Aminobenzosuberone Scaffold as a Modular Chemical Tool for the Inhibition of Therapeutically Relevant M1 Aminopeptidases

 ,
,  ,
,
Abstract
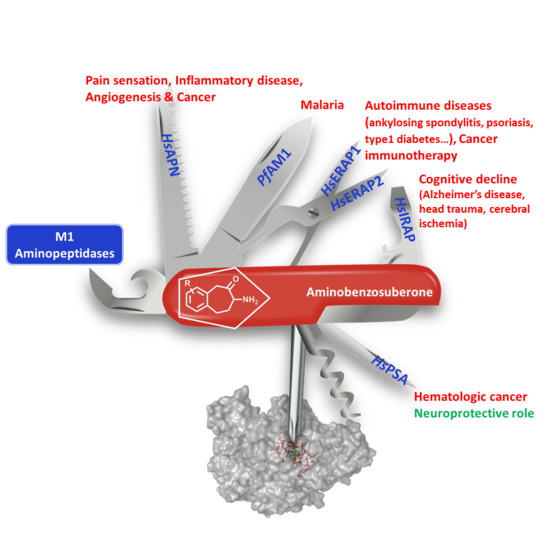
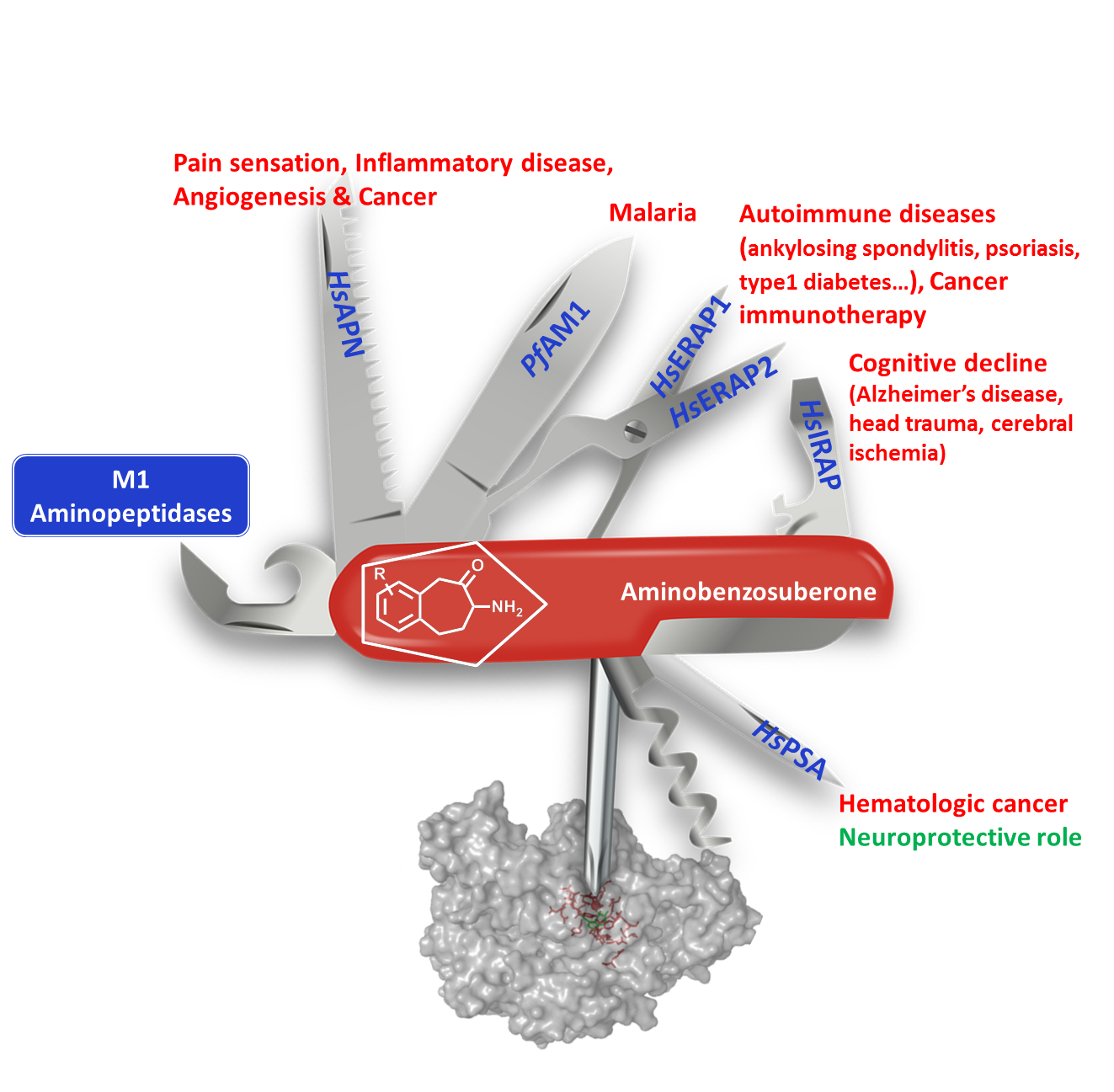
:
1. Introduction
2. Results
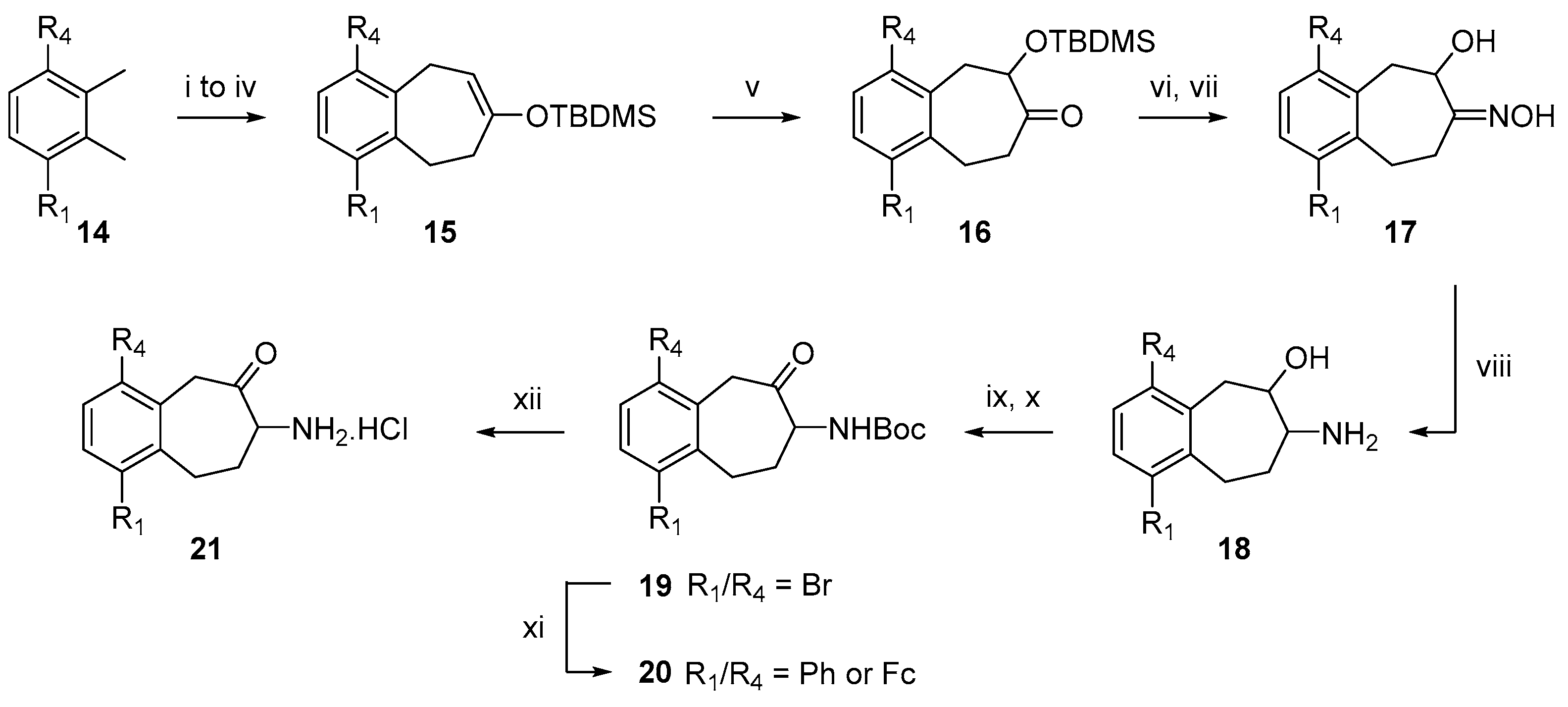
2.1. Chemistry
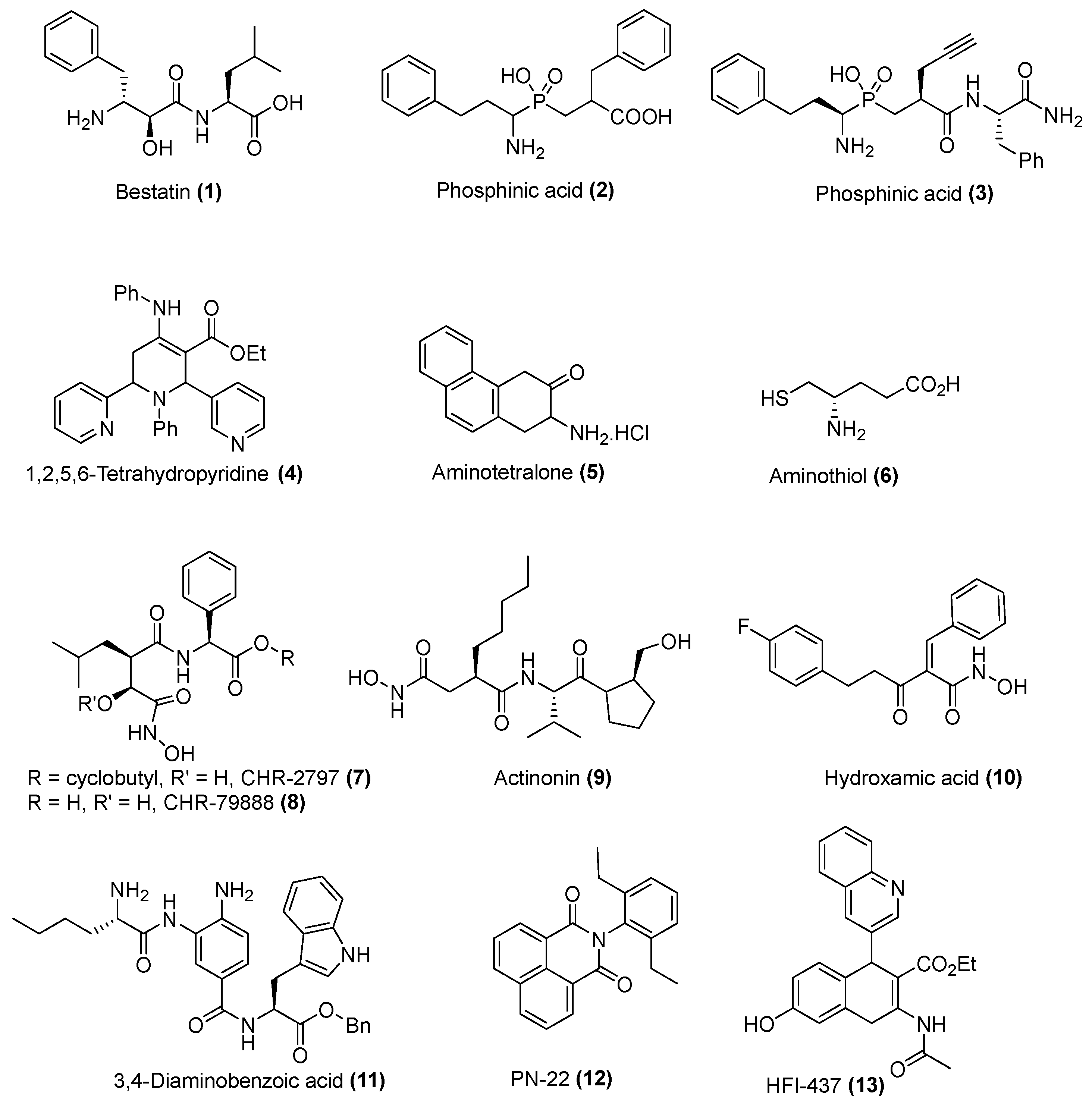
2.2. In Vitro Inhibition of A Representative Panel of M1 Aminopeptidases
3. Discussion
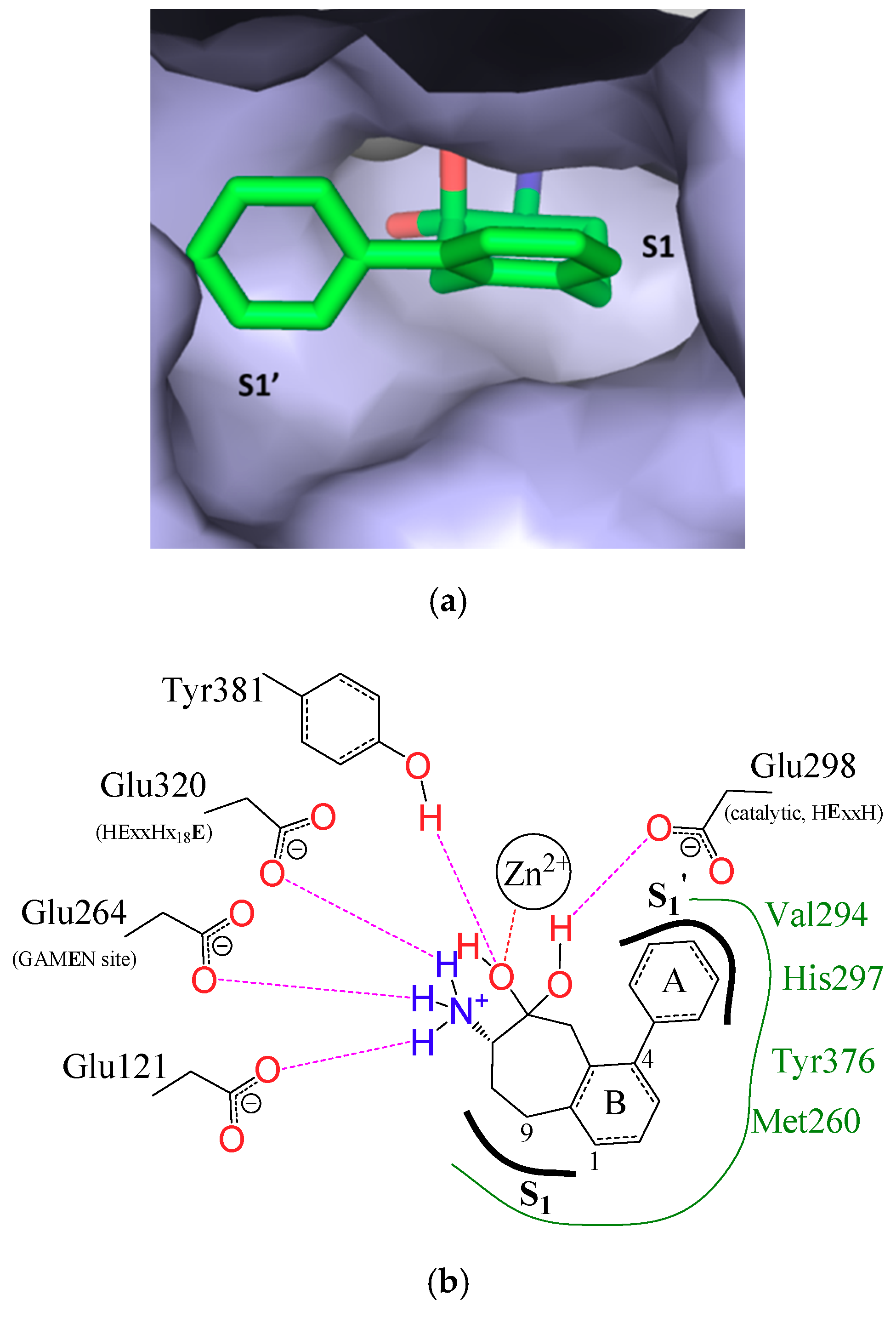
3.1. Structure-Based Analysis of the Structure-Activity Relationships
3.2. S1 Subsite
3.3. S1′ Subsite
3.4. Intra/Inter-Domain Movements
3.5. ADME-Tox Properties
4. Materials and Methods
4.1. General Information
4.2. General Procedure for Rubottom Oxidation
4.3. General Procedure for Oxime Reduction
4.4. Production and Purification of Recombinant Aminopeptidases
4.4.1. PfAM1 and PfAM17
4.4.2. HsERAP1, HsERAP2 and HsIRAP
4.4.3. EcPepN and HsPSA
4.5. Enzymatic Assays and Kinetic Analysis
4.5.1. PfAM1 and PfAM17
4.5.2. LTA4H
4.5.3. EcPepN and HsPSA
4.5.4. HsERAP1, HsERAP2 and HsIRAP
4.6. Measurement of Octanol/Water Partition Coefficient (logD7.4)
4.7. In Silico Prediction of ADMET Properties
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rawlings, N.D.; Barret, A.J. Metallopeptidases and their clans. In Handbook of Proteolytic Enzymes; Rawlings, N.D., Salvesen, G.S., Eds.; Academic Press: New York, NY, USA, 2013; Volume 1, pp. 325–370. ISBN 978-0-12-407744-7. [Google Scholar]
- Barret, A.J.; McDonald, J.K. Mammalian Proteases: A Glossary and Bibliography, Vol. 2: Exopeptidases; Academic Press: London, UK, 1986. [Google Scholar]
- Lowther, W.T.; Matthews, B.W. Metalloaminopeptidases: Common functional themes in disparate structural surroundings. Chem. Rev. 2002, 102, 4581–4608. [Google Scholar] [CrossRef] [PubMed]
- Straeter, N.; Lipscomb, W.N. Two-metal ion mechanism of bovine lens leucine aminopeptidase: Active site solvent structure and binding mode of L-Leucinal, a gem-diolate transition state analog, by X-ray crystallography. Biochemistry 1995, 34, 14792–14800. [Google Scholar] [CrossRef]
- Hooper, N.M. Families of zinc metalloproteases. FEBS Lett. 1994, 354, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Barrett, A.J. Evolutionary families of metallopeptidases. In Methods in Enzymology; Academic Press: San Diego, CA, USA, 1995; Volume 248, pp. 183–228. ISBN 978-0-12-182149-4. [Google Scholar]
- Taylor, A. Aminopeptidases: Structure and function. FASEB J. 1993, 7, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Maynard, K.B.; Smith, S.A.; Davis, A.C.; Trivette, A.; Seipelt-Thiemann, R.L. Evolutionary analysis of the mammalian M1 aminopeptidases reveals conserved exon structure and gene death. Gene 2014, 552, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Peer, W.A. The role of multifunctional M1 metallopeptidases in cell cycle progression. Ann. Bot. 2011, 107, 1171–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, N.M.; Lendeckel, U. Aminopeptidases in Biology and Disease; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2004; Volume 2, ISBN 0-306-48465-X. [Google Scholar]
- Drinkwater, N.; Lee, J.; Yang, W.; Malcolm, T.R.; McGowan, S. M1 aminopeptidases as drug targets: Broad applications or therapeutic niche? FEBS J. 2017, 284, 1473–1488. [Google Scholar] [CrossRef] [PubMed]
- Saveanu, L.; Carroll, O.; Weimershaus, M.; Guermonprez, P.; Firat, E.; Lindo, V.; Greer, F.; Davoust, J.; Kratzer, R.; Keller, S.R.; et al. IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science 2009, 325, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Li, Y.; Zuo, X.-B.; Tang, H.-Y.; Tang, X.-F.; Gao, J.-P.; Sheng, Y.-J.; Yin, X.-Y.; Zhou, F.-S.; Zhang, C.; et al. Identification of a missense variant in LNPEP that confers psoriasis risk. J. Investig. Dermatol. 2014, 134, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Stratikos, E. Modulating antigen processing for cancer immunotherapy. Oncoimmunology 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratikos, E.; Stamogiannos, A.; Zervoudi, E.; Fruci, D. A Role for naturally occurring alleles of endoplasmic reticulum aminopeptidases in tumor immunity and cancer pre-disposition. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Keller, C.; Kühl, A.A.; Textor, A.; Seifert, U.; Blankenstein, T.; Willimsky, G.; Kloetzel, P.-M. ERAP1-dependent antigen cross-presentation determines efficacy of adoptive T-cell therapy in mice. Cancer Res. 2018, 78, 3243–3254. [Google Scholar] [CrossRef] [PubMed]
- Azimzadeh, O.; Sow, C.; Gèze, M.; Nyalwidhe, J.; Florent, I. Plasmodium falciparum PfA-M1 aminopeptidase is trafficked via the parasitophorous vacuole and marginally delivered to the food vacuole. Malar. J. 2010, 9, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef] [PubMed]
- MEROPS: The Peptidase Database. Available online: https://www.ebi.ac.uk/merops/cgi-bin/famsum?family=M1 (accessed on 19 July 2018).
- Chen, L.; Lin, Y.-L.; Peng, G.; Li, F. Structural basis for multifunctional roles of mammalian aminopeptidase N. Proc. Natl. Acad. Sci. USA 2012, 109, 17966–17971. [Google Scholar] [CrossRef] [PubMed]
- Kochan, G.; Krojer, T.; Harvey, D.; Fischer, R.; Chen, L.; Vollmar, M.; von Delft, F.; Kavanagh, K.L.; Brown, M.A.; Bowness, P.; et al. Crystal structures of the endoplasmic reticulum aminopeptidase-1 (ERAP1) reveal the molecular basis for N-terminal peptide trimming. Proc. Natl. Acad. Sci. USA 2011, 108, 7745–7750. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Chang, S.-C.; Evnouchidou, I.; York, I.A.; Zikos, C.; Rock, K.L.; Goldberg, A.L.; Stratikos, E.; Stern, L.J. Structural basis for antigenic peptide precursor processing by the endoplasmic reticulum aminopeptidase ERAP1. Nat. Struct. Mol. Biol. 2011, 18, 604–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, C.; Mudgal, G.; Reguera, J.; Recacha, R.; Albrecht, S.; Enjuanes, L.; Casasnovas, J.M. Allosteric inhibition of aminopeptidase N functions related to tumour growth and virus infection. Sci. Rep. 2017, 7, 46045. [Google Scholar] [CrossRef] [PubMed]
- Mpakali, A.; Saridakis, E.; Harlos, K.; Zhao, Y.; Kokkala, P.; Georgiadis, D.; Giastas, P.; Papakyriakou, A.; Stratikos, E. Ligand-induced conformational change of insulin-regulated aminopeptidase: Insights on catalytic mechanism and active site plasticity. J. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.H.M.; Zhou, D.; Rini, J.M. The X-ray crystal structure of human aminopeptidase N reveals a novel dimer and the basis for peptide processing. J. Biol. Chem. 2012, 287, 36804–36813. [Google Scholar] [CrossRef] [PubMed]
- Hermans, S.J.; Ascher, D.B.; Hancock, N.C.; Holien, J.K.; Michell, B.J.; Chai, S.Y.; Morton, C.J.; Parker, M.W. Crystal structure of human insulin-regulated aminopeptidase with specificity for cyclic peptides. Protein Sci. 2015, 24, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Wallis, M.G.; Lankford, M.F.; Keller, S.R. Vasopressin is a physiological substrate for the insulin-regulated aminopeptidase IRAP. Am. J. Physiol.-Endocrinol. Metab. 2007, 293, E1092–E1102. [Google Scholar] [CrossRef] [PubMed]
- Bauvois, B.; Dauzonne, D. Aminopeptidase-N/CD13 (EC 3.4.11.2) inhibitors: Chemistry, biological evaluations and therapeutic prospects. Med. Res. Rev. 2006, 26, 88–130. [Google Scholar] [CrossRef] [PubMed]
- Mucha, A.; Drag, M.; Dalton, J.P.; Kafarski, P. Metallo-aminopeptidase inhibitors. Biochimie 2010, 92, 1509–1529. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Adhikari, N.; Jha, T. Design of aminopeptidase N inhibitors as anti-cancer agents. J. Med. Chem. 2018, 61, 6468–6490. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Bacerio, J.; Fando, R.; del Monte-Martinez, A.; Charli, J.-L.; de los A Chavez, M. Plasmodium falciparum M1-aminopeptidase: A promising target for the development of antimalarials. Curr. Drug Targets 2014, 15, 1144–1165. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, H.; Aoyagi, T.; Suda, H.; Hamada, M.; Takeuchi, T. Bestatin, an inhibitorof aminopeptidase B, produced by actinomycetes. J. Antibiot. (Tokyo) 1976, 29, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. The most potent organophosphorus inhibitors of leucine aminopeptidase. structure-based design, chemistry and activity. J. Med. Chem. 2003, 46, 2641–2655. [Google Scholar] [CrossRef] [PubMed]
- Kannan Sivaraman, K.; Paiardini, A.; Sieńczyk, M.; Ruggeri, C.; Oellig, C.A.; Dalton, J.P.; Scammells, P.J.; Drag, M.; McGowan, S. Synthesis and structure–activity relationships of phosphonic arginine mimetics as inhibitors of the M1 and M17 aminopeptidases from Plasmodium falciparum. J. Med. Chem. 2013, 56, 5213–5217. [Google Scholar] [CrossRef] [PubMed]
- Flipo, M.; Beghyn, T.; Leroux, V.; Florent, I.; Deprez, B.P.; Deprez-Poulain, R.F. Novel selective inhibitors of the zinc plasmodial aminopeptidase PfA-M1 as potential antimalarial agents. J. Med. Chem. 2007, 50, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Drinkwater, N.; Bamert, R.S.; Sivaraman, K.K.; Paiardini, A.; McGowan, S. X-ray crystal structures of an orally available aminopeptidase inhibitor, Tosedostat, bound to anti-malarial drug targets P f A-M1 and P f A-M17: Structures of Pf A-M1/M17 Bound to Tosedostat. Proteins Struct. Funct. Bioinform. 2015, 83, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Krige, D.; Needham, L.A.; Bawden, L.J.; Flores, N.; Farmer, H.; Miles, L.E.C.; Stone, E.; Callaghan, J.; Chandler, S.; Clark, V.L.; et al. CHR-2797: An antiproliferative aminopeptidase inhibitor that leads to amino acid deprivation in human leukemic cells. Cancer Res. 2008, 68, 6669–6679. [Google Scholar] [CrossRef] [PubMed]
- Schalk, C.; Dorchymont, H.; Jauch, M.F.; Tarnus, C. 3-Amino-2-tetralone derivatives: Novel potent and selective inhibitors of aminopeptidase-M (EC-3.4.11.2). Arch. Biochem. Biophys. 1994, 311, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Harbeson, S.L.; Rich, D.H. Inhibition of aminopeptidases by peptides containing ketomethylene and hydroxyethylene amide bond replacements. J. Med. Chem. 1989, 32, 1378–1392. [Google Scholar] [CrossRef] [PubMed]
- Tieku, S.; Hooper, N.M. Inhibition of aminopeptidases N, A and W: A re-evaluation of the actions of bestatin and inhibitors of angiotensin converting enzyme. Biochem. Pharmacol. 1992, 44, 1725–1730. [Google Scholar] [CrossRef]
- Lee, J.; Shim, J.S.; Jung, S.-A.; Lee, S.-T.; Kwon, H.J. N-hydroxy-2-(naphthalene-2-ylsulfanyl)-acetamide, a novel hydroxamic acid-based inhibitor of aminopeptidase N and its anti-angiogenic activity. Bioorg. Med. Chem. Lett. 2005, 15, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, S.; Al-Lakkis-Wehbe, M.; Orsini, A.; Defoin, A.; Pale, P.; Salomon, E.; Tarnus, C.; Weibel, J.-M. Amino-benzosuberone: A novel warhead for selective inhibition of human aminopeptidase-N/CD13. Bioorg. Med. Chem. 2011, 19, 1434–1449. [Google Scholar] [CrossRef] [PubMed]
- Gumpena, R.; Kishor, C.; Ganji, R.J.; Addlagatta, A. Discovery of α,β- and α,γ-diamino acid scaffolds for the inhibition of M1 family aminopeptidases. ChemMedChem 2011, 6, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Velmourougane, G.; Harbut, M.B.; Dalal, S.; McGowan, S.; Oellig, C.A.; Meinhardt, N.; Whisstock, J.C.; Klemba, M.; Greenbaum, D.C. Synthesis of new (−)-bestatin-based inhibitor libraries reveals a novel binding mode in the S1 pocket of the essential malaria M1 metalloaminopeptidase. J. Med. Chem. 2011, 54, 1655–1666. [Google Scholar] [CrossRef] [PubMed]
- Skinner-Adams, T.S.; Lowther, J.; Teuscher, F.; Stack, C.M.; Grembecka, J.; Mucha, A.; Kafarski, P.; Trenholme, K.R.; Dalton, J.P.; Gardiner, D.L. Identification of Phosphinate Dipeptide Analog Inhibitors Directed against the Plasmodium falciparum M17 Leucine Aminopeptidase as Lead Antimalarial Compounds. J. Med. Chem. 2007, 50, 6024–6031. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, S.; Węglarz-Tomczak, E.; Berlicki, Ł.; Pawełczak, M.; Nocek, B.; Mulligan, R.; Joachimiak, A.; Mucha, A. Structure-guided, single-point modifications in the phosphinic dipeptide structure yield highly potent and selective inhibitors of neutral aminopeptidases. J. Med. Chem. 2014, 57, 8140–8151. [Google Scholar] [CrossRef] [PubMed]
- McGowan, S.; Porter, C.J.; Lowther, J.; Stack, C.M.; Golding, S.J.; Skinner-Adams, T.S.; Trenholme, K.R.; Teuscher, F.; Donnelly, S.M.; Grembecka, J.; et al. Structural basis for the inhibition of the essential Plasmodium falciparum M1 neutral aminopeptidase. Proc. Natl. Acad. Sci. USA 2009, 106, 2537–2542. [Google Scholar] [CrossRef] [PubMed]
- Kokkala, P.; Mpakali, A.; Mauvais, F.-X.; Papakyriakou, A.; Daskalaki, I.; Petropoulou, I.; Kavvalou, S.; Papathanasopoulou, M.; Agrotis, S.; Fonsou, T.-M.; et al. Optimization and structure–activity relationships of phosphinic pseudotripeptide inhibitors of aminopeptidases that generate antigenic peptides. J. Med. Chem. 2016, 59, 9107–9123. [Google Scholar] [CrossRef] [PubMed]
- Aeluri, R.; Ganji, R.J.; Marapaka, A.K.; Pillalamarri, V.; Alla, M.; Addlagatta, A. Highly functionalized tetrahydropyridines are cytotoxic and selective inhibitors of human puromycin sensitive aminopeptidase. Eur. J. Med. Chem. 2015, 106, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Chauvel, E.N.; Coric, P.; Llorens-Cortes, C.; Wilk, S.; Roques, B.P.; Fournie-Zaluski, M.-C. Investigation of the active site of aminopeptidase A using a series of new thiol-containing inhibitors. J. Med. Chem. 1994, 37, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, H.; Aoyagi, T.; Tanaka, T.; Suda, H.; Okuyama, A.; Naganawa, H.; Hamada, M.; Takeuchi, T. Production of actinonin, an inhibitor of aminopeptidase M, by actinomycetes. J. Antibiot. (Tokyo) 1985, 38, 1629–1630. [Google Scholar] [CrossRef] [PubMed]
- Ansorge, S.; Neubert, K.; Bank, U.; Reichstein, I.; Faust, J.; Täger, M.; Fuchs, P.; Senns, B. Novel Dual Peptidase Inhibitors as Prodrugs for the Therapy of Inflammatory and Other Disorders. 2007. Google Patent. Available online: https://patents.google.com/patent/WO2007057128A1/en (accessed on 5 April 2018).
- Deprez-Poulain, R.; Flipo, M.; Piveteau, C.; Leroux, F.; Dassonneville, S.; Florent, I.; Maes, L.; Cos, P.; Deprez, B. Structure–activity relationships and blood distribution of antiplasmodial aminopeptidase-1 inhibitors. J. Med. Chem. 2012, 55, 10909–10917. [Google Scholar] [CrossRef] [PubMed]
- Papakyriakou, A.; Zervoudi, E.; Tsoukalidou, S.; Mauvais, F.-X.; Sfyroera, G.; Mastellos, D.C.; van Endert, P.; Theodorakis, E.A.; Vourloumis, D.; Stratikos, E. 3,4-Diaminobenzoic acid derivatives as inhibitors of the oxytocinase subfamily of M1 aminopeptidases with immune-regulating properties. J. Med. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kakuta, H.; Tanatani, A.; Nagasawa, K.; Hashimoto, Y. Specific nonpeptide inhibitors of puromycin-sensitive aminopeptidase with a 2,4(1H,3H)-quinazolinedione skeleton. Chem. Pharm. Bull. (Tokyo) 2003, 51, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Mountford, S.J.; Albiston, A.L.; Charman, W.N.; Ng, L.; Holien, J.K.; Parker, M.W.; Nicolazzo, J.A.; Thompson, P.E.; Chai, S.Y. Synthesis, structure–activity relationships and brain uptake of a novel series of benzopyran inhibitors of insulin-regulated aminopeptidase. J. Med. Chem. 2014, 57, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Mina-Osorio, P. The moonlighting enzyme CD13: Old and new functions to target. Trends Mol. Med. 2008, 14, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Maiereanu, C.; Schmitt, C.; Schifano-Faux, N.; Le Nouën, D.; Defoin, A.; Tarnus, C. A novel amino-benzosuberone derivative is a picomolar inhibitor of mammalian aminopeptidase N/CD13. Bioorg. Med. Chem. 2011, 19, 5716–5733. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, S.; Salomon, E.; Defoin, A.; Tarnus, C. Rapid and efficient synthesis of a novel series of substituted aminobenzosuberone derivatives as potent, selective, non-peptidic neutral aminopeptidase inhibitors. Bioorg. Med. Chem. 2012, 20, 4942–4953. [Google Scholar] [CrossRef] [PubMed]
- Revelant, G.; Al-Lakkis-Wehbe, M.; Schmitt, M.; Alavi, S.; Schmitt, C.; Roux, L.; Al-Masri, M.; Schifano-Faux, N.; Maiereanu, C.; Tarnus, C.; et al. Exploring S1 plasticity and probing S1′ subsite of mammalian aminopeptidase N/CD13 with highly potent and selective aminobenzosuberone inhibitors. Bioorg. Med. Chem. 2015, 23, 3192–3207. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.; Voegelin, M.; Marin, A.; Schmitt, M.; Schegg, F.; Hénon, P.; Guenot, D.; Tarnus, C. Selective aminopeptidase-N (CD13) inhibitors with relevance to cancer chemotherapy. Bioorg. Med. Chem. 2013, 21, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Adam, W.; Hadjiarapoglou, L.; Wang, X. Epoxidation of silyl enol ethers, phthalides and enol esters by dimethyldioxirane. Tetrahedron Lett. 1989, 30, 6497–6500. [Google Scholar] [CrossRef]
- Solladié-Cavallo, A.; Lupattelli, P.; Jierry, L.; Bovicelli, P.; Angeli, F.; Antonioletti, R.; Klein, A. Asymmetric oxidation of silyl enol ethers using chiral dioxiranes derived from α-fluoro cyclohexanones. Tetrahedron Lett. 2003, 44, 6523–6526. [Google Scholar] [CrossRef]
- Abiraj, K.; Gowda, D.C. Magnesium-catalyzed proficient reduction of oximes to amines using ammonium formate. Synth. Commun. 2004, 34, 599–605. [Google Scholar] [CrossRef]
- Denis, J.-N.; Jolivalt, C.M.; Maurin, M.M.L.; Jeanty, M. Novel Bis-Indolic Derivatives, a Process for Preparing the Same and Their Uses as a Drug. 2013. Google Patent. Available online: https://patents.google.com/patent/EP2548864A1/un (accessed on 11 October 2013).
- Groneberg, R.; Zhan, J.; Askew, B.; D’Amico, D.; Han, N.; Fotsch, C.; Liu, Q.; Riahi, B.; Zhu, J.; Yang, K.; et al. Cyclic Amine Derivatives and Methods of Use. 2005. Google Patent. Available online: https://patents.google.com/patent/US7199244B2/en (accessed on 11 October 2013).
- Binet, J.; Guffroy, C.; Kasai, H.; Wagatsuma, N. 2-Ureido-Benzamide Derivatives. 1999. Google Patent. Available online: https://patents.google.com/patent/US5872115A/en (accessed on 11 October 2013).
- Ipaktschi, J. Reductive displacement of the acetate group in allyl, propargyl and benzyl acetates by NaBH4/NiCl2·6·H2O. Chem. Ber. 1984, 117, 3320–3324. [Google Scholar] [CrossRef]
- Bounaadja, L.; Schmitt, M.; Albrecht, S.; Mouray, E.; Tarnus, C.; Florent, I. Selective inhibition of PfA-M1, over PfA-M17, by an amino-benzosuberone derivative blocks malaria parasites development in vitro and in vivo. Malar. J. 2017, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papakyriakou, A.; Stratikos, E. The Role of conformational dynamics in antigen trimming by intracellular aminopeptidases. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Addlagatta, A.; Gay, L.; Matthews, B.W. Structure of aminopeptidase N from Escherichia coli suggests a compartmentalized, gated active site. Proc. Natl. Acad. Sci. USA 2006, 103, 13339–13344. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.H.; Ferraz, F.A.; Honorato, R.V.; Xavier-Neto, J.; Sobreira, T.J.; de Oliveira, P.S. KVFinder: Steered identification of protein cavities as a PyMOL plugin. BMC Bioinform. 2014, 15, 197. [Google Scholar] [CrossRef] [PubMed]
- Ganji, R.J.; Reddi, R.; Gumpena, R.; Marapaka, A.K.; Arya, T.; Sankoju, P.; Bhukya, S.; Addlagatta, A. Structural basis for the inhibition of M1 family aminopeptidases by the natural product actinonin: Crystal structure in complex with E. coli aminopeptidase N. Protein Sci. 2015, 24, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; McEwen, A.G.; Olieric, V.; Schmitt, C.; Albrecht, S.; Cavarelli, J.; Tarnus, C. Insight into the remarkable affinity and selectivity of the aminobenzosuberone scaffold for the M1 aminopeptidases family based on structure analysis. Proteins Struct. Funct. Bioinform. 2017, 85, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Dalal, S.; Ragheb, D.R.T.; Schubot, F.D.; Klemba, M. A Naturally variable residue in the S1 subsite of M1 family aminopeptidases modulates catalytic properties and promotes functional specialization. J. Biol. Chem. 2013, 288, 26004–26012. [Google Scholar] [CrossRef] [PubMed]
- Addlagatta, A.; Gay, L.; Matthews, B.W. Structural basis for the unusual specificity of Escherichia coli aminopeptidase N. Biochemistry 2008, 47, 5303–5311. [Google Scholar] [CrossRef] [PubMed]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; Pennington, L.D.; Meanwell, N.A. A Survey of the role of noncovalent sulfur interactions in drug design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [CrossRef] [PubMed]
- Rosati, M.; Dalal, S.; Klemba, M. Two cap residues in the S1 subsite of a Plasmodium falciparum M1-family aminopeptidase promote broad specificity and enhance catalysis. Mol. Biochem. Parasitol. 2017, 217, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Zervoudi, E.; Papakyriakou, A.; Georgiadou, D.; Evnouchidou, I.; Gajda, A.; Poreba, M.; Salvesen, G.S.; Drag, M.; Hattori, A.; Swevers, L.; et al. Probing the S1 specificity pocket of the aminopeptidases that generate antigenic peptides. Biochem. J. 2011, 435, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Węglarz-Tomczak, E.; Berlicki, Ł.; Pawełczak, M.; Nocek, B.; Joachimiak, A.; Mucha, A. A structural insight into the P1 S1 binding mode of diaminoethylphosphonic and phosphinic acids, selective inhibitors of alanine aminopeptidases. Eur. J. Med. Chem. 2016, 117, 187–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamogiannos, A.; Maben, Z.; Papakyriakou, A.; Mpakali, A.; Kokkala, P.; Georgiadis, D.; Stern, L.J.; Stratikos, E. Critical role of interdomain interactions in the conformational change and catalytic mechanism of endoplasmic reticulum aminopeptidase 1. Biochemistry 2017, 56, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Cadel, S.; Darmon, C.; Pernier, J.; Hervé, G.; Foulon, T. The M1 family of vertebrate aminopeptidases: Role of evolutionarily conserved tyrosines in the enzymatic mechanism of aminopeptidase B. Biochimie 2015, 109, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Blomster, M.; Wetterholm, A.; Mueller, M.J.; Haeggström, J.Z. Evidence for a catalytic role of tyrosine 383 in the peptidase reaction of leukotriene A4 hydrolase. Eur. J. Biochem. 1995, 231, 528–534. [Google Scholar] [CrossRef] [PubMed]
- ADMETlab. Available online: http://admet.scbdd.com/calcpre/index_sys (accessed on 19 July 2018).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Fournie-Zaluski, M.-C.; Fassot, C.; Valentin, B.; Djordjijevic, D.; Goazigo, A.R.-L.; Corvol, P.; Roques, B.P.; Llorens-Cortes, C. Brain renin-angiotensin system blockade by systemically active aminopeptidase A inhibitors: A potential treatment of salt-dependent hypertension. Proc. Natl. Acad. Sci. USA 2004, 101, 7775–7780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.W.; Harding, J.W. The brain renin–angiotensin system: A diversity of functions and implications for CNS diseases. Pflüg. Arch. Eur. J. Physiol. 2013, 465, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Farag, E.; Sessler, D.I.; Ebrahim, Z.; Kurz, A.; Morgan, J.; Ahuja, S.; Maheshwari, K.; John Doyle, D. The renin angiotensin system and the brain: New developments. J. Clin. Neurosci. 2017, 46, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pal Khaket, T.; Singh, J.; Attri, P.; Dhanda, S. Enkephalin degrading enzymes: Metalloproteases with high potential for drug development. Curr. Pharm. Des. 2012, 18, 220–230. [Google Scholar] [CrossRef]
- Karsten, S.L.; Sang, T.-K.; Gehman, L.T.; Chatterjee, S.; Liu, J.; Lawless, G.M.; Sengupta, S.; Berry, R.W.; Pomakian, J.; Oh, H.S.; et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron 2006, 51, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Horowitz, P.M.; Karsten, S.L.; Jackson, G.R.; Geschwind, D.H.; Fu, Y.; Berry, R.W.; Binder, L.I. Degradation of tau protein by puromycin-sensitive aminopeptidase in vitro. Biochemistry 2006, 45, 15111–15119. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, N.; Venkatraman, P.; Goldberg, A.L. Puromycin-sensitive aminopeptidase is the major peptidase responsible for digesting polyglutamine sequences released by proteasomes during protein degradation. EMBO J. 2007, 26, 1385–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, F.M.; Hourez, R.; Imarisio, S.; Raspe, M.; Sadiq, O.; Chandraratna, D.; O’Kane, C.; Rock, K.L.; Reits, E.; Goldberg, A.L.; et al. Puromycin-sensitive aminopeptidase protects against aggregation-prone proteins via autophagy. Hum. Mol. Genet. 2010, 19, 4573–4586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, L.C.; Parfenova, L.; Ren, G.; Vi, N.; Hui, M.; Ma, Z.; Lau, K.; Gray, M.; Bardag-Gorce, F.; Wiedau-Pazos, M.; et al. Puromycin-sensitive aminopeptidase (PSA/NPEPPS) impedes development of neuropathology in hPSA/TAUP301L double-transgenic mice. Hum. Mol. Genet. 2011, 20, 1820–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruppa, A.J.; Ott, S.; Chandraratna, D.S.; Irving, J.A.; Page, R.M.; Speretta, E.; Seto, T.; Camargo, L.M.; Marciniak, S.J.; Lomas, D.A.; et al. Suppression of Aβ toxicity by puromycin-sensitive aminopeptidase is independent of its proteolytic activity. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 2115–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voorhis, W.C.V.; Adams, J.H.; Adelfio, R.; Ahyong, V.; Akabas, M.H.; Alano, P.; Alday, A.; Resto, Y.A.; Alsibaee, A.; Alzualde, A.; et al. Open source drug discovery with the malaria box compound collection for neglected diseases and beyond. PLoS Pathog. 2016, 12, e1005763. [Google Scholar] [CrossRef] [PubMed]
- Mpakali, A.; Saridakis, E.; Harlos, K.; Zhao, Y.; Papakyriakou, A.; Kokkala, P.; Georgiadis, D.; Stratikos, E. Crystal structure of insulin-regulated aminopeptidase with bound substrate analogue provides insight on antigenic epitope precursor recognition and processing. J. Immunol. 2015, 195, 2842–2851. [Google Scholar] [CrossRef] [PubMed]
- Mpakali, A.; Giastas, P.; Mathioudakis, N.; Mavridis, I.M.; Saridakis, E.; Stratikos, E. Structural basis for antigenic peptide recognition and processing by endoplasmic reticulum (ER) aminopeptidase 2. J. Biol. Chem. 2015, 290, 26021–26032. [Google Scholar] [CrossRef] [PubMed]
- Golich, F.C.; Han, M.; Crowder, M.W. Over-expression, purification and characterization of aminopeptidase N from Escherichia coli. Protein Expr. Purif. 2006, 47, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Minick, D.J.; Frenz, J.H.; Patrick, M.A.; Brent, D.A. A comprehensive method for determining hydrophobicity constants by reversed-phase high-performance liquid chromatography. J. Med. Chem. 1988, 31, 1923–1933. [Google Scholar] [CrossRef] [PubMed]
- Pomper, M.G.; VanBrocklin, H.; Thieme, A.M.; Thomas, R.D.; Kiesewetter, D.O.; Carlson, K.E.; Mathias, C.J.; Welch, M.J.; Katzenellenbogen, J.A. 11.beta.-Methoxy-, 11.beta.-ethyl and 17.alpha.-ethynyl-substituted 16.alpha.-fluoroestradiols: Receptor-based imaging agents with enhanced uptake efficiency and selectivity. J. Med. Chem. 1990, 33, 3143–3155. [Google Scholar] [CrossRef] [PubMed]
- Hitzerd, S.M.; Verbrugge, S.E.; Ossenkoppele, G.; Jansen, G.; Peters, G.J. Positioning of aminopeptidase inhibitors in next generation cancer therapy. Amino Acids 2014, 46, 793–808. [Google Scholar] [CrossRef] [PubMed]
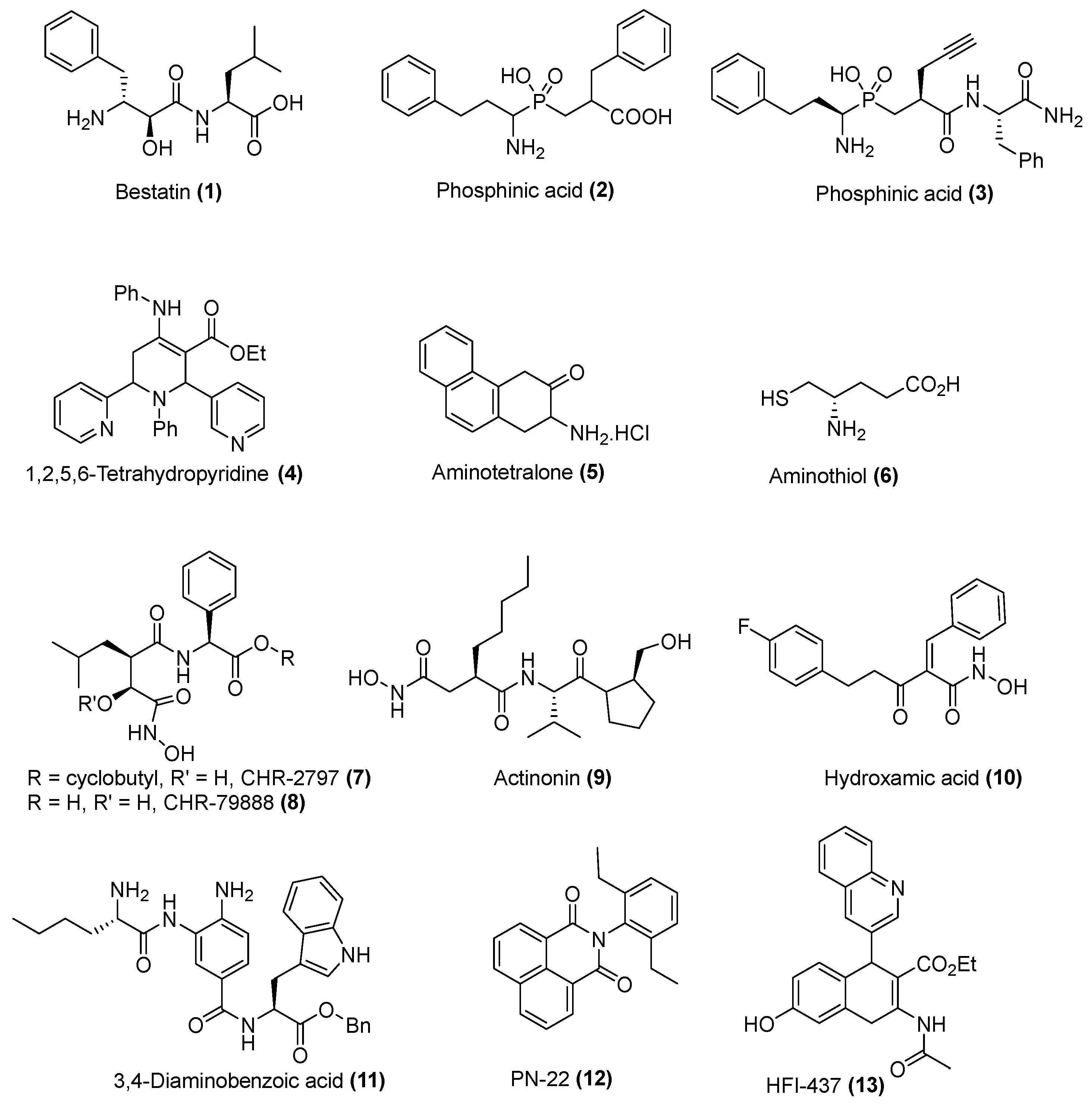
Sample Availability: Samples of the compounds 21a–j are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | M1 Family | Biological Roles | Associated Diseases |
|---|---|---|---|
| H. sapiens | APN | Metabolism of regulatory peptides of diverse cell types Processing of peptide hormones (Angiotensin III and IV, neuropeptides and chemokines Regulation of angiogenesis, cell motility, cell-cell adhesion Coronavirus receptor | Pain sensation, Inflammatory diseases, cancer & upper respiratory tract infections |
| APA | Activation or inactivation of various components of the angiotensin system | Hypertension | |
| LTA4H | Biosynthesis of the proinflammatory mediator LTB-4 | Inflammatory & allergic diseases | |
| TRHDE | Inactivation of Thyrotropin Releasing Hormone (brain) | ||
| PSA | Regulation of neuropeptide activity, digestion of polyQ peptides Antigen processing pathway for MHC class I molecules | hematologic cancer Impedes the development of neuropathology | |
| IRAP | Peptide hormone degradation (oxytocin, vasopressin, angiotensin III) Maintain homeostasis during pregnancy Inactivation of neuronal peptide (enkephalin, dynorphin) Generation of antigenic peptides in dendritic cells [12] | cognitive impairment (Alzheimer’s disease, head trauma, cerebral ischemia) Inflammatory autoimmunity [13] | |
| APB | Biosynthesis of the proinflammatory mediator LTB-4 | ||
| ERAP1 | Regulation of blood pressure Antigen processing pathway for MHC class I molecules | Autoimmune diseases (ankylosing spondylitis, psoriasis, type 1 diabetes, Crohn…) Cancer [14,15,16] | |
| ERAP2 | Antigen processing pathway for MHC class I molecules | ||
| APQ | Placentation: regulation of biological activity of key peptides at the embryo-maternal interface | Pre-eclampsia | |
| APO | Activation or inactivation of various components of the angiotensin system | ||
| P. falciparum | PfAM1 | Catabolism of host haemoglobin in the food vacuole of Plasmodium | Malaria [17] |
| E. coli | PepN | Cytosolic peptide catabolism and adaptation to nutritional downshift and high temperature stress |
| M1 Aminopeptidase | Substrate Specificity: Favoured N-Terminus Amino-Acid |
|---|---|
| APN | Ala, Phe, Tyr, Leu, most of aa including Pro (slow) |
| APA | Glu and to a lesser extent Asp |
| LTA4H | Ala, Arg, Leu, Pro |
| TRHDE | pGlu (pyroglutamyl) |
| PSA | Ala, Leu, Lys, most of aa (except Gly and Pro) |
| IRAP | Cys, Leu, Arg, Ala and most of aa (except Asp, Glu) including cyclic peptides [27] |
| APB | Arg, Lys |
| ERAP1 | Leu and most of aa including Met, Cys, Phe |
| ERAP2 | Arg, Lys |
| RNPL1 | Ala, Lys, Ser, Ile, Met, most of aa |
| APQ | Leu, Arg, Lys, Met, most of aa |
| APO | Arg and to a lesser extent Asn |
| PfAM1 | Ala, Leu, Lys, Arg and most of aa (except Pro, Asp, Glu) |
| PepN | Arg, Ala, most of aa including Pro |
| Compound | M1 Aminopeptidases ‘Monometallic’ | M17 & M28 Aminopeptidases ‘Bimetallic’ | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HsAPA | HsAPB | mAPN | HsERAP1 | HsERAP2 | HsIRAP | HsLTA4H | HsPSA | EcPepN | PfAM1 | AAP | mLAPc | PfAM17 | |
| 1 | NI [38] | 0.014 [39]–6.0 [38] | 0.3 * [37]–89.0 [40] | 11.2 [41] | 0.2 * [37] 0.5 [42] | 0.35 * [37] | 7.3 * [43] | 0.19 [44] | 0.0016 [42] | 0.0006 [38]–0.020 [39] | 0.025 [45] | ||
| 2 | - | - | 0.002 [46] | - | - | - | - | - | - | 0.079 [47] | - | 0.066 [33] | 0.013 [45] |
| 3 [48] | 0.043 * | 0.037 * | 0.002 * | ||||||||||
| 4 [49] | >100 | 1.79 * | >100 | >100 | |||||||||
| 5 [38] | >1000 | >1000 | 0.02 | 3.0 | |||||||||
| 6 [50] | 0.14 | 0.12 | |||||||||||
| 7 [37] 8 [37] | - | >1 * | 0.22 * | >5 * | - | - | >10 * 0.008 * | 0.15 * 0.85 * | - | 6 [36] | - | 0.1 * 0.03 * | 0.079 [36] - |
| 9 | >100 [40] | >100 [51] | 0.3 [52]–2.0 * [40] | 2.6 * [51] | |||||||||
| 10 [35,53] | 1.37 * | 0.006 * | |||||||||||
| 11 [54] | 0.92 * | 1.6 * | 0.105 * | ||||||||||
| 12 [55] | >100 | 0.8 * | |||||||||||
| 13 [56] | 0.03 | ||||||||||||
 | R1 | R4 | M1 Aminopeptidases ‘Monometallic’ | M17 & M28 Aminopeptidases ‘Bimetallic’ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Papn k [58] | rPfAM1 l | REcPepN m | rHsERAP1 n * | rHsERAP2 o * | rHsPSA p | rHsIRAP q * | rHsLTA4H s [58] | rPfAM17 t | AAP u [58] | |||
| 21a | H | H | 1 | 15 ± 0.07 | 50 ± 3.0 | - | - | - | - | >100 | - | >100 |
| 21b | H | Br | 0.04 | 0.04 ± 0.009 | 1.07 ± 0.11 | 120 ± 11 | 49 ± 3.8 | 1.63 ± 0.08 | 1.0 ± 0.08 | >100 | >100 | >100 |
| 21c | H | Ph | 0.007 | 0.05 ± 0.005 [69] | 0.16 ± 0.012 | 63 ± 3 | 18 ± 1.3 | 0.05 ± 0.004 | 2.4 ± 0.3 | 19 | >100 [69] | 28 |
| 21d | H | Fc | 0.004 [60] | >50 | 0.06 ± 0.008 | - | - | - | - | - | - | - |
| 21e | Br | H | 0.02 | 1 ± 0.04 | 4.59 ± 0.17 | 56 ± 7 | 104 ± 7 | 1.07 ± 0.063 | 2.6 ± 0.2 | >100 | >100 | >100 |
| 21f | Ph | H | 0.25 | 2 ± 0.07 | - | 137 ± 5 | >200 | - | 2.2 ± 0.13 | >100 | >100 | >100 |
| 21g | Benzo [1,2] | 0.04 | 15 ± 1 | - | - | - | - | - | >100 | - | >100 | |
| 21h | Br | Br | 0.006 | 0.005 ± 0.001 | - | 5.5 ± 1.1 | 2.8 ± 0.3 | - | 0.034 ± 0.001 | >100 | - | >100 |
| 21i | Br | Ph | 0.00006 | 0.03 ± 0.005 | 0.05 ± 0.003 | 1.6 ± 0.1 | 0.39 ± 0.013 | 0.021 ± 0.003 | 0.12 ± 0.015 | 68% # | - | 39 |
| 21j | Ph | Br | 0.07 | 4 ± 0.7 | - | - | - | - | - | 54% # | - | >100 |
| M1 AP | PDB | Amino Acid Composition of the S1 Subsite | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HsAPN | 4FYT | Q211 | Q213 | N350 | A351 | G352 | A353 | M354 | E355 | H392 | E411 | F472 | Y477 | F896 | |||||||
| 4FYR | Q211 | Q213 | A214 | D216 | N350 | A351 | G352 | A353 | M354 | E355 | H392 | E411 | S469 | F472 | Y477 | S895 | S897 | ||||
| HsERAP1 | 2YD0 | H160 | Q181 | E183 | P184 | A186 | F314 | Q315 | S316 | G317 | A318 | M319 | E320 | H357 | E376 | R430 | F433 | Y438 | E865 | S868 | S869 |
| HsERAP2 | 4JBS | E177 | D198 | E200 | P201 | Q203 | F331 | A332 | P333 | G334 | A335 | M336 | E337 | H374 | E393 | Q447 | F450 | Y455 | Y892 | R895 | |
| HsIRAP | 5MJ6 | Y272 | Q293 | E295 | P296 | F425 | E426 | A427 | G428 | A429 | M430 | E431 | H468 | E487 | E541 | F544 | Y549 | P957 | Y961 | ||
| HsLTA4H | 1HS6 | Q134 | Q136 | A137 | Y267 | G268 | G269 | M270 | E271 | H299 | E318 | D375 | Y378 | Y383 | |||||||
| EcPepN | 5MFS | Q119 | E121 | A122 | M260 | G261 | A262 | M263 | E264 | H301 | E320 | Y376 | Y381 | R825 | |||||||
| 2DQM | Q119 | E121 | A122 | N259 | M260 | G261 | A262 | M263 | E264 | H301 | E320 | N373 | Y376 | Y381 | Q821 | R825 | |||||
| PfAM1 | 3EBH | T305 | Q317 | E319 | A320 | T321 | F457 | N458 | V459 | G460 | A461 | M462 | E463 | H500 | E519 | E572 | Y575 | Y580 | M1034 | ||
| M1 AP | Amino Acid Composition of the S1′ Subsite | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HsAPN | G352 | A353 | R381 | T384 | V385 | H388 | E389 | S415 | E418 | Y477 | |||
| HsERAP1 | G317 | A318 | G346 | M349 | T350 | H353 | E354 | K380 | E383 | Y438 | |||
| HsERAP2 | G334 | A335 | S348 | W363 | R366 | V367 | H370 | E371 | K397 | E400 | Y455 | ||
| HsIRAP | G428 | A429 | T442 | L457 | K460 | I461 | H464 | E465 | T491 | E494 | Y549 | ||
| HsLTA4H | G268 | G269 | N291 | V292 | H295 | E296 | V322 | E325 | Y383 | R563 | K565 | ||
| EcPepN | G261 | A262 | Y275 | R293 | V294 | H297 | E298 | V324 | D327 | Y381 | |||
| PfAM1 | G460 | A461 | R489 | T492 | V493 | H496 | E497 | V523 | E526 | Y580 | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salomon, E.; Schmitt, M.; Marapaka, A.K.; Stamogiannos, A.; Revelant, G.; Schmitt, C.; Alavi, S.; Florent, I.; Addlagatta, A.; Stratikos, E.; et al. Aminobenzosuberone Scaffold as a Modular Chemical Tool for the Inhibition of Therapeutically Relevant M1 Aminopeptidases. Molecules 2018, 23, 2607. https://doi.org/10.3390/molecules23102607
Salomon E, Schmitt M, Marapaka AK, Stamogiannos A, Revelant G, Schmitt C, Alavi S, Florent I, Addlagatta A, Stratikos E, et al. Aminobenzosuberone Scaffold as a Modular Chemical Tool for the Inhibition of Therapeutically Relevant M1 Aminopeptidases. Molecules. 2018; 23(10):2607. https://doi.org/10.3390/molecules23102607
Chicago/Turabian StyleSalomon, Emmanuel, Marjorie Schmitt, Anil Kumar Marapaka, Athanasios Stamogiannos, Germain Revelant, Céline Schmitt, Sarah Alavi, Isabelle Florent, Anthony Addlagatta, Efstratios Stratikos, and et al. 2018. "Aminobenzosuberone Scaffold as a Modular Chemical Tool for the Inhibition of Therapeutically Relevant M1 Aminopeptidases" Molecules 23, no. 10: 2607. https://doi.org/10.3390/molecules23102607