All reactions involving water-sensitive reagents were conducted under anhydrous conditions and a dry atmosphere of nitrogen. Reagents were used as purchased from commercial suppliers. Anhydrous
N,
N-dimethylformamide (DMF) was purchased and stored over 4 Å molecular sieves. All other anhydrous solvents were purified and dried using a solvent purification system (MB SPS-800 from Braun) and stored over appropriate molecular sieves. Solvents for normal phase (NP) chromatography were distilled prior to use. Acetonitrile was purchased in HPLC grade for reversed phase (RP) chromatography and HPLC. Evaporation of solvents was performed under reduced pressure on a rotary evaporator or using a high vacuum pump. Reactions were monitored via thin layer chromatography (TLC) carried out on pre-coated Macherey-Nagel TLC plates Alugram
® Xtra SIL G/UV
254, and compounds stained with Vanillin (Vanillin (5 g), 1000 mL MeOH/AcOH 9:1, 35 mL H
2SO
4) under heating. For automated NP or RP chromatography two flash systems (Interchim Puriflash 430 (Montlucon, France) or Büchi Sepacore
® Flash System (Essen, Germany), combined with Macherey-Nagel Chromabond
® Flash RS 80 SiOH (NP) or RS40 C
18 ec (RP) columns (Düren, Germany)) were used. For purifications of phosphordiamidites, a chromatotron (Harrison Research 7924T) with glass plates coated with 2 or 4 mm layers of VWR60 PF
254 silica gel containing a fluorescent indicator (VWR no. 7749) was used. Analytical RP-High Performance Liquid Chromatography-Mass Spectrometry (RP-HPLC/MS) was performed with an Agilent 1260 Infinity instrument, Waldbronn, Germany (pump G1311B, autosampler G1329B, column compartment G1316A, diode array detector G4221B, column Agilent Poroshell 120 EC-C18, 2.7 mm, 4.6 × 50 mm) coupled with single-quad MS (Advion expression
L CMS, Harlow, U.K.). Ultrapure water was generated by a Sartorius Arium
® pro unit (Sartopore 0.2 µm, UV, Göttingen, Germany). As elution buffer served a tetra-
n-butylammonium acetate solution (10 mM, pH 7.2). HPLC/MS runs were performed according to the following method: 0–15 min: water/acetonitrile gradient (2–98% B) with a flow of 0.5 mL/min, 20 °C column temperature and UV detection at 259 nm and 270 nm, MS scans from 150 to 1100
m/
z. Nuclear magnetic resonance (NMR) spectra were recorded at room temperature on Bruker Fourier 300 (300 MHz for
1H acquisitions), Bruker AMX 400 (Karlsruhe, Germany) (400 MHz for
1H 101 MHz for
13C and 152 MHz for
31P acquisitions) or Bruker AVIII 600 (Karlsruhe, Germany) (600 MHz for
1H and 151 MHz for
13C acquisitions) spectrometers in automation mode. All chemical shifts (δ) are given in parts per million (ppm) with the solvent resonance as internal standard. Coupling constants
J are given in Hertz (Hz). Two-dimensional NMR experiments (HSQC, HMBC) were used for the assignment of quaternary carbons. For mass spectrometric (MS) analysis, spectra were acquired on an Agilent 6224 ESI-TOF spectrometer (Waldbronn, Germany) in positive and negative mode as required. Infrared spectroscopy (IR) was carried out with a Bruker Alpha P FT-IR (Bremen, Germany) in attenuated total reflection (ATR) mode at room temperature ranging from 400 cm
−1 to 4000 cm
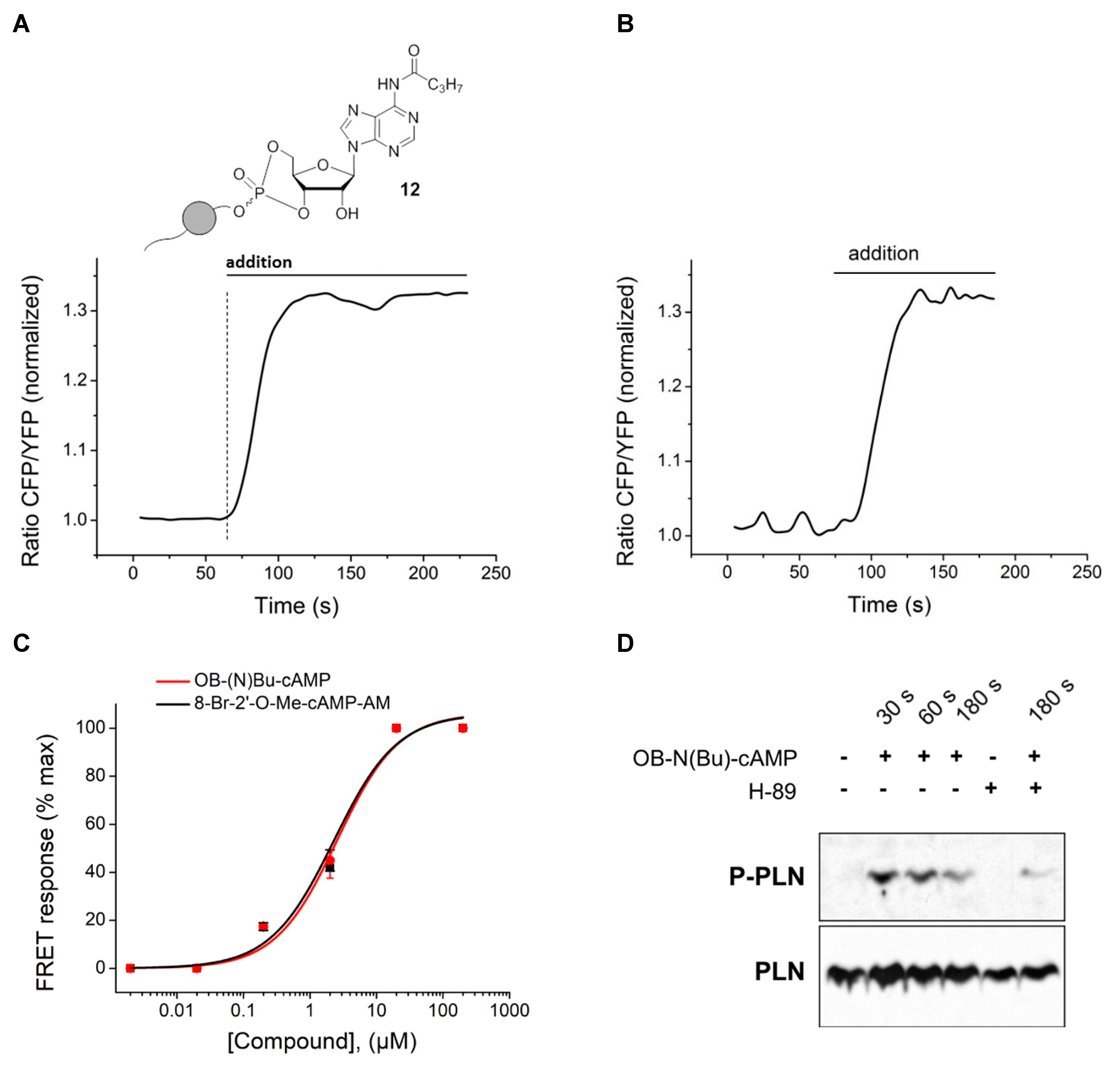
−1. For FRET measurements, primary mouse ventricular cardiomyocytes were isolated from Epac1-camps biosensor expressing transgenic mice [
29] as described [
28] and plated onto laminin coated glass cover slides. Measurements were performed 1–2 h after plating using a Nikon Ti microscope based FRET imaging system containing pE-100 440 nm light source (CoolLED), DV2 Dual View and ORCA-03G charge-coupled device camera (Hamamatsu, Japan), and analyzed as described [
28]. Cells were kept in a buffer containing 144 mM NaCl, 5.4 mM KCl, 1 mM MgSO
4, 1 mM CaCl
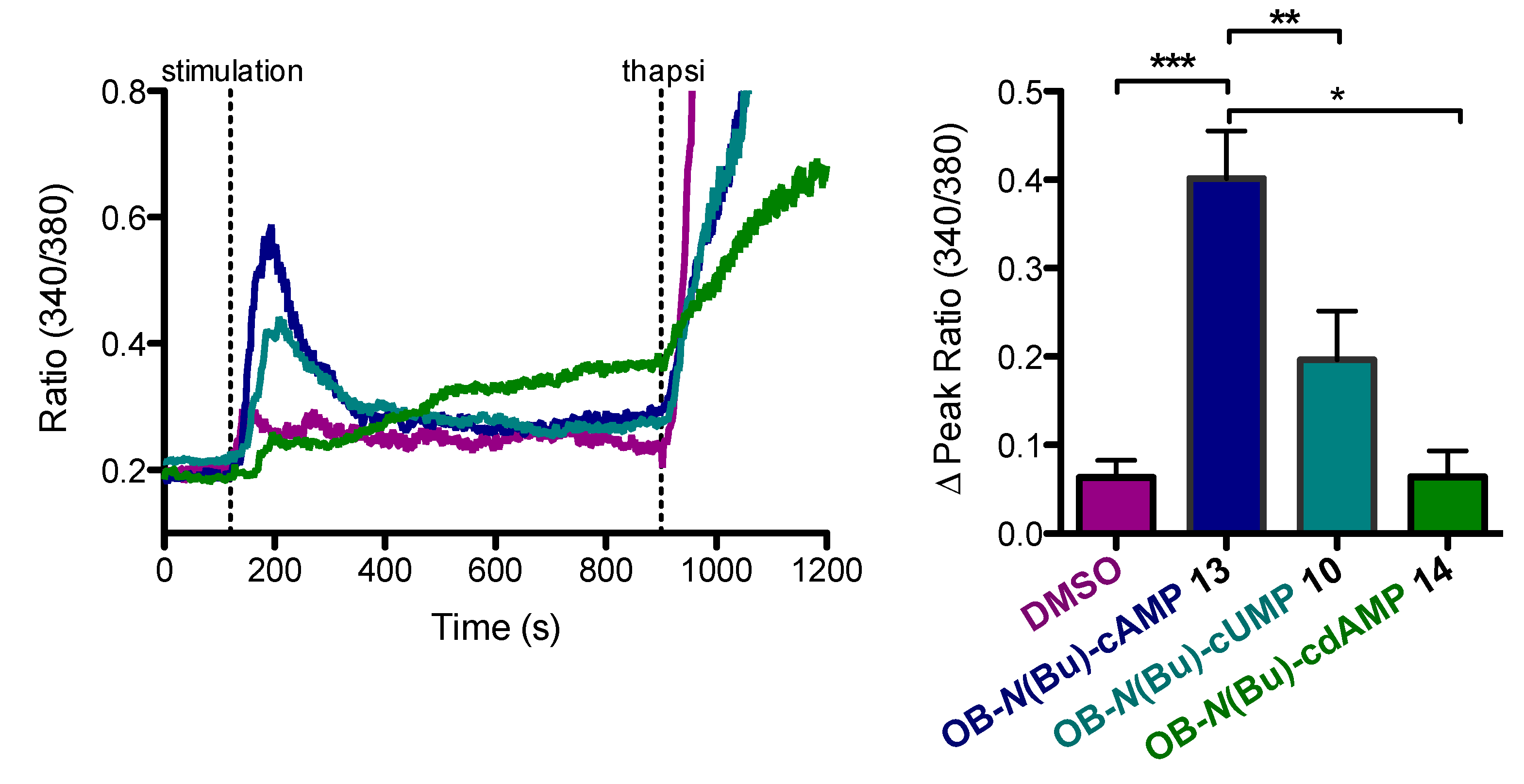
2, 10 mM Hepes (pH 7.3) and stimulated with OB-cNMPs or 8-Br-2′-O-Me-cAMP-AM (purchased from BIOLOG Life Science Institute, Bremen, Germany) dissolved 1:1000 in the same buffer from a freshly made 20 mM DMSO stock solution. For Ca
2+ mobilization assays, Jurkat T cells were incubated with the membrane-permeable AM ester of the Ca
2+ dye Fura-2 (4 µM, Calbiochem). Therefore, about 2 × 10
6 cells were centrifuged at 500 g for 5 min and resuspended in 1 mL of freshly supplemented RPMI medium containing Fura-2 AM. Cells were incubated for 30 min at 37 °C. After centrifugation, cells were washed and resuspended in Ca
2+ buffer (140 mM NaCl, 5 mM KCl, 1 mM MgSO
4, 1 mM CaCl
2, 20 mM Hepes (pH 7.4), 1 mM NaH
2PO
4, 5 mM glucose) and kept for 20 min at rt for de-esterification. Cells were added on prepared coverslips and allowed to adhere before measurement. Slides were mounted onto a Leica IRBE microscope (100-fold magnification) and after 120 s the respective OB-cNMPs (20 µM) or DMSO (as control) were added. As positive control, Thapsigagarin (1.67 µM, Calbiochem) was added after 900 s. A Sutter DG-4 was used as a light source, and frames were acquired with an electron-multiplying charge-coupled device camera (C9100-13, Hamamatsu). Images (512 × 512 pixels) were acquired in 16-bit mode with the following filter sets (AHF Analysentechnik) (excitation (ex), beam splitter (bs), and emission (em), all in nanometers): Fura-2 (ex, HC 340/26, HC 387/11; bs, 400DCLP; em, 510/84).
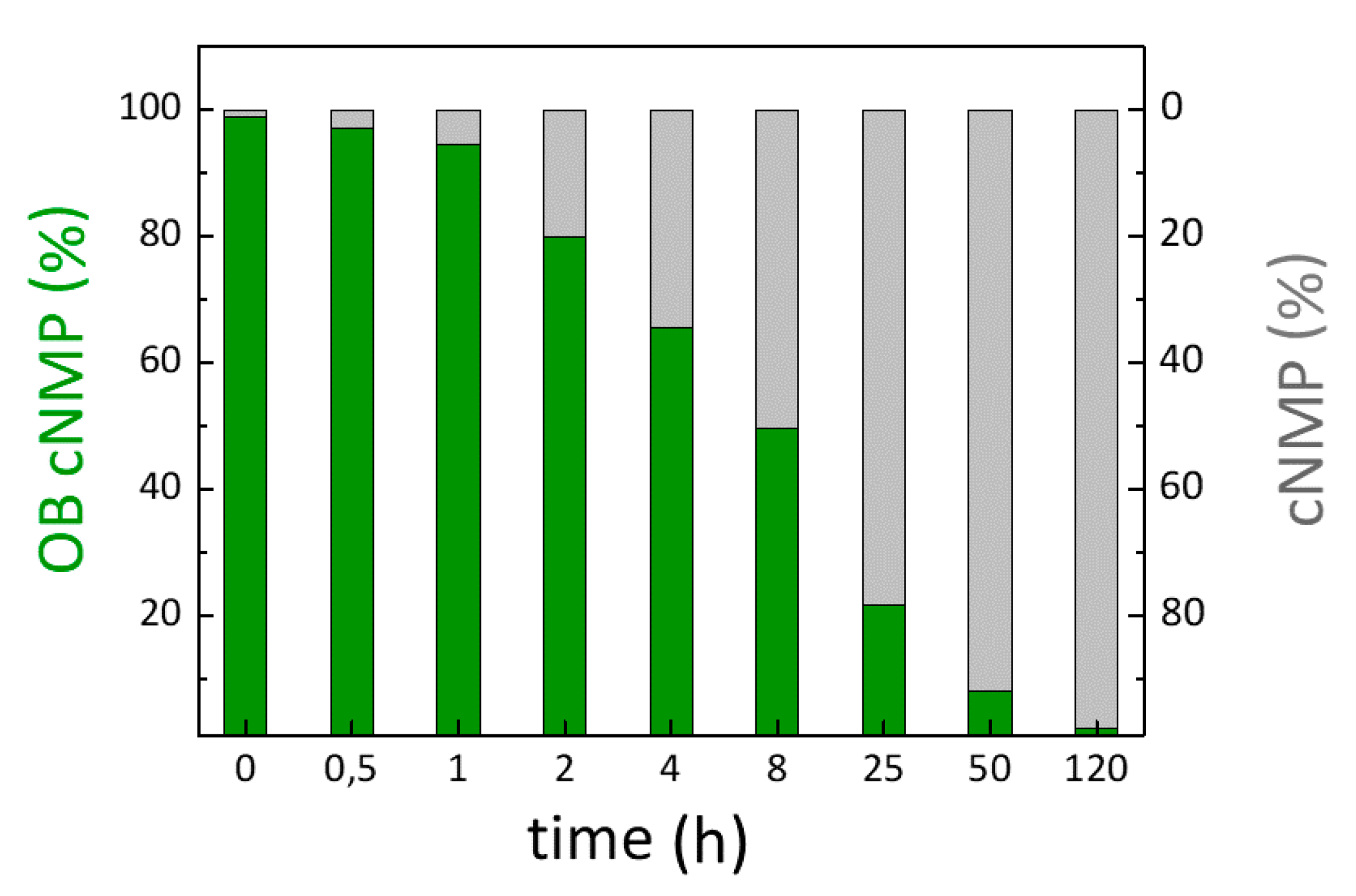
For immunoblot analysis, freshly isolated cardiomyocytes sedimented in stop buffer without serum were preincubated for 30 min either with vehicle or with 10 µM H-89 (Sigma). Next, 20 µM of OB-N(Bu)-cAMP were added to various time points (30, 90, and 180 s), after which cells were lysed and processed for immunoblot analysis. Homogenized proteins were quantified using BCA Protein Assay (Pierce). Samples (20 µg protein per lane) were boiled at 95 °C for 5 min to disrupt phospholamban pentamers, loaded onto 15% SDS–polyacrylamide gel and immunoblotting using the anti-P-Ser16 phospholamban (Badrilla 1:5.000), and total anti-PLN A-1 antibodies (Badrilla).
3.2. Syntheses
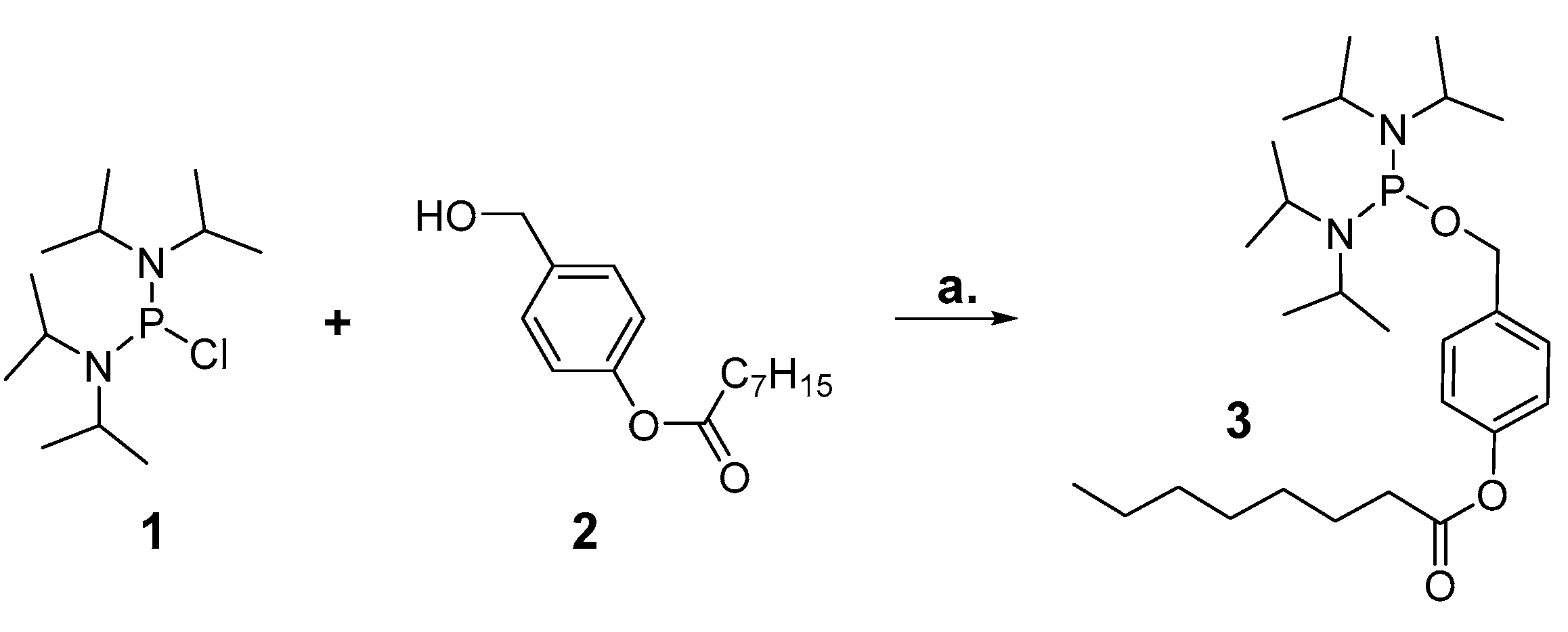
3.2.1. Synthesis of bis(N,N-diisopropylamino)-4-octanoyloxybenzyl phosphordiamidite 3
1.00 g (3.75 mmol) bis(N,N-diisopropylamino)chlorophosphine 1 were dissolved in 15 mL anhydrous THF. In a separate flask, 0.68 mL (4.87 mmol, 1.3 equiv.) NEt3 and 0.94 g (3.75 mmol, 1 equiv.) 4-(hydroxymethyl)phenyloctanoate 2 were mixed with 7 mL anhydrous THF, and the mixture was added dropwise to the chlorophosphine. The reaction mixture was stirred at rt for 20 h, then filtrated and the filtrate concentrated to dryness in vacuum. The remaining residue was purified by NP chromatography on silica gel with PE/TEA 98:2 as eluents, and the desired product obtained as colorless syrup.
Yield: 1.28 g (2.67 mmol, 71%).
1H-NMR (600 MHz, chloroform-d): δ [ppm] = 7.36 (d, 2JH,H = 8.2 Hz, 2H), 7.14–6.95 (m, 2H), 4.63 (d, 2JH,H = 7.2 Hz, 2H), 3.66–3.51 (m, 4H), 2.54 (t, 2,3JH,H = 7.5 Hz, 2H), 1.75 (p, 2,3JH,H = 7.4 Hz, 2H), 1.51–1.23 (m, 8 H), 1.21 (d, 2JH,H = 2.7 Hz, 12H), 1.20 (d, 2JH,H = 2.8 Hz, 12H) 0.88 (t, 2,3JH,H = 7.3 Hz, 3H); 13C{1H}-NMR (151 MHz, chloroform-d): δ [ppm] = 172.6, 149.7, 138.2, 127.9, 121.5, 65.8, 44.69, 44.56, 34.6, 31.8, 29.2, 29.1, 25.1, 24.8, 24.7, 24.1, 24.0, 22.75, 14.22; 31P{1H}-NMR (162 MHz, chloroform-d): δ [ppm] = 123.5. IR (ATR): ṽ in [cm−1] = 2963.3, 2927.8, 2861.4, 2079.0, 2025.5, 1761.3, 1607.8, 1507.4, 1457.5, 1416.5, 1390.1, 1361.6, 1300.3, 1194.2, 1184.8, 1162.9, 1140.3, 1116.2, 1045.2, 1016.9, 952.7, 916.5, 866.3, 779.6, 748.7, 706.7, 642.7, 566.1, 527.6. MS (MALDI): m/z [M-H] calcd. for C27H48N2O3P-: 479.340, found: 479.245.
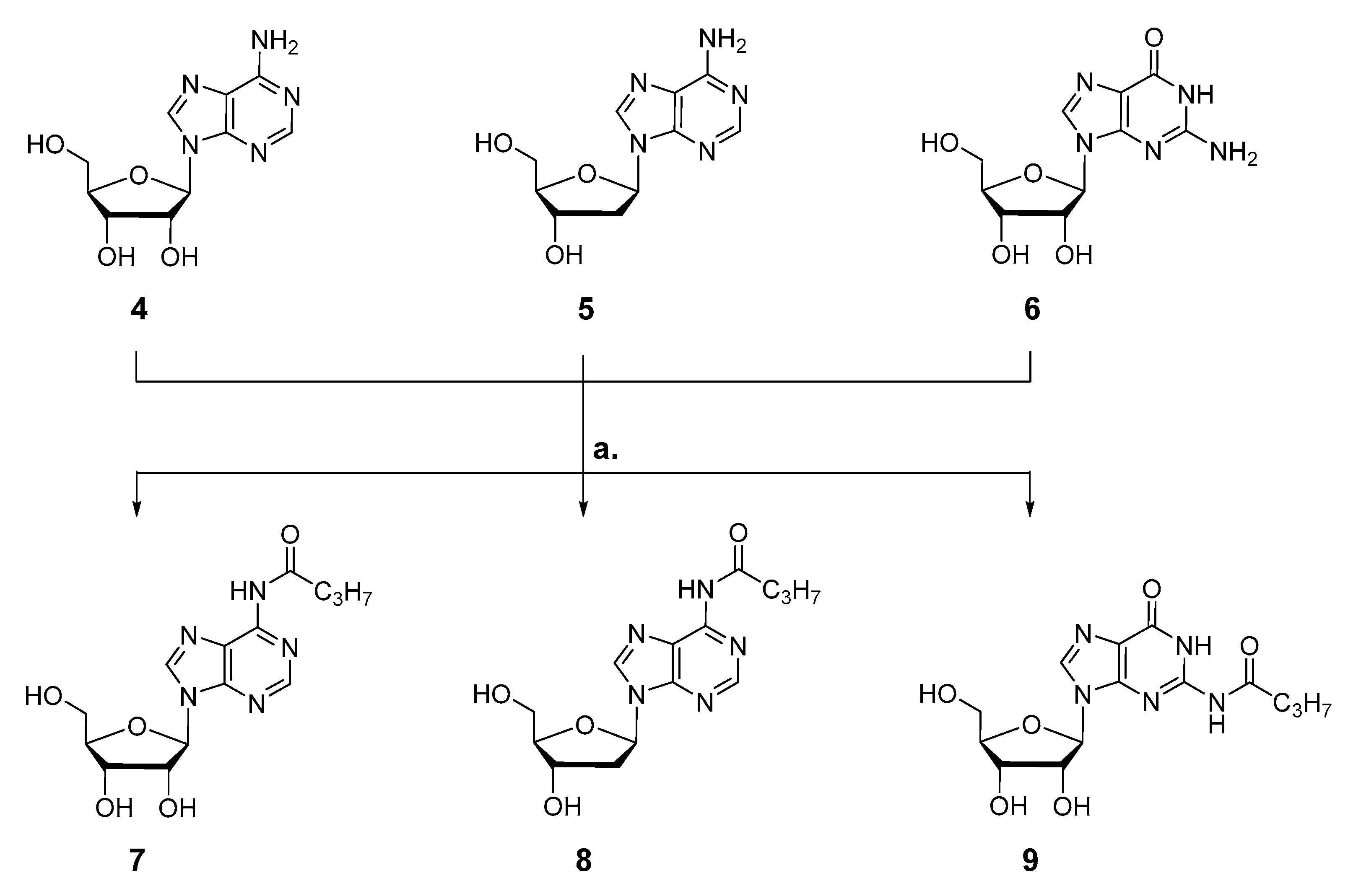
3.2.2. Synthesis of 6-N-butanoyl-adenosine 7
In accordance with GP I, 1.40 g (5.26 mmol) adenosine 4 were dissolve in 34 mL pyridine/THF 1:1 and converted with 2.11 mL (16.5 mmol, 3.2 equiv.) TMSCl and 0.60 mL (5.78 mmol, 1.1 equiv.) butyryl chloride. After 6 h stirring at rt, 2.5 mL 1 M HCl (aq.) was added to cleave the TMS ethers, and after 5 min, all volatile components were removed under vacuum. Upon final purification of the crude product via automated RP flash column chromatography on C18 modified silica gel with an CH3CN gradient in water (0% to 100%), the product was obtained as colorless powder.
Yield: 1.09 g (3.22 mmol, 61%).
1H-NMR (500 MHz, DMSO-d6): δ [ppm] = 10.63 (s, 1H), 8.69 (s, 1H), 8.65 (s, 1H), 6.01 (d, 3JH,H = 5.8 Hz, 1H), 5.53 (d, 3JH,H = 5.8 Hz, 1H), 5.23 (d, 3JH,H = 4.8 Hz, 1H), 5.12 (t, 3JH,H = 5.6 Hz, 1H), 4.63 (q, 3JH,H = 5.4 Hz, 1H), 4.19 (q, 3JH,H = 4.3 Hz, 1H), 3.98 (q, 3JH,H = 3.9 Hz, 1H), 3.76–3.64 (m, 1H), 3.58 (ddd, 2JH,H = 11.9 Hz, 3JH,H = 6.1 Hz, 3JH,H = 4.0 Hz, 1H), 2.55 (t, 2,3JH,H = 7.3 Hz, 2H), 1.63 (h, 2,3JH,H = 7.4 Hz, 2H), 0.94 (t, 2,3JH,H = 7.4 Hz, 3H); 13C-NMR (126 MHz, DMSO-d6): δ [ppm] = 171.5, 151.7, 151.6, 149.7, 142.7, 123.9, 87.6, 85.7, 73.7, 70.3, 61.3, 38.0, 18.2, 13.5. IR (ATR): ṽ in [cm−1] = 3273.2, 2963.3, 2934.0, 2875.0, 1716.3, 1685.0, 1613.3, 1587.0, 1521.9, 1460.3, 1409.0, 1356.6, 1327.0, 1224.3, 1124.1, 1082.2, 1056.7, 984.6, 902.4, 866.3, 799.6, 745.0, 704.7, 643.7, 547.3. MS (ESI-HR): m/z [M + H]+ calcd. for C14H20N5O5+: 338.1459, found: 338.1469.
3.2.3. Synthesis of 6-N-butanoyl-2′-deoxyadenosine 8
Following GP I, 1.14 g (4.24 mmol) 2′-deoxyadenosine 5 were dissolve in 24 mL pyridine/CH2Cl2 1:2. At 0 °C, 1.13 mL (8.91 mmol, 2.1 equiv.) TMSCl were added, and the reaction mixture was stirred for 18 h at rt. Successively, 0.48 mL (4.67 mmol, 1.1 equiv.) butyryl chloride were added. After further 3 h stirring at rt, the TMS ethers were removed by addition of 8 mL CH3OH at 0 °C. After further 5 h at rt, the reaction was terminated by removing all volatile components under vacuum. The crude product was taken up in water containing little amount of CH3CN and purified via automated RP flash column chromatography on C18 modified silica gel with an CH3CN gradient in water (0% to 100%) to afford the desired product as colorless powder.
Yield: 0.43 g (1.35 mmol, 32%).
1H-NMR (400 MHz, methanol-d4): δ [ppm] = 8.66 (s, 1H), 8.62 (s, 1H), 6.61–6.52 (m, 1 H), 4.64 (dt, 3JH,H = 6.1 Hz, 3JH,H = 3.1 Hz, 1H), 4.10 (q, 3JH,H = 3.4 Hz, 1H), 3.88 (dd, 2JH,H = 12.2 Hz, 3JH,H = 3.4 Hz, 1H), 3.79 (dd, 2JH,H = 12.2 Hz, 3JH,H = 3.9 Hz, 1H), 2.88 (ddd, 2JH,H = 13.4 Hz, 3JH,H = 7.4 Hz, 3JH,H = 6.0 Hz, 1H), 2.67 (t, 2,3JH,H = 7.4 Hz, 2H), 2.51 (ddd, 2JH,H = 13.5 Hz, 3JH,H = 6.2 Hz, 3JH,H = 3.3 Hz, 1H), 1.82 (h, 2,3JH,H = 7.4 Hz, 2H), 1.08 (t, 2,3JH,H = 7.4 Hz, 3H); 13C{1H}-NMR (101 MHz, methanol-d4): δ [ppm] = 174.4, 152.9, 150.7, 144.3, 123.2, 89.7, 86.7, 72.7, 63.4, 41.5, 39.9, 19.6, 14.0. IR (ATR): ṽ in [cm−1] = 3336.8, 2964.6, 2933.1, 2875.3, 2592.1, 2330.9, 1682.2, 1612.5.1585.9, 1522.5, 1459.6, 1402.8, 1354.8, 1329.2, 1223.8, 1093.3, 1058.5, 993.8, 941.1, 867.1, 799.6, 749.6, 984.8, 644.4, 585.3, 561.2, 542.4, 527.4, 509.6, 464.7. MS (ESI-HR): m/z [M + H]+ calcd. for C14H20N5O4+: 322.1510, found: 322.1521.
3.2.4. Synthesis of 2-N-butanoyl-guanosine 9
According to GP I, 1.55 g (5.48 mmol) guanosine 6 were co-evaporated with pyridine three times and then dissolved in 81 mL pyridine/CH2Cl2 1:2. At 0 °C, 6.28 mL (49.4 mmol, 9 equiv.) TMSCl were added, and the reaction mixture was stirred for 4 h at rt. Successively, 0.62 mL (6.03 mmol, 1.1 equiv.) butyryl chloride were added. After 3 h stirring at rt, the cleavage of the TMS ethers was induced by addition of 27 mL CH3OH, and after further 12 h at rt, the reaction was terminated and all volatile components were removed under vacuum. The crude residue was taken up in water containing little amount of CH3CN and finally purified via automated RP flash column chromatography on C18 modified silica gel with an CH3CN gradient in water (0% to 100%) to afford the desired product as colorless powder.
Yield: 0.92 g (2.61 mmol, 48%).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.07 (bs, 1H), 11.71 (bs, 1H), 8.26 (s, 1 H), 5.80 (d, 3JH,H = 5.7 Hz, 1H), 5.47 (d, 3JH,H = 5.7 Hz, 1H), 5.17 (d, 3JH,H = 4.5 Hz, 1H), 5.03 (t, 3JH,H = 5.4 Hz, 1H), 4.43 (d, 3JH,H = 5.2 Hz, 1H), 4.20–4.05 (m, 1H), 3.90 (q, J = 3.9 Hz, 1H), 3.64 (dt, 2JH,H = 11.9 Hz, 3JH,H = 4.8 Hz, 1H), 3.55 (dt, 2JH,H = 11.9 Hz, 3JH,H = 4.7 Hz, 1H), 2.45 (t, 2,3JH,H = 7.3 Hz, 2H), 1.62 (h, 2,3JH,H = 7.4 Hz, 2H), 0.92 (t, 2,3JH,H = 7.5 Hz, 3H); 13C{1H}-NMR (101 MHz, DMSO-d6): δ [ppm] = 176.2, 154.8, 148.8, 148.0, 137.6, 120.1, 86.6, 85.3, 73.9, 61.1, 37.8, 17.9, 13.4. IR (ATR): ṽ in [cm−1] = 3364.4, 3279.5, 2968.3, 2941.1, 1680.2, 1608.8, 1564.3, 1554.1, 1536.0, 1481.4, 1469.2, 1449.9, 1402.5, 1251.7, 1204.2, 1179.4, 1127.9, 1089.0, 1060.2, 993.3, 976.4, 901.1, 863.3, 818.6, 801.0, 762.7, 737.4, 717.0, 680.4, 643.7, 607.0, 589.1, 562.3, 510.2, 484.2. MS (ESI-HR): m/z [M + H]+ calcd. for C14H19N5O6+: 354.1408, found: 354.1400.
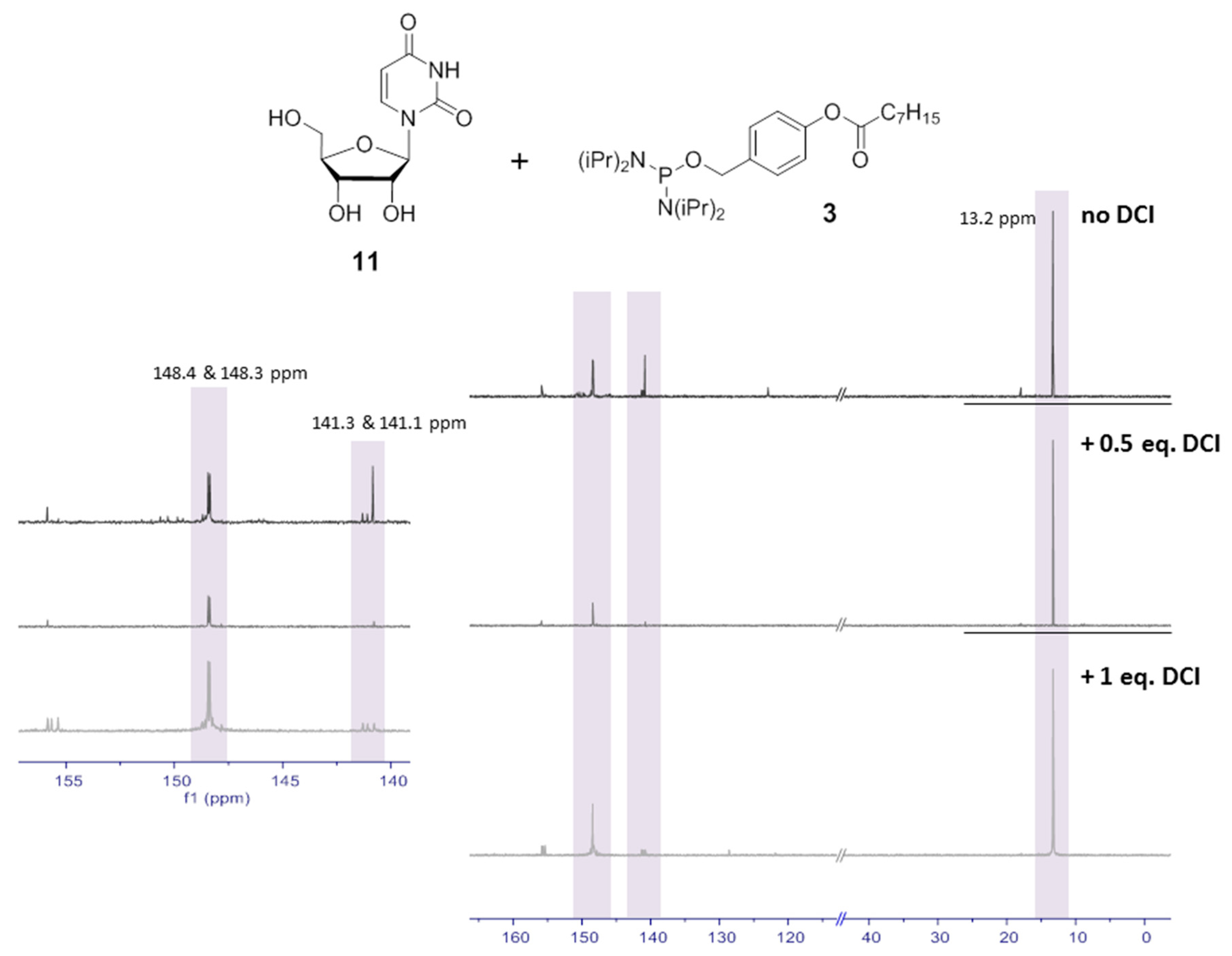
3.2.5. Synthesis of Uridine-3′,5′-(4-octanoyloxybenzyl)cyclophosphate 10
According to GP II, 26 mg (0.11 mmol) uridine 10 were dissolved in 2 mL DMF and reacted with 57 mg (0.12 mmol, 1.1 equiv.) OB-PA2 3, dissolved in 2.5 mL CH3CN, in the presence of 0.54 mL (0.13 mmol, 1.3 equiv.) DCI (0.25 M in CH3CN) and 0.45 mL (0.13 mmol, 1.3 equiv.) BTT (0.3 M in CH3CN). The addition of 32 µL (0.16 mmol, 1.5 equiv.) tBuOOH (5.5 M in n-decane) followed successively. After purification by automated RP flash column chromatography on C18 modified silica gel with a CH3CN gradient in water (0% to 100%) and lyophilization, the product was obtained as colorless cotton and in two fractions containing each of the stereoisomers.
Yield: 11 mg (0.02 mmol, 19%).
1H-NMR (400 MHz, methanol-d4): δ [ppm] = 7.66–7.51 (m, 2H), 7.47 (d, 3JH,H = 8.1 Hz, 1H), 7.22–7.08 (m, 2H), 5.69 (d, 3JH,H = 8.0 Hz, 1H), 5.64 (d, 3JH,H = 0.9 Hz, 1H), 5.24–5.14 (m, 2H), 4.64 (ddd, 3JH,P = 22.5 Hz, 2JH,H = 9.3 Hz, 3JH,H = 4.6 Hz, 1H), 4.39 (ddd, 3JH,H = 10.2, 2JH,H = 9.4, 3JH,H = 0.8 Hz, 1H), 4.35–4.25 (m, 2H), 4.25–4.10 (m, 1H), 2.58 (t, 2,3JH,H = 7.4 Hz, 2H), 1.73 (p, 2,3JH,H = 7.4 Hz, 2H), 1.53–1.22 (m, 8H), 1.05–0.82 (m, 3H)
and
7.63 (d, 3JH,H = 8.1 Hz, 1H), 7.53–7.47 (m, 2H), 7.19–7.10 (m, 2H), 5.73 (d, 3JH,H = 1.2 Hz, 1H), 5.71 (d, 3JH,H = 8.1 Hz, 1H), 5.19 (d, 2JH,H = 8.7 Hz, 2H), 4.81 (ddd, 3JH,P = 9.8 Hz, 3JH,H = 5.1 Hz, 3JH,H = 1.0 Hz, 1H), 4.67 (ddd, 3JH,P = 14.2 Hz, 2JH,H = 9.2 Hz, 3JH,H = 5.5 Hz, 1H), 4.62–4.47 (m, 2H), 4.42 (td, 2JH,H = 10.1, 3JH,H = 5.5 Hz, 1H), 2.60 (t, 2,3JH,H 7.4 Hz, 2H), 1.74 (p, 2,3JH,H 7.4 Hz, 2H), 1.50–1.26 (m, 8H), 1.02–0.84 (m, 3H); 13C{1H}-NMR (101 MHz, methanol-d4): δ [ppm] = 173.8, 165.9, 151.5, 150.1, 143.7, 134.3, 131.2, 123.3, 103.0, 97.5, 80.1, 72.4, 71.5, 71.2, 70.3, 35.0, 32.9, 30.2, 30.1, 25.0, 23.7, 14.4
and
172.8, 164.9, 151.7, 150.5, 143.5, 133.0, 130.2, 122.8, 103.1, 97.7, 79.3, 72.4, 71.2, 71.1, 70.6, 35.0, 32.8, 30.2, 30.1, 26.0, 23.7, 14.4; 31P{1H}-NMR (162 MHz, methanol-d4): δ [ppm] = −3.93, −5.01. MS (ESI-HR): m/z [M + Na]+ calcd. for C24H31N2O10PNa+: 561.1609, found: 561.1445.
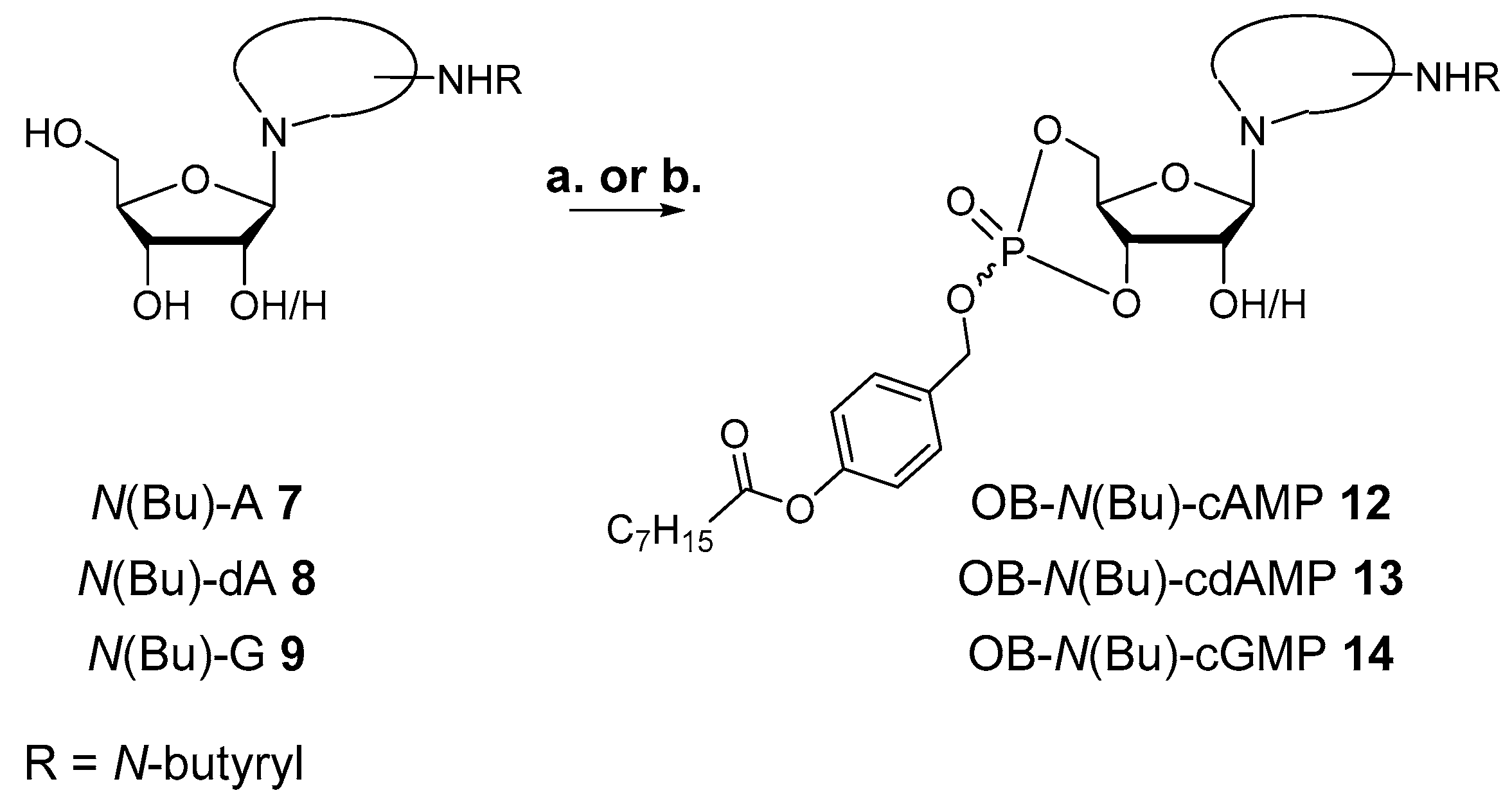
3.2.6. Synthesis of 6-N-butanoyl-adenosine-3′,5′-(4-octanoyloxybenzyl)cyclophosphate 12
According to GP II, 48 mg (0.14 mmol) 6-N-butanoyl-adenosine 7 were dissolved in 4 mL DMF and treated with a solution of 76 mg (0.16 mmol, 1.1 equiv.) OB-PA2 3 in 4 mL CH3CN and 0.97 mL (0.24 mmol, 1.7 equiv.) DCI (0.25 M in CH3CN) as well as 0.62 mL (0.19 mmol, 1.3 equiv.) BTT (0.3 M in CH3CN). Then, 43 µL (0.21 mmol, 1.5 equiv.) tBuOOH (5.5 M in n-decane) were added. After purification by automated RP flash chromatography on C18 modified silica with a CH3CN gradient in water (0% to 100%), the product was obtained as colorless cotton and mixture of two diastereomers.
Yield: 13 g (0.02 mmol, 14%).
1H-NMR (400 MHz, methanol-d4): δ [ppm] = 8.68, 8.58 (2× s, 2H), 8.49, 8.38 (2× s, 2H), 7.62–7.56, 7.55–7.49 (2× m, 4H), 7.17–7.12, 7.13–7.06 (2× m, 4H), 6.20, 6.12 (2× s, 2H), 5.47 (dd, 3JH,P = 9.1 Hz, 3JH,H = 5.1 Hz, 1H) 5.23 (2× d, 2JH,H = 10.4 Hz & 8.1 Hz, 4H), 5.05 (ddd, 3JH,P = 9.6 Hz, 3JH,H = 5.1 Hz, 3JH,H = 1.6 Hz, 1H), 4.89–4.86 (m, 1H), 4.73–4.34 (m, 7H), 2.71–2.55 (m, 6H,), 2.51 (t, 2,3JH,H = 7.4 Hz, 2H), 1.86–1.70 (m, 6H), 1.68–1.57 (m, 2H), 1.53–1.25 (m, 16H), 1.05 (2× t, 2,3JH,H = 7.4 Hz, 6H), 0.98–0.95 (m, 6H); 13C{1H}-NMR (151 MHz, methanol-d4): δ [ppm] = 173.8, 173.6, 152.8, 152.4, 150.8, 143.1, 133.3, 133.2, 130.7, 130.6, 124.5, 123.1, 123.0, 88.8, 81.7, 80.4, 69.3, 69.2, 68.5, 59.8, 39.9, 34.5, 31.8, 29.2, 29.1, 25.0, 22.8, 18.5,14.2, 13.9; 31P{1H}-NMR (162 MHz, methanol-d4): δ [ppm] = −3.80, −4.90. MS (ESI-HR): m/z [M + H]+ calcd. for C29H39N5O9P+: 632.2848, found: 632.2848.
3.2.7. Synthesis of 6-N-butanoyl-2′-deoxyadenosine-3′,5′-(4-octanoyloxybenzyl)cyclophosphate 13
According to GP II, 50 mg (0.15 mmol) 6-N-butanoyl-2’-deoxyadenosine 8 were dissolved in 4 mL DMF and reacted with 82 mg (0.17 mmol, 1.1 equiv.) OB-PA2 3, dissolved in 4.3 mL CH3CN, in the presence of 1.60 mL (0.40 mmol, 2.6 equiv.) DCI (0.25 M in CH3CN) in total and 50 µL (0.25 mmol, 1.6 equiv.) tBuOOH (5.5 M in n-decane). After purification by automated RP flash column chromatography on C18 modified silica gel with a CH3CN gradient in water (0% to 100%) and lyophilization, the product was obtained as colorless cotton and mixture of two diastereomers.
Yield: 12 mg (0.02 mmol, 13%).
1H-NMR (400 MHz, methanol-d4): δ [ppm] = 8.69 (s, 1H), 8.61 (s, 1H), 8.47 (s, 1H), 8.37 (s, 1H), 7.65–7.57, 7.55–7.48 (2× m, 2H), 7.19–7.10 (m, 4H), 6.59 (dd, 3JH,H = 9.0 Hz, 3JH,H = 2.0 Hz, 1H), 6.55 (dd, 3JH,H = 6.7 Hz, 3JH,H = 4.0 Hz, 1H), 5.64 (q, 3JH,H/P = 9.2 Hz, 1H), 5.33 (q, 3JH,H/P = 9.2 Hz, 1H), 5.23 (2× d, 2JH,H = 13.1 Hz & 3JH,H = 12.9 Hz, 4H), 4.70–4.40 (m, 4H), 4.22 (td, 3JH,H = 9.9 Hz, 3JH,H = 5.6 Hz, 1H), 4.08 (td, 3JH,H = 9.9 Hz, 3JH,H = 4.7 Hz, 1H), 2.98 (ddd, 2JH,H = 13.1 Hz, 3JH,H = 7.7 Hz, 3JH,H = 1.9 Hz, 1H), 2.83–2.70 (m, 3H), 2.69–2.57 (m, 6H), 2.54 (t, 2,3JH,H = 7.4 Hz, 2H), 1.90–1.70 (m, 6H), 1.66 (q, 2,3JH,H = 7.3 Hz, 2H), 1.51–1.26 (m, 16H), 1.06 (2 x t, 2,3JH,H = 7.4, 6H), 0.98–0.86 (m, 6H); 13C{1H}-NMR (101 MHz, methanol-d4): δ [ppm] = 174.5, 174.4, 173.8, 173.6, 153.4, 153.2, 152.8, 152.7, 152.4, 152.3, 150.8, 150.7, 144.6, 144.5, 134.4, 134.3, 131.1, 130.7, 123.2, 123.1, 122.6, 85.8, 85.6, 79.3, 79.2, 75.7, 75.6, 71.7, 71.6, 71.4, 71.3, 40.0, 39.9, 35.0, 34.9, 32.8, 30.1, 30.0, 25.9, 25.9, 23.7, 19.7, 19.6, 14.4, 14.1, 14.0; 31P{1H}-NMR (162 MHz, methanol-d4): δ [ppm] = −4.14, −5.50. MS (ESI-HR): m/z [M + H]+ calcd. for C29H39N5O8P+: 616.2531, found: 616.2534.
3.2.8. Synthesis of 2-N-butanoyl-guanosine-3′,5′-(4-octanoyloxybenzyl)cyclophosphate 14
In accordance with GP II, 52 mg (0.15 mmol) 6-N-butanoyl-guanosine 9 were dissolved in 4 mL DMF and treated with a solution of 77 mg (0.16 mmol, 1.1 equiv.) OB-PA2 3 in 4 mL CH3CN, 1.75 mL (0.44 mmol, 3 equiv.) DCI (0.25 M in CH3CN) in total and finally, 44 µL (0.21 mmol, 1.5 equiv.) tBuOOH (5.5 M in n-decane). After purification by automated RP flash column chromatography on C18 modified silica gel with a CH3CN gradient in water (0% to 100%) and lyophilization, the product was obtained as colorless cotton and a single diastereomer.
Yield: 4 mg (5.6 µmol, 4%).
1H-NMR (400 MHz, methanol-d4): δ [ppm] = 7.92 (s, 1H), 7.56 (d, 3JH,H = 8.2 Hz, 2H), 7.07 (d, 3JH,H = 8.3 Hz, 2H), 6.01 (s, 1H), 5.23 (d, 2JH,H = 11.6 Hz, 2H), 4.69 (ddd, 3JH,H = 22.2 Hz, 3JH,P = 9.4 Hz, 3JH,H = 4.8 Hz, 1H), 4.55 (t, 3JH,H = 10.0 Hz, 1H), 4.35 (dd, 3JH,H = 10.0 Hz, 3JH,H = 4.8 Hz, 1H), 4.32 (d, 3JH,H = 4.7 Hz, 1H), 4.18 (dd, 3JH,P = 9.9 Hz, 3JH,H = 4.7 Hz, 1H), 2.51 (t, 2,3JH,H = 7.4 Hz, 2H), 2.46 (t, 2,3JH,H = 7.1 Hz, 2H), 1.74 (h, 2,3JH,H = 7.4 Hz, 2H), 1.62 (t, 2,3JH,H = 7.1 Hz, 2H), 1.49–1.25 (m, 8H,), 1.02 (t, 2,3JH,H = 7.4 Hz, 3H), 0.98–0.86 (m, 3H); 13C{1H}-NMR (101 MHz, methanol-d4): δ [ppm] = 177.7, 173.6, 152.2, 150.0, 149.8, 137.5, 134.4, 131.4, 123.3, 120.2, 93.4, 80.3, 73.3, 71.6, 71.5, 70.5, 39.4, 34.8, 32.9, 30.1, 25.8, 23.7, 19.3, 14.4, 13.9; 31P{1H}-NMR (162 MHz, methanol-d4): δ [ppm] = −4.93. MS (ESI-HR): m/z [M + H]+ calcd. for C29H39N5O10P+: 648.2429, found: 648.2720.
3.2.9. Synthesis of adenosine-3′,5′-(4-octanoyloxybenzyl)cyclophosphate 15
Following GP II, 21 mg (78 µmol) adenosine were dissolved in 2 mL DMF and treated with a solution of 41 mg (85 µmol, 1.1 equiv.) (OB)PA2 101 in 2 mL CH3CN as well as 0.74 mL (0.19 mmol, 2.4 equiv.) DCI (0.25 M in CH3CN). Lastly, 23 µL (0.12 mmol, 1.5 equiv.) tBuOOH (5.5 M in n-decane) were added for oxidation. After purification by automated RP flash column chromatography on C18 modified silica gel with a CH3CN gradient in water (0% to 100%) and lyophilization, the product was obtained as a colorless cotton and mixture of two diastereomers.
Yield: 5 mg (10 µmol, 12%).
1H-NMR (400 MHz, acetonitrile-d3): δ [ppm] = 8.19 (s, 1H), 8.09 (s, 1 H), 7.93 (s, 1H), 7.88 (s, 1H), 7.55–7.46 (m, 2H), 7.45–7.41 (m, 2H), 7.12–7.02 (m, 4H), 5.99 (bs, 4H), 5.98 (s, 1H), 5.93 (s, 1H), 5.47 (dd, 3JH,P = 9.1 Hz, 3JH,H = 5.1 Hz, 1H), 5.27 (ddd, 3JH,P = 9.4 Hz, 3JH,H = 4.9 Hz, 3JH,H = 1.0 Hz, 1H), 5.19 (ddd, 3JH,P = 9.3 Hz, 3JH,H = 5.0 Hz, 3JH,H = 1.6 Hz, 1H), 5.14–5.06 (m, 4H), 4.72 (d, 3JH,H = 4.9 Hz, 1H), 4.60 (d, 3JH,H = 5.1 Hz, 1H), 4.58–4.14 (m, 6H), 2.50 (2 x t, 3JH,H = 7.5 Hz, 4H), 1.64 (h, 3JH,H = 7.6 Hz, 4H), 1.47–1.14 (m, 16H), 0.89–0.78 (m, 6H); 13C{1H}-NMR (151 MHz, acetonitrile-d3): δ [ppm] = 173.3, 156.9, 153.9, 151.3, 149.4, 140.9, 133.3, 130.6, 130.7, 123.1, 119.8, 93.6, 81.0, 72.7, 71.5, 69.4, 34.7, 32.4, 29.6, 25.5, 23.3, 20.1, 14.3; 31P{1H}-NMR (162 MHz, acetonitrile-d3): δ [ppm] = −4.37, −6.03. MS (ESI-HR): m/z [M + H]+ calcd. for C25H33N5O8P+: 562.2061, found: 562.2068.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












