
Structure-Based Identification of Potent Natural Product Chemotypes as Cannabinoid Receptor 1 Inverse Agonists

 , ,
, ,
Abstract
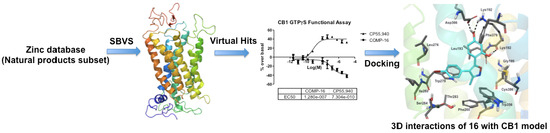
:
1. Introduction
2. Results and Discussion
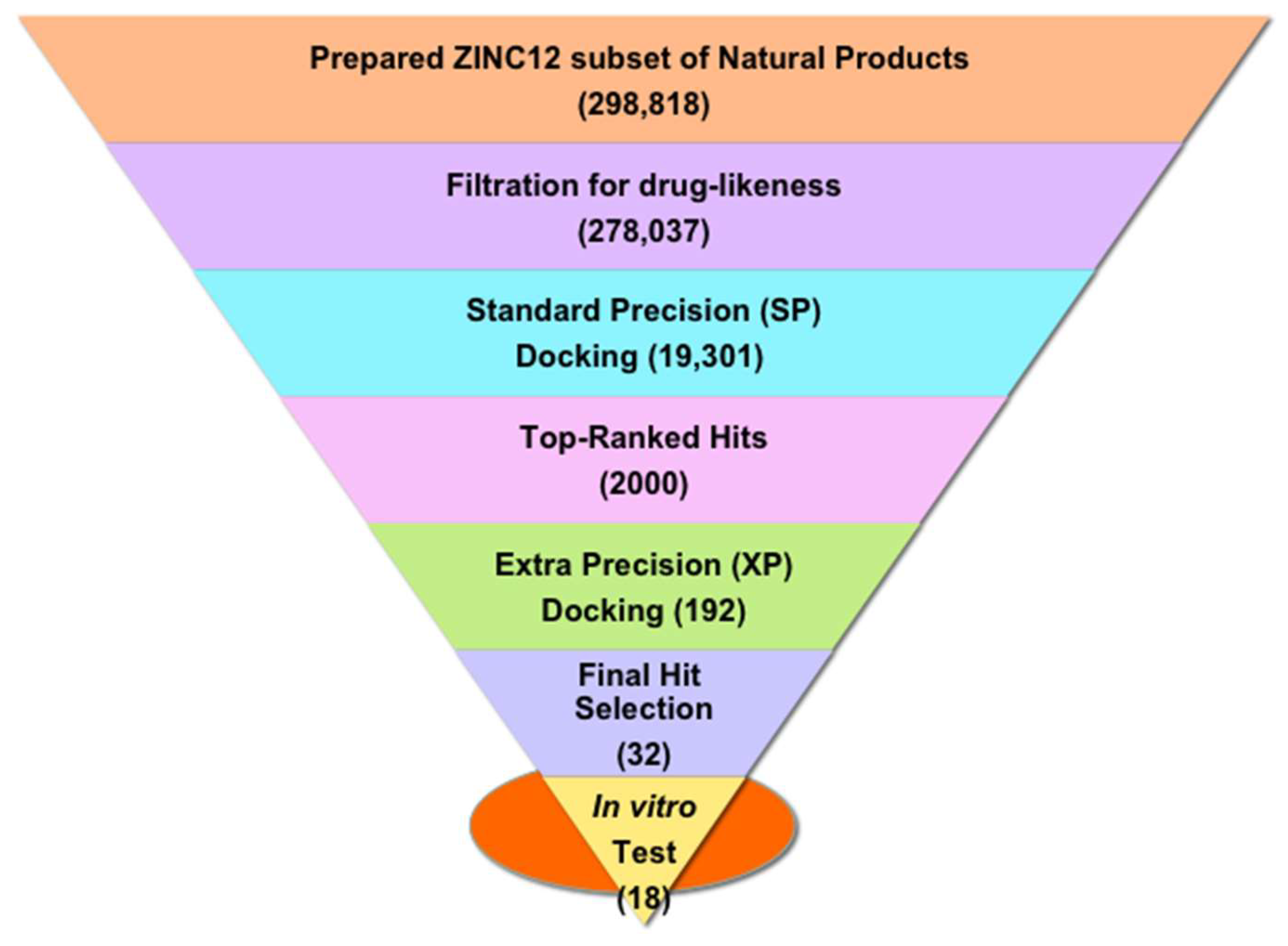
2.1. Structure-Based Virtual Screening of the ZINC12 Subset of Natural Products
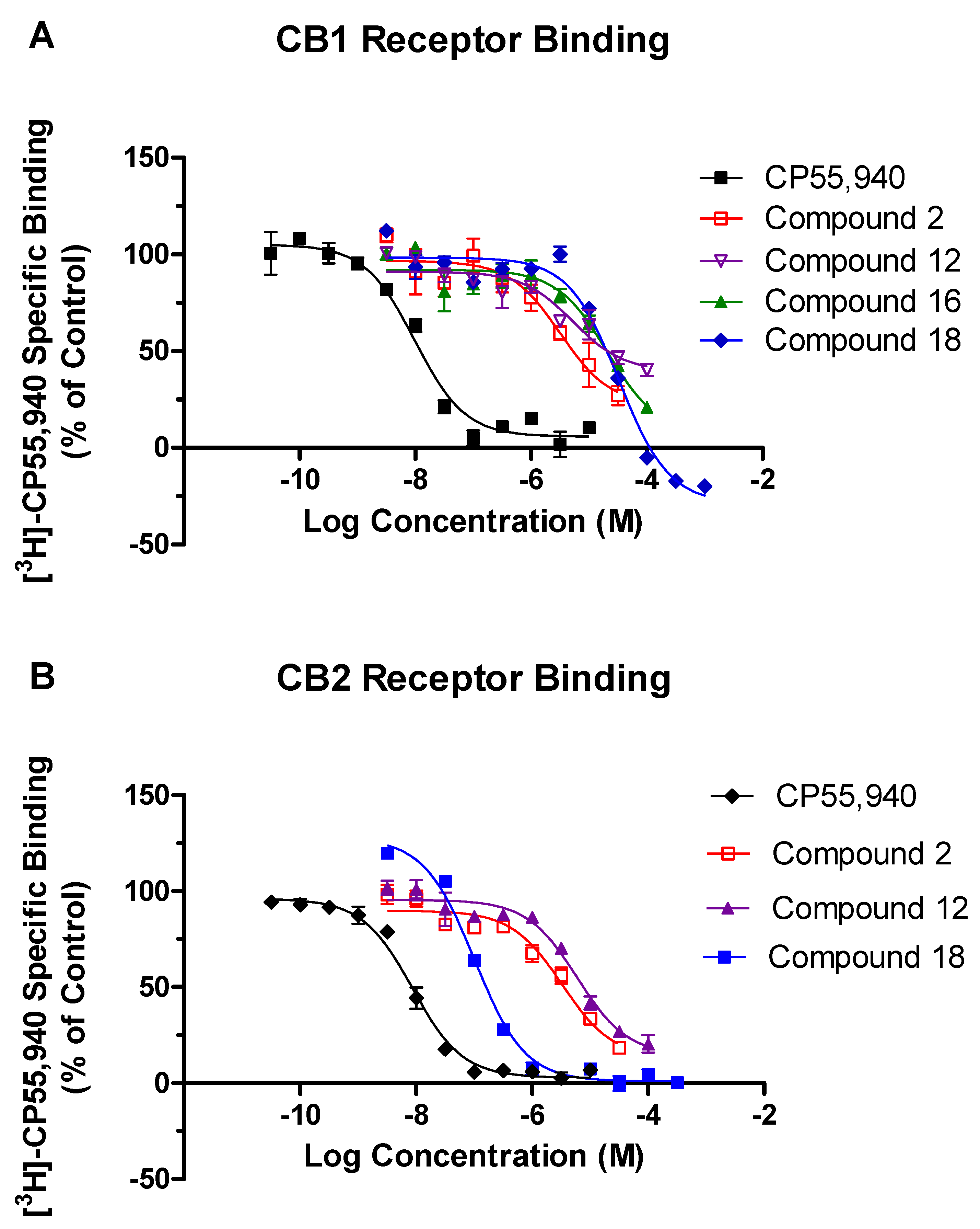
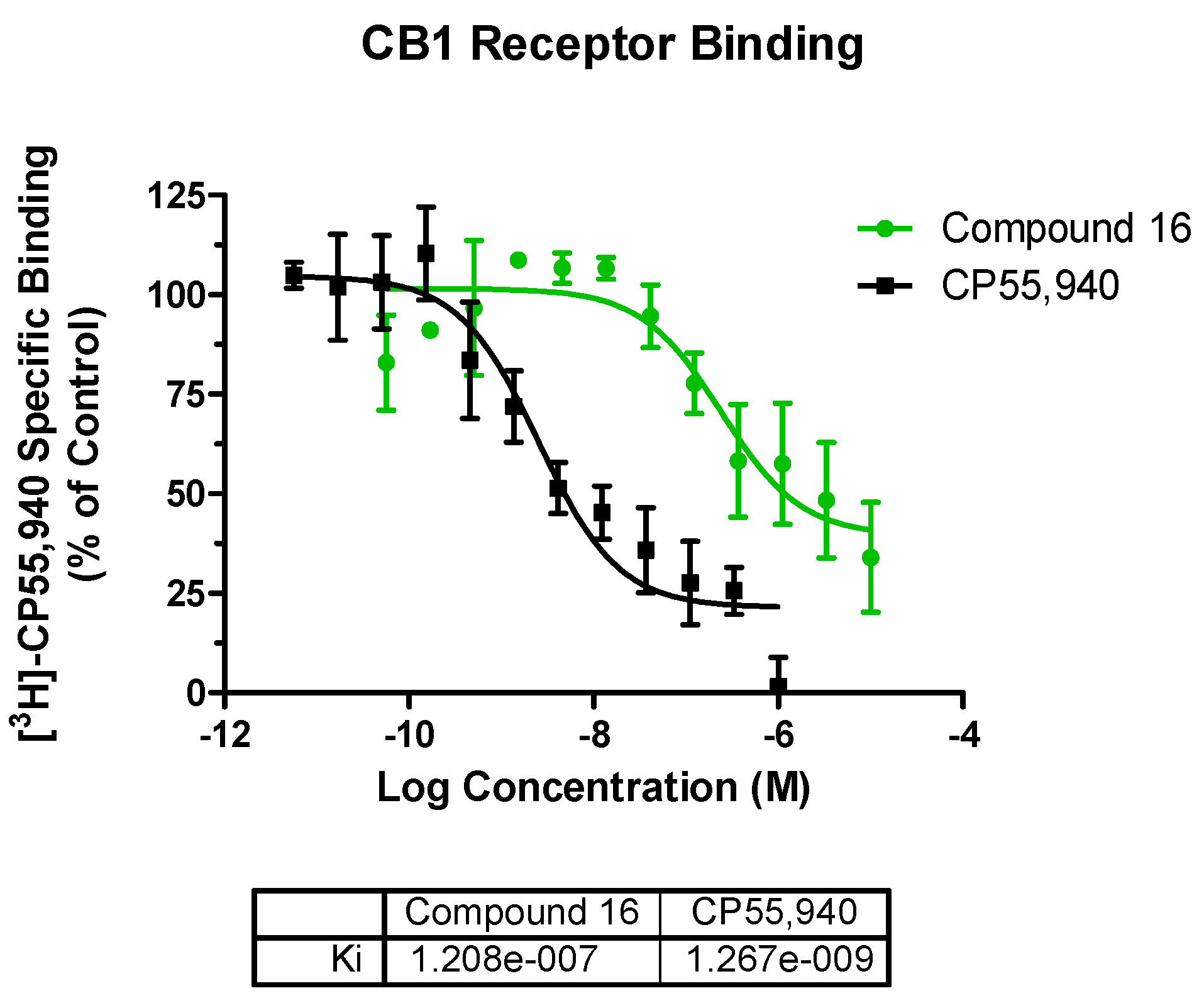
2.2. In Vitro Screening in the Competitive Radioligand Binding Assay
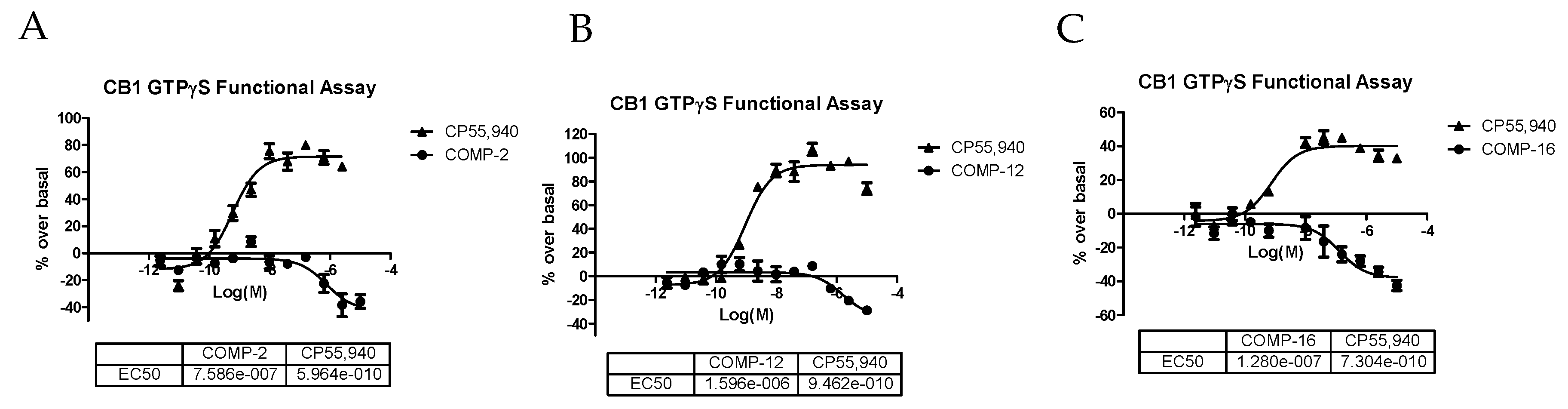
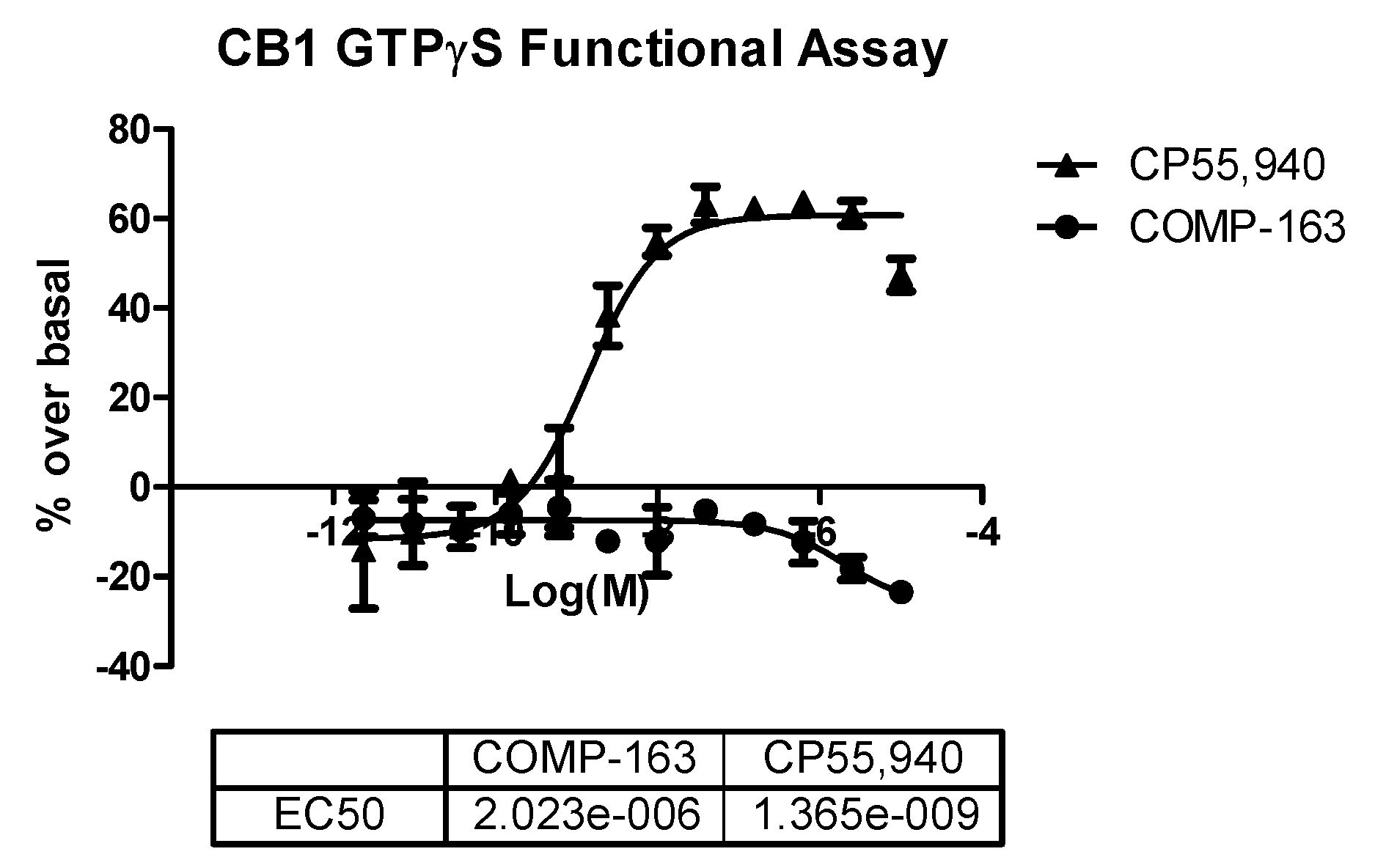
2.3. In Vitro GTPγS Functional Assays for CB1 and CB2 Receptors
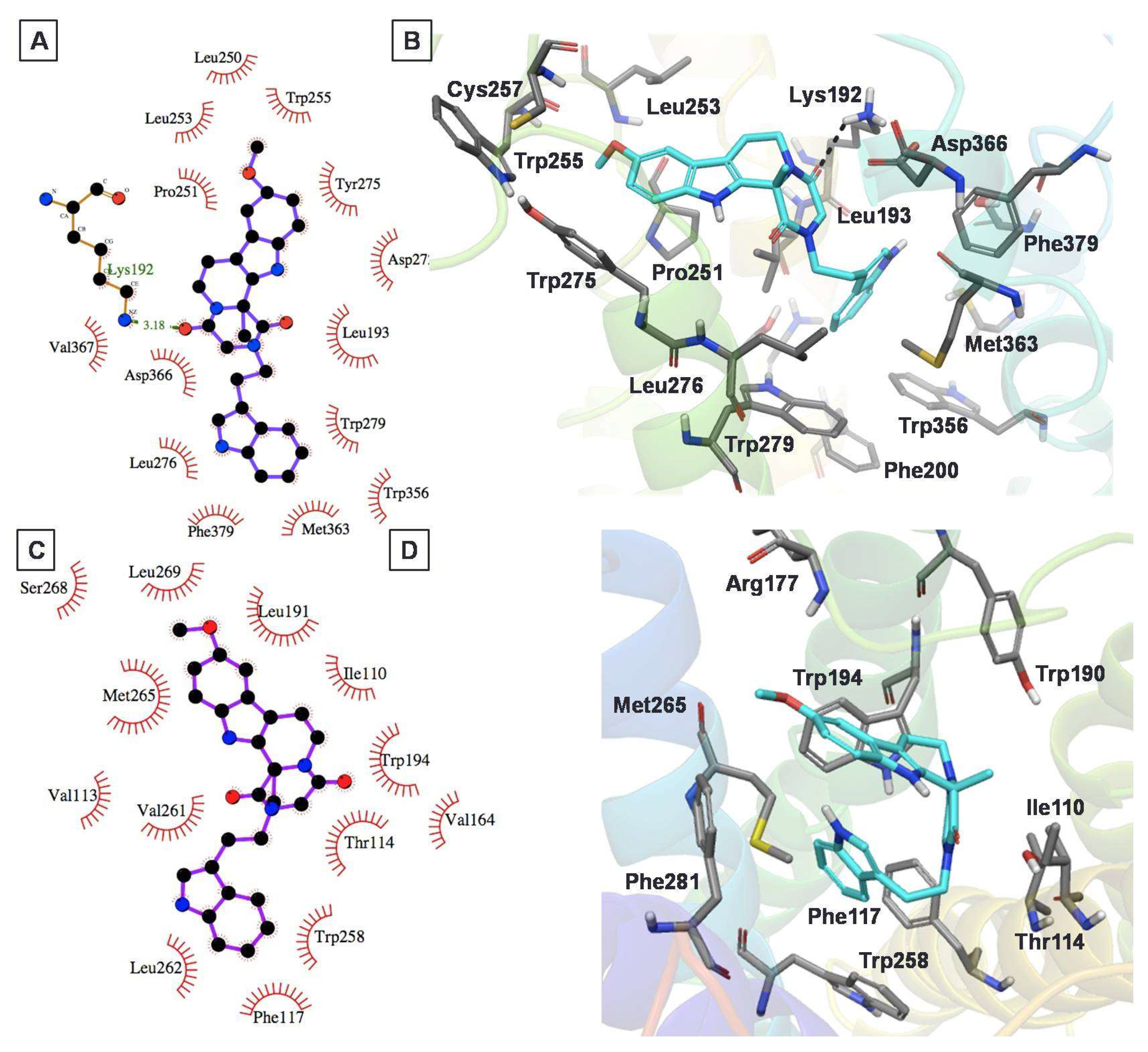
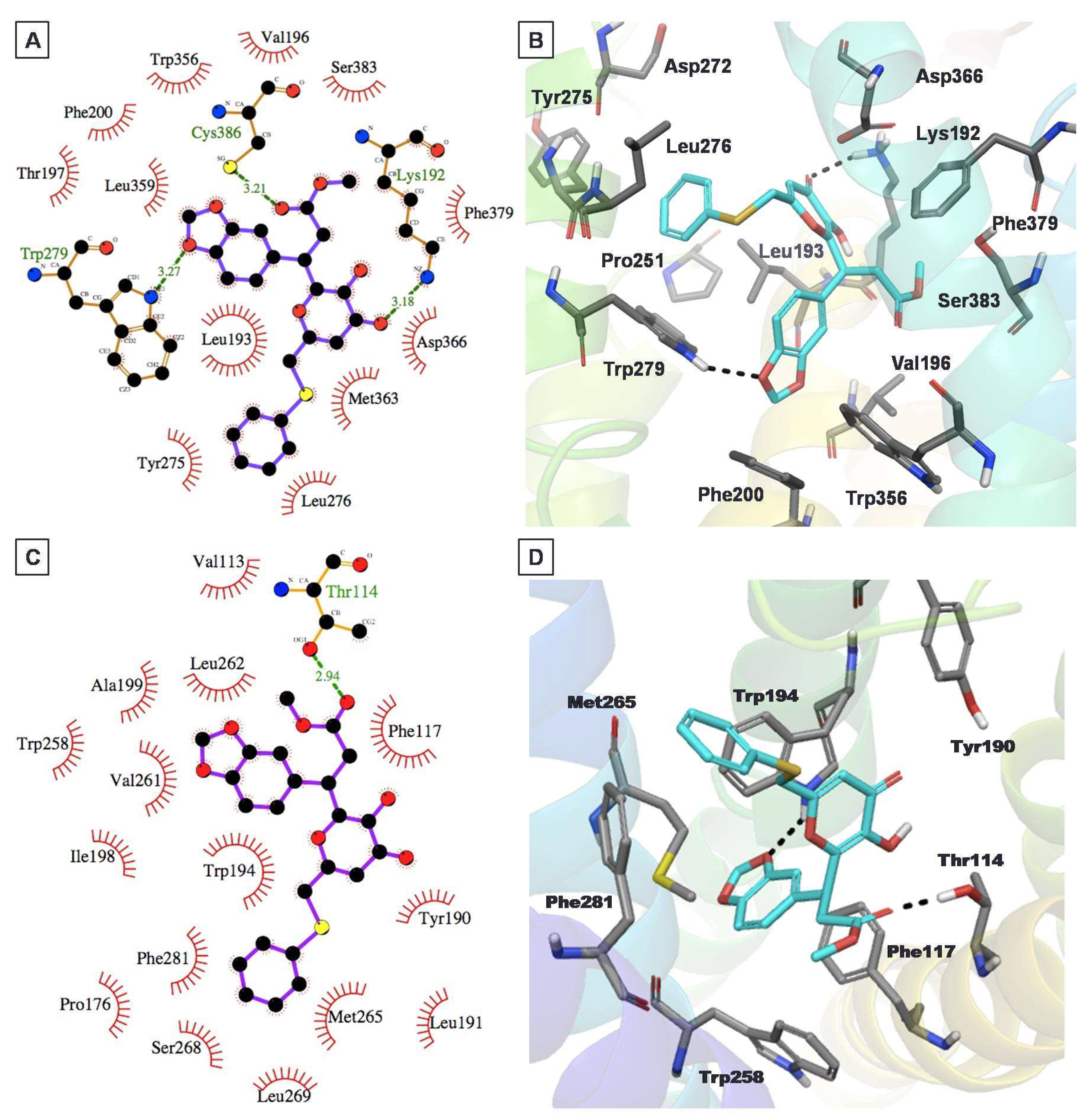
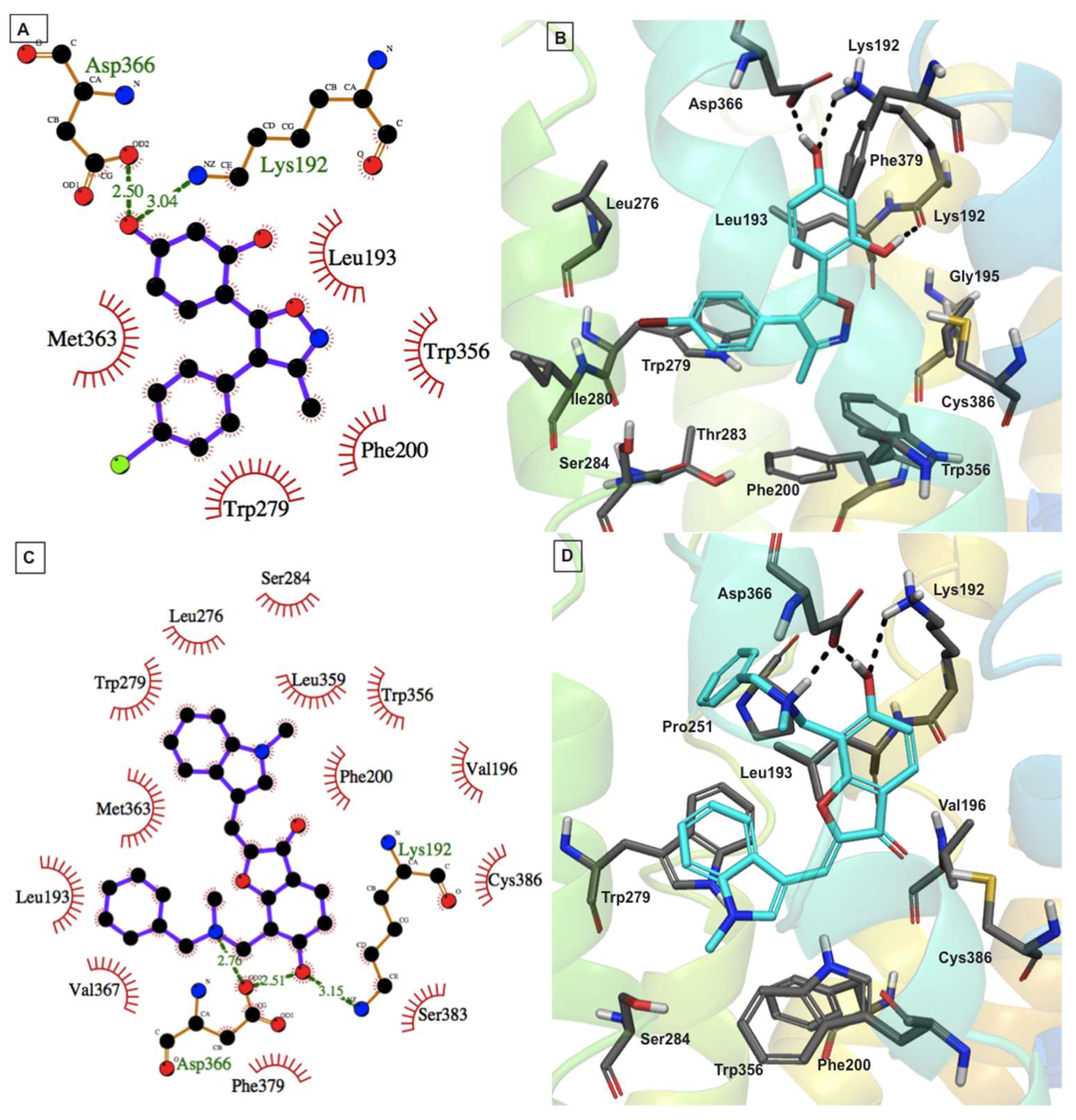
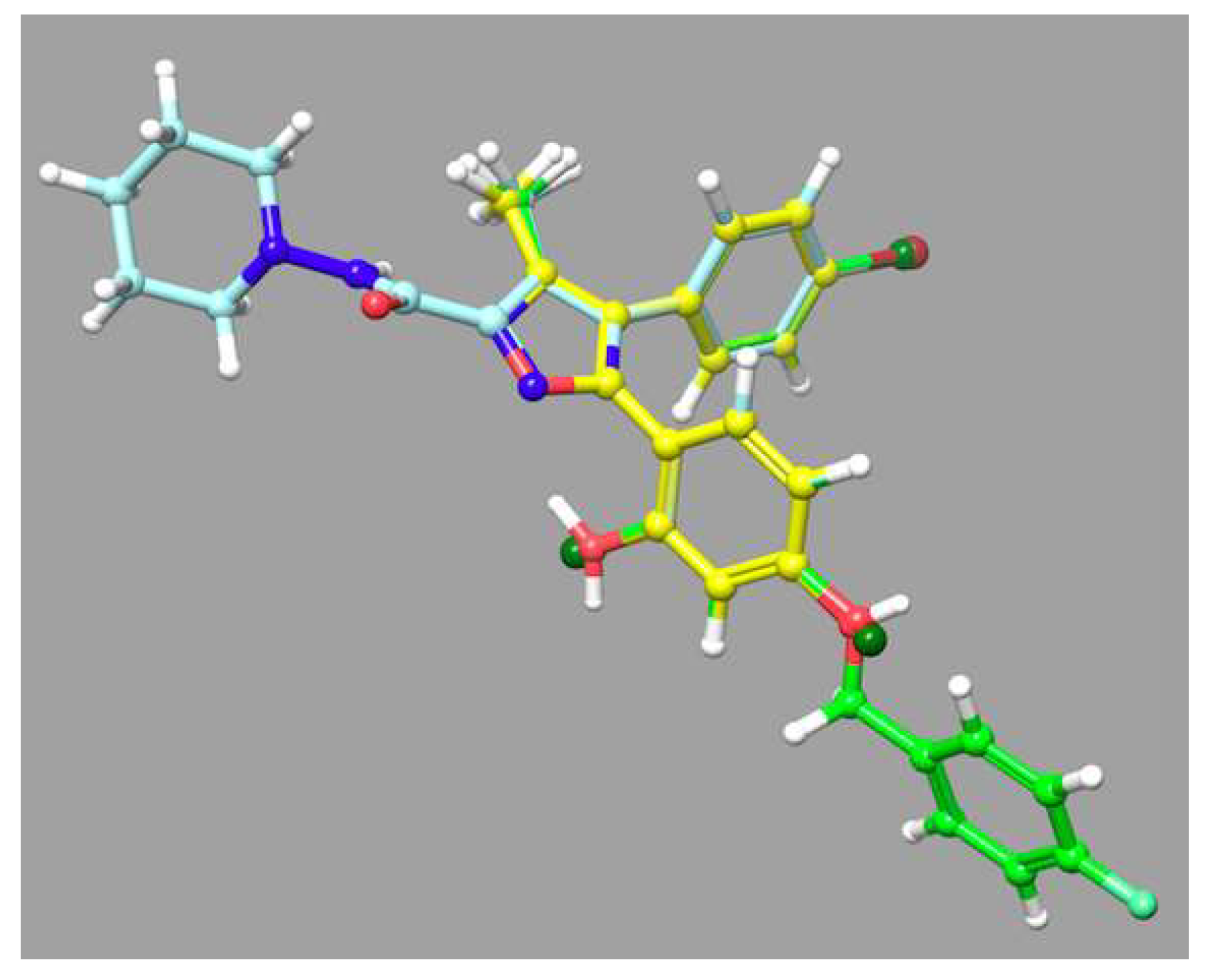
2.4. Protein–Ligand Interactions
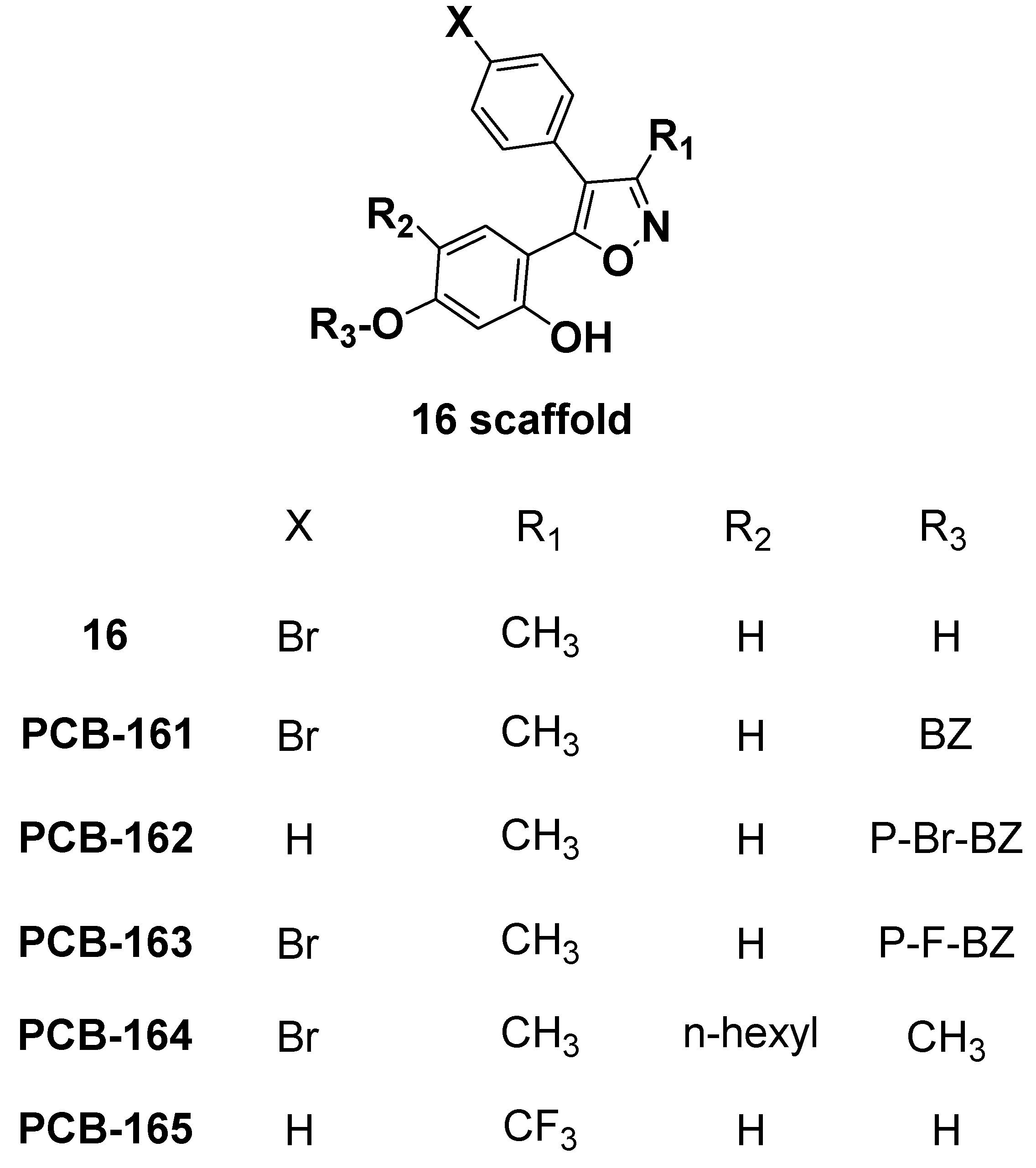
2.5. Analog Exploration
3. Materials and Methods
3.1. Computational Methods and Approaches
3.1.1. Protein Preparation, Receptor Grid Generation, and Amino Acid Numbering System
3.1.2. Database Preparation
3.1.3. Structure-Based Virtual Screening: Docking and Scoring
3.1.4. Hit Postprocessing and Selection of Hits
3.1.5. Procurement, Purity Assessments, and Characterization of Selected Hits
3.2. Biological Methods
3.2.1. Reagents
3.2.2. Cell Culture
3.2.3. Transfection and Stable Expression of CB1 and CB2 Receptors in Mammalian Cell Lines
3.2.4. Membrane Preparation
3.2.5. Radioligand Receptor Binding Studies
3.2.6. CB1 GTPγS Functional Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stevens, R.C.; Cherezov, V.; Katritch, V.; Abagyan, R.; Kuhn, P.; Rosen, H.; Wuthrich, K. The GPCR Network: A large-scale collaboration to determine human GPCR structure and function. Nat. Rev. Drug Discov. 2013, 12, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.H.; Grossmann, M.; Ji, I. G protein-coupled receptors. I. Diversity of receptor-ligand interactions. J. Biol. Chem. 1998, 273, 17299–17302. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Song, C.; Berglund, B.A.; Wilken, G.H.; Pigg, J.J. Characterization of CB1 cannabinoid receptors using receptor peptide fragments and site-directed antibodies. Mol. Pharmacol. 1998, 53, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef]
- Galiegue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carriere, D.; Carayon, P.; Bouaboula, M.; Shire, D.; Le Fur, G.; Casellas, P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur. J. Biochem. 1995, 232, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. An overview of the cannabinoid type 2 receptor system and its therapeutic potential. Curr. Opin. Anaesthesiol. 2018, 31, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Gonsiorek, W.; Hesk, D.; Chen, S.C.; Kinsley, D.; Fine, J.S.; Jackson, J.V.; Bober, L.A.; Deno, G.; Bian, H.; Fossetta, J.; et al. Characterization of peripheral human cannabinoid receptor (hCB2) expression and pharmacology using a novel radioligand, [35S]SCH225336. J. Biol. Chem. 2006, 281, 28143–28151. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.P.; Patel, S.; Perchuk, A.; Meozzi, P.A.; Myers, L.; Mora, Z.; Tagliaferro, P.; Gardner, E.; et al. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann. N.Y. Acad. Sci. 2006, 1074, 514–536. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Tang, Y. The central cannabinoid receptor type-2 (CB2) and chronic pain. Int. J. Neurosci. 2017, 127, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.J.; Gao, M.; Gao, F.F.; Su, Q.X.; Wu, J. Brain cannabinoid receptor 2: Expression, function and modulation. Acta Pharmacol. Sin. 2017, 38, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Furjanic, M.A.; Ferrara, J.J.; McAndrew, N.R.; Ardino, E.L.; Ngondara, A.; Bernstein, Y.; Thomas, K.J.; Kim, E.; Walker, J.M.; et al. The endocannabinoid system and rimonabant: A new drug with a novel mechanism of action involving cannabinoid CB1 receptor antagonism–or inverse agonism–as potential obesity treatment and other therapeutic use. J. Clin. Pharm. Ther. 2007, 32, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Kunos, G.; Tam, J. The case for peripheral CB(1) receptor blockade in the treatment of visceral obesity and its cardiometabolic complications. Br. J. Pharmacol. 2011, 163, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; Chowen, J.; Rocio, A.C.M.; del Arco, I.; Villanua, M.A.; Martin, Y.; Roberts, A.J.; Koob, G.F.; de Fonseca, F.R. CB1 cannabinoid receptor antagonist-induced opiate withdrawal in morphine-dependent rats. Neuroreport 1998, 9, 3397–3402. [Google Scholar] [CrossRef] [PubMed]
- Beardsley, P.M.; Thomas, B.F.; McMahon, L.R. Cannabinoid CB1 receptor antagonists as potential pharmacotherapies for drug abuse disorders. Int. Rev. Psychiatry. 2009, 21, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi-Carmona, M.; Barth, F.; Heaulme, M.; Shire, D.; Calandra, B.; Congy, C.; Martinez, S.; Maruani, J.; Neliat, G.; Caput, D.; et al. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994, 350, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Van Gaal, L.F.; Rissanen, A.M.; Scheen, A.J.; Ziegler, O.; Rossner, S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet 2005, 365, 1389–1397. [Google Scholar] [CrossRef] [Green Version]
- Despres, J.P.; Golay, A.; Sjostrom, L. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N. Engl. J. Med. 2005, 353, 2121–2134. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, F.X.; Aronne, L.J.; Heshmati, H.M.; Devin, J.; Rosenstock, J. Effect of rimonabant, a cannabinoid-1 receptor blocker, on weight and cardiometabolic risk factors in overweight or obese patients: RIO-North America: A randomized controlled trial. JAMA 2006, 295, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Christopoulou, F.D.; Kiortsis, D.N. An overview of the metabolic effects of rimonabant in randomized controlled trials: Potential for other cannabinoid 1 receptor blockers in obesity. J. Clin. Pharm. Ther. 2011, 36, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Cinar, R.; Liu, J.; Godlewski, G.; Wesley, D.; Jourdan, T.; Szanda, G.; Mukhopadhyay, B.; Chedester, L.; Liow, J.S.; et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012, 16, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Shui, F.; Liu, C.; Zhou, X.; Li, W.; Zheng, Z.; Fu, W.; Wang, L. Novel peripherally restricted cannabinoid 1 receptor selective antagonist TXX-522 with prominent weight-loss efficacy in diet induced obese mice. Front. Pharmacol. 2017, 8, 707. [Google Scholar] [CrossRef] [PubMed]
- Ashton, J.C.; Glass, M. The cannabinoid CB2 receptor as a target for inflammation-dependent neurodegeneration. Curr. Neuropharmacol. 2007, 5, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Toguri, J.T.; Lehmann, C.; Laprairie, R.B.; Szczesniak, A.M.; Zhou, J.; Denovan-Wright, E.M.; Kelly, M.E. Anti-inflammatory effects of cannabinoid CB(2) receptor activation in endotoxin-induced uveitis. Br. J. Pharmacol. 2014, 171, 1448–1461. [Google Scholar] [CrossRef] [PubMed]
- Defer, N.; Wan, J.; Souktani, R.; Escoubet, B.; Perier, M.; Caramelle, P.; Manin, S.; Deveaux, V.; Bourin, M.C.; Zimmer, A.; et al. The cannabinoid receptor type 2 promotes cardiac myocyte and fibroblast survival and protects against ischemia/reperfusion-induced cardiomyopathy. FASEB J. 2009, 23, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Thatte, J.; Buzard, D.J.; Jones, R.M. Therapeutic utility of cannabinoid receptor type 2 (CB(2)) selective agonists. J. Med. Chem. 2013, 56, 8224–8256. [Google Scholar] [CrossRef] [PubMed]
- Pressly, J.D.; Mustafa, S.M.; Adibi, A.H.; Alghamdi, S.; Pandey, P.; Roy, K.K.; Doerksen, R.J.; Moore, B.M., Jr.; Park, F. Selective cannabinoid 2 receptor stimulation reduces tubular epithelial cell damage after renal ischemia-reperfusion injury. J. Pharmacol. Exp. Ther. 2018, 364, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal structure of the human cannabinoid receptor CB1. Cell 2016, 167, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB 1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhang, F.F.; Xie, X.; Chen, J.Z. Design, synthesis, biological evaluation, and comparative docking study of 1,2,4-triazolones as CB1 receptor selective antagonists. Eur. J. Med. Chem. 2014, 74, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Methods for the Development of in Silico GPCR Models. Methods Enzymol. 2017, 593, 405–448. [Google Scholar] [PubMed]
- Gatley, S.J.; Gifford, A.N.; Volkow, N.D.; Lan, R.; Makriyannis, A. 123I-labeled AM251: A radioiodinated ligand which binds in vivo to mouse brain cannabinoid CB1 receptors. Eur. J. Pharmacol. 1996, 307, 331–338. [Google Scholar] [CrossRef]
- Walentiny, D.; Vann, R.; Mahadevan, A.; Kottani, R.; Gujjar, R.; Wiley, J. Novel 3-substituted rimonabant analogues lack Δ9-tetrahydrocannabinol-like abuse-related behavioural effects in mice. Br. J. Pharmacol. 2013, 169, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.L.; Selley, D.E.; Wang, P.; Kottani, R.; Gadthula, S.; Mahadeven, A. 3-Substituted pyrazole analogs of the cannabinoid type 1 (CB(1)) receptor antagonist rimonabant: Cannabinoid agonist-like effects in mice via non-CB(1), non-CB(2) mechanism. J. Pharmacol. Exp. Ther. 2012, 340, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.N.; Kim, K.R.; Ahn, S.H.; Bae, M.A.; Kang, N.S. Discovery of cannabinoid-1 receptor antagonists by virtual screening. Bioorg. Med. Chem. Lett. 2010, 20, 5130–5132. [Google Scholar] [CrossRef] [PubMed]
- Shrinivasan, M.; Skariyachan, S.; Aparna, V.; Kolte, V.R. Homology modelling of CB1 receptor and selection of potential inhibitor against obesity. Bioinformation 2012, 8, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Roy, K.K.; Liu, H.; Elokely, K.M.; Pettaway, S.; Cutler, S.J.; Doerksen, R.J. Search for cannabinoid receptor 1 antagonists using structure-based virtual screening: Identification of natural product hits. Planta Med. 2014, 80, PF11. [Google Scholar] [CrossRef]
- Wang, H.; Duffy, R.A.; Boykow, G.C.; Chackalamannil, S.; Madison, V.S. Identification of novel cannabinoid CB1 receptor antagonists by using virtual screening with a pharmacophore model. J. Med. Chem. 2008, 51, 2439–2446. [Google Scholar] [CrossRef] [PubMed]
- Foloppe, N.; Allen, N.H.; Bentley, C.H.; Brooks, T.D.; Kennett, G.; Knight, A.R.; Leonardi, S.; Misra, A.; Monck, N.J.; Sellwood, D.M. Discovery of a novel class of selective human CB1 inverse agonists. Bioorg. Med. Chem. Lett. 2008, 18, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Foloppe, N.; Benwell, K.; Brooks, T.D.; Kennett, G.; Knight, A.R.; Misra, A.; Monck, N.J. Discovery and functional evaluation of diverse novel human CB(1) receptor ligands. Bioorg. Med. Chem. Lett. 2009, 19, 4183–4190. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Doerksen, R.J. New drugs from natural products around the world. Pharm. Sci. 2016, 22, 215–216. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Patel, R.Y.; Doerksen, R.J. Structure of the cannabinoid receptor 1: Homology modeling of its inactive state and enrichment study based on CB1 antagonist docking. Med. Chem. Comm. 2014, 5, 1297–1302. [Google Scholar] [CrossRef]
- Topiol, S.; Sabio, M. X-ray structure breakthroughs in the GPCR transmembrane region. Biochem. Pharmacol. 2009, 78, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Dysarz, F.A., 3rd; Johnson, M.R.; Melvin, L.S.; Howlett, A.C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988, 34, 605–613. [Google Scholar] [PubMed]
- Dean, B.; Sundram, S.; Bradbury, R.; Scarr, E.; Copolov, D. Studies on [3H]CP-55940 binding in the human central nervous system: Regional specific changes in density of cannabinoid-1 receptors associated with schizophrenia and cannabis use. Neuroscience 2001, 103, 9–15. [Google Scholar] [CrossRef]
- Brents, L.K.; Medina-Bolivar, F.; Seely, K.A.; Nair, V.; Bratton, S.M.; Nopo-Olazabal, L.; Patel, R.Y.; Liu, H.; Doerksen, R.J.; Prather, P.L.; et al. Natural prenylated resveratrol analogs arachidin-1 and -3 demonstrate improved glucuronidation profiles and have affinity for cannabinoid receptors. Xenobiotica 2012, 42, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.D.; Rizvi, G.; Anavi-Goffer, S.; Hurst, D.P.; Barnett-Norris, J.; Lynch, D.L.; Reggio, P.H.; Abood, M.E. An aromatic microdomain at the cannabinoid CB(1) receptor constitutes an agonist/inverse agonist binding region. J. Med. Chem. 2003, 46, 5139–5152. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.Y.; Bertalovitz, A.C.; Kendall, D.A. Probing the interaction of SR141716A with the CB1 receptor. J. Biol. Chem. 2012, 287, 38741–38754. [Google Scholar] [CrossRef] [PubMed]
- Hurst, D.P.; Lynch, D.L.; Barnett-Norris, J.; Hyatt, S.M.; Seltzman, H.H.; Zhong, M.; Song, Z.H.; Nie, J.; Lewis, D.; Reggio, P.H. N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole -3-carboxamide (SR141716A) interaction with LYS 3.28(192) is crucial for its inverse agonism at the cannabinoid CB1 receptor. Mol. Pharmacol. 2002, 62, 1274–1287. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Meth. Neurosci. 1995, 25, 366–428. [Google Scholar]
- Release, S. Suite 2012: LigPrep; Schrödinger, LLC: New York, NY, USA, 2012. [Google Scholar]
- Sastry, M.; Lowrie, J.F.; Dixon, S.L.; Sherman, W. Large-scale systematic analysis of 2D fingerprint methods and parameters to improve virtual screening enrichments. J. Chem. Inf. Model 2010, 50, 771–784. [Google Scholar] [CrossRef] [PubMed]
- InterBioScreen Ltd. Available online: http://www.ibscreen.com (accessed on 6 June 2013).
- Ma, G.; Bavadekar, S.A.; Davis, Y.M.; Lalchandani, S.G.; Nagmani, R.; Schaneberg, B.T.; Khan, I.A.; Feller, D.R. Pharmacological effects of ephedrine alkaloids on human alpha(1)- and alpha(2)-adrenergic receptor subtypes. J. Pharmacol. Exp. Ther. 2007, 322, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.C.; Veluz, J.S.; Williams, H.L.; Briley, E.M.; Matsuda, L.A. Cannabinoid agonists stimulate both receptor- and non-receptor-mediated signal transduction pathways in cells transfected with and expressing cannabinoid receptor clones. Mol. Pharmacol. 1992, 42, 838–845. [Google Scholar] [PubMed]
- Gao, J.; Leon, F.; Radwan, M.M.; Dale, O.R.; Husni, A.S.; Manly, S.P.; Lupien, S.; Wang, X.; Hill, R.A.; Dugan, F.M.; et al. Benzyl derivatives with in vitro binding affinity for human opioid and cannabinoid receptors from the fungus Eurotium repens. J. Nat. Prod. 2011, 74, 1636–1639. [Google Scholar] [CrossRef] [PubMed]
- Husni, A.S.; McCurdy, C.R.; Radwan, M.M.; Ahmed, S.A.; Slade, D.; Ross, S.A.; ElSohly, M.A.; Cutler, S.J. Evaluation of Phytocannabinoids from high potency using bioassays to determine structure-activity relationships for cannabinoid receptor 1 and cannabinoid receptor 2. Med. Chem. Res. 2014, 23, 4295–4300. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.L.; Horswill, J.G.; Anavi-Goffer, S.; Reggio, P.H.; Bolognini, D.; Abood, M.E.; McAllister, S.; Strange, P.G.; Stephens, G.J.; Pertwee, R.G.; et al. CB(1) receptor allosteric modulators display both agonist and signaling pathway specificity. Mol. Pharmacol. 2013, 83, 322–338. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.; Traynor, J.R. The [35S]GTPgammaS binding assay: Approaches and applications in pharmacology. Life Sci. 2003, 74, 489–508. [Google Scholar] [CrossRef] [PubMed]
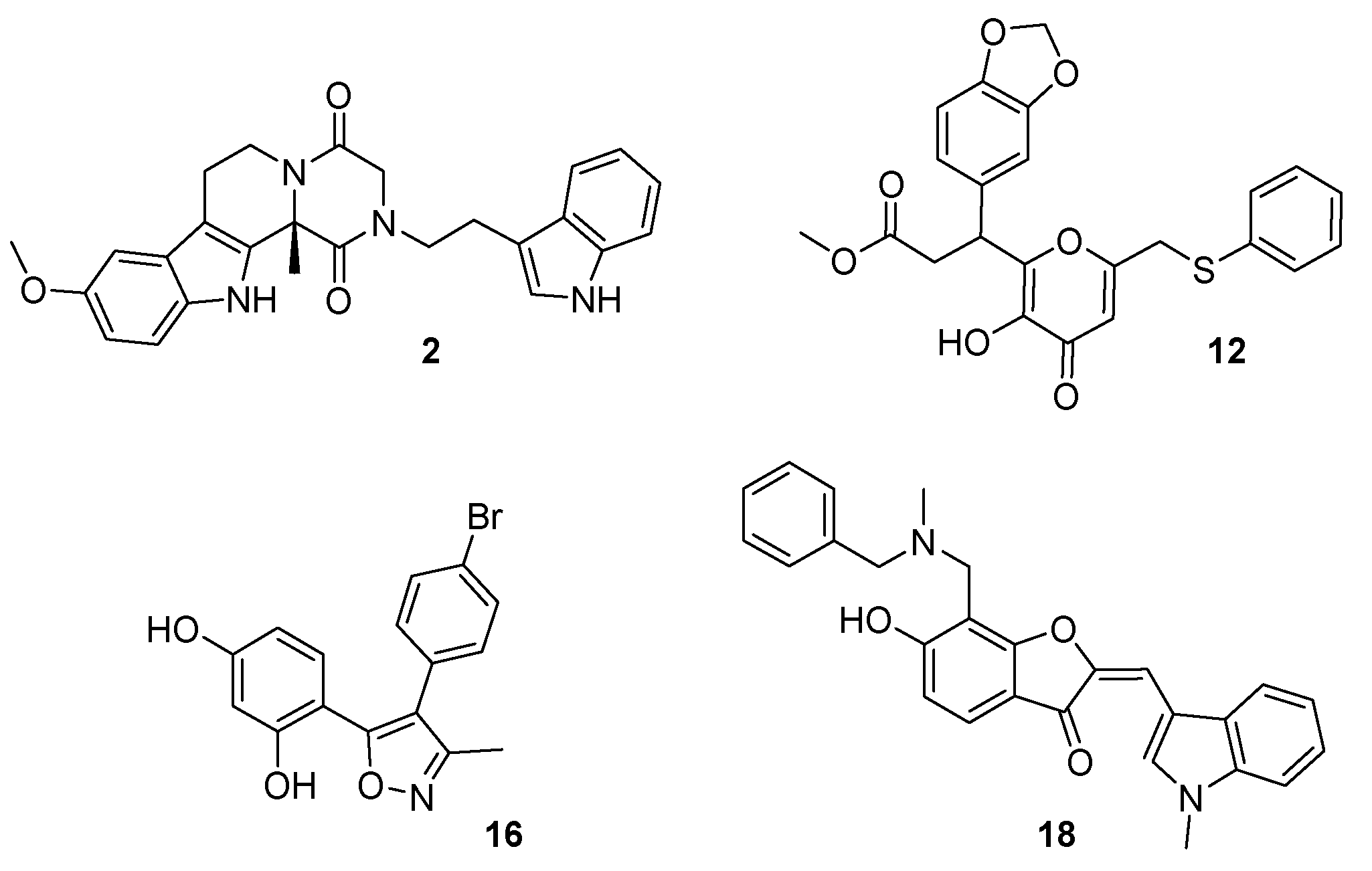
Sample Availability: The PDB coordinates of all the compounds [2, 12, 18 and 16] with the CB1 receptor are available on request. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 ± SEM (μM) | Ki ± SEM (μM) | ||
|---|---|---|---|---|
| CB1 | CB2 | CB1 | CB2 | |
| 2a | 3.2 ± 1.0 | 3.25 ± 0.5 | 1.62 ± 0.5 | 1.62 ± 0.51 |
| 12ab | >5.3 ± 1.4 | >5.93 ± 0.8 | >2.6 ± 0.7 | >2.97 ± 0.4 |
| 16 | 20.8 ± 4.9 | ND c | 10.4 ± 2.5 | ND c |
| 16a | 0.242 ± 0.07 d | ND c | 0.121 ± 0.04 d | ND c |
| 18e | 24.6 ± 2.6 | 0.101 ± 0.035 | 12.3 ± 1.3 | 0.051 ± 0.01 |
| CP55,940 | 9.84 nM | 8.62 nM | 4.9 nM | 4.31 nM |
| Compound | MW | (% Displacement at 10 µM) | Ki ± SEM (μM) | ||
|---|---|---|---|---|---|
| CB1 | CB2 | CB1 | CB2 | ||
| PCB-161 | 436.3 | 37.4 | 44.6 | >10.0 | >10.0 |
| PCB-162 | 436.3 | 3.9 | 28.4 | ND | >10.0 |
| PCB-163 | 454.3 | 72.0 | 81.1 | 0.4870 ± 0.1516 | 1.084 ± 0.141 |
| PCB-164 | 444.4 | 29.8 | 97.2 | NC # | 0.1556 ± 0.0173 |
| PCB-165 | 321.3 | 33.0 | - | >10.0 | - |
| CP55,940 | 376.6 | 0.002162 ± 0.000420 | 0.001185 ± 0.000128 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, P.; Roy, K.K.; Liu, H.; Ma, G.; Pettaway, S.; Alsharif, W.F.; Gadepalli, R.S.; Rimoldi, J.M.; McCurdy, C.R.; Cutler, S.J.; et al. Structure-Based Identification of Potent Natural Product Chemotypes as Cannabinoid Receptor 1 Inverse Agonists. Molecules 2018, 23, 2630. https://doi.org/10.3390/molecules23102630
Pandey P, Roy KK, Liu H, Ma G, Pettaway S, Alsharif WF, Gadepalli RS, Rimoldi JM, McCurdy CR, Cutler SJ, et al. Structure-Based Identification of Potent Natural Product Chemotypes as Cannabinoid Receptor 1 Inverse Agonists. Molecules. 2018; 23(10):2630. https://doi.org/10.3390/molecules23102630
Chicago/Turabian StylePandey, Pankaj, Kuldeep K. Roy, Haining Liu, Guoyi Ma, Sara Pettaway, Walid F. Alsharif, Rama S. Gadepalli, John M. Rimoldi, Christopher R. McCurdy, Stephen J. Cutler, and et al. 2018. "Structure-Based Identification of Potent Natural Product Chemotypes as Cannabinoid Receptor 1 Inverse Agonists" Molecules 23, no. 10: 2630. https://doi.org/10.3390/molecules23102630