5 (C-11B’), 162.2 (C-13A), 170.1 (C(17A)-OC(O)CH3), 170.5 (C(17B’)-OC(O)CH3), 171.1 (C(17B)-OC(O)CH3), 171.79 (C(16A)-COOCH3), 171.83 (C(16B’)-COOCH3), 172.1 (C(17B)-COOCH3), 199.1 (C-11A).
HRMS: [(M + 2H)/2]2+ = 676.32271 (C74H92O18N6, Δ = –0.2 ppm). ESI-MS-MS (CID = 45%, rel. int. %): 646(100), 637(14), 616(17), 595(45), 565(16), 542(39), 512(16), 457(16).
Compound 29:
TLC (dichloromethane-methanol 7:1), Rf = 0.11.
1H NMR (799.7 MHz, DMSO-d6:CD3CN:D2O = 3:1:1 (v/v)) δ (ppm) 1.02 (t, J = 7.3 Hz, 3H, H3-18), 1.93–1.97 (m, 1H, Hx-6), 2.04–2.09 (m, 1H, Hx-19), 2.07–2.10 (m, 1H, Hx-17), 2.18–2.24 (m, 1H, Hy-19), 2.27–2.31 (m, 1H, Hy-17), 2.55–2.60 (m, 1H, Hy-6), 2.92–2.95 (m, 1H, Hx-3), 3.04 (s, 3H, N(4)-CH3), 3.14–3.16 (m, 1H, H-14), 3.23 (s, 3H, C(16)-COOCH3), 3.58–3.61 (m, 1H, Hx-5), 3.65–3.68 (m, 1H, Hy-3), 4.10–4.14 (m, 1H, Hy-5), 4.94 (d, J = 1.6 Hz, 1H, H-21), 6.41–6.44 (m, 1H, H-15), 6.80–6.84 (m, 1H, H-10), 6.87–6.90 (m, 1H, H-12), 7.48–7.50 (m, 1H, H-9), 7.50–7.53 (m, 1H, H-11).
13C NMR (201.1 MHz, DMSO-d6:CD3CN:D2O = 3:1:1 (v/v)) δ (ppm) 10.5 (C-18), 25.0 (C-6), 27.1 (C-19), 28.2 (C-17), 29.1 (C-14), 52.6 (C-16), 52.9 (C(16)-COOCH3), 55.6 (N(4)-CH3), 56.8 (C-5), 60.5 (C-3), 63.5 (C-2), 63.7 (C-21), 112.8 (C-12), 118.9 (C-8), 119.2 (C-10), 124.6 (C-9), 130.7 (C-15), 138.9 (C-11), 140.8 (C-20), 160.8 (C-13), 169.9 (C(16)-COOCH3), 213.1 (C-7).
HRMS: M + H = 367.20122 (C22H27O3N2, Δ = –1.1 ppm). ESI-MS-MS (CID = 55%, rel. int. %): 203(100), 160(7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










