3.1. Chemistry
IR spectra (thin films or solutions in CHCl
3) were obtained with use of a Vertex 70v spectrometer (Bruker, Karlsruhe, Germany).
1H- and
13C-NMR spectra were recorded in CDCl
3, in MeOD or in
d6-DMSO with Me
4Si as the internal standard on an AVANCE–500 instrument (500.13 (
1H), 125.78 MHz (
13C), 470.59 MHz (
19F)) or on an AVANCE-400 (400.13 (
1H), 100.62 MHz (
13C), 376.50 MHz (
19F)) (Bruker). Mass spectra of new compounds were recorded on a Bruker–Autoflex III spectrometer (MALDI TOF, positive ion mode, sinapic acid as the matrices). Optical rotation was determined on a 141 polarimeter (Perkin–Elmer, Beaconsfield, UK). Specific rotation [α]
D is expressed in (deg·mL)/(g·dm)
−1; the concentration of the solution
c is expressed in g/100 mL. Elemental analysis was carried out on a 1106 analyzer (Carlo Erba, Milan, Italy). TLC was carried out on Sorbfil plates (Sorbpolimer, Krasnodar, Russia) in hexane–EtOAc and chloroform–methanol, spots were visualized with anisaldehyde. Silica gel L (KSKG grade, 50–160 μm) was employed for column chromatography. All reagents and solvents were of the purest grade available, and generally were used without further treatment. The starting compounds ursolic, oleanolic acids and reagents: sodium borohydride (NaBH
4), acetyl chloride, 10% Pd/C, oxalyl chloride, 1,2-diaminoethane, 1,4-diaminobutane, tris(2-aminoethyl)amine, 1,4-bis(3-aminopropyl)piperazine, tris(hydroxymethyl)aminomethane, 1,3-di-Boc-2-(trifluoromethylsulfonyl)guanidine, triethylamine (Et
3N), dimethylaminopyridine (DMAP), trifluoroacetic acid (TFA) were purchased from Acros Organics (Geel, Belgium). Dihydrobetulonic and dihydrobetulinic acids were obtained from betulin according to the known procedures [
41]. Acetates of oleanolic, ursolic, dihydrobetulonic and dihydrobetulinic acids were synthesized according to the typical procedures. Mono-Boc-protected bis-aminopropylpiperazine and compounds
19–
23 were prepared by a reported methods [
43,
45]. NMR
1H and
13C spectra of all new compounds are in
Supplementary Materials.
3.1.1. General Procedure for the Synthesis of Amines 4–7
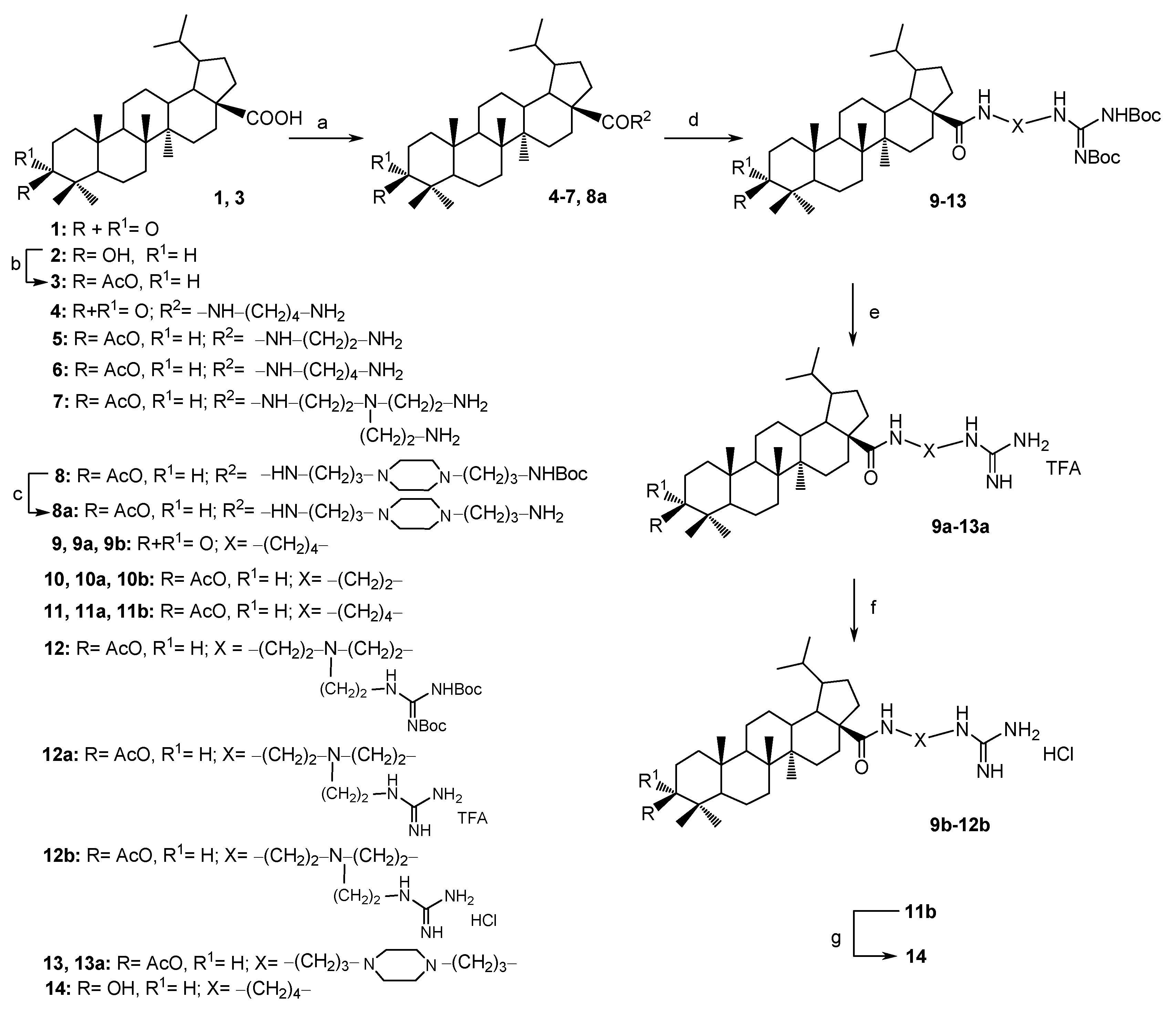
Oxalyl chloride (0.13 mL, 1.5 mmol) was added with stirring to a solution of compounds 1 or 3 (0.5 mmol) in dry CH2Cl2 (5 mL) precooled to 0 °C, and stirring of the reaction mixture was continued at room temperature for 2 h. Then the solvent and excess oxalyl chloride were removed under vacuum. The amine (1.5 mmol) was dissolved in dry CH2Cl2 (2 mL) and under vigorous stirring were added Et3N (0.2 mL, 1.5 mmol) and a solution of the acid chloride of 1 or 3 (0.5 mmol) in dry CH2Cl2 (4 mL). The mixture was stirred for 24 h (monitoring by TLC). The mixture was then poured into cold H2O and extracted with CH2Cl2 (2 × 15 mL each). The organic phases washed brine and were dried over Na2SO4 and evaporated under reduced pressure. The residue was chromatographed on silica gel, using CH2Cl2/MeOH 30:1→1:1 (v/v), to obtain pure compounds 4–7.
N-(4-Aminobuthyl)-3-oxolupane-28-amide (4), White powder, 78% yield; mp 170–172 °C (EtOH); : +4° (c 0.23, CH2Cl2); IR (CHCl3) νmax 1641, 1702 (C=O), 3357 (NH) cm−1; 1H-NMR (500 MHz, CDCl3) δ: 5.86 (t, J = 5.5 Hz, 1H, CONH), 3.32–3.20 (m, 2H, H-1′), 2.76 (t, J = 6.5 Hz, 2H, H-4′), 2.55–1.14 (m, 26H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 1.08, 1.03, 0.98, 0.97, 0.94 (all s, 3H each, H-23–H-27), 0.87 (d, J = 6.5 Hz, 3H, H-29), 0.75 (d, J = 7.0 Hz, 3H, H-30); 13C-NMR (125 MHz, CDCl3) δ: 218.2 (C-3), 176.3 (C-28), 56.0 (C-17), 55.0 (C-5), 49.8 (C-9), 49.5 (C-19), 47.4 (C-4), 44.2 (C-18), 42.7 (C-14), 41.6 (C-4′), 40.7 (C-8), 39.6 (C-1), 39 (C-22), 38.8 (C-1′), 37.7 (C-13), 36.9 (C-10), 34.2 (C-2), 33.8 (C-16), 33.6 (C-7), 30.6 (C-3′), 29.9 (C-20), 29.4 (C-15), 27.2 (C-23), 27.0 (C-2’), 26.6 (C-12), 23.1 (C-29), 23.0 (C-21), 21.5 (C-24), 21.1 (C-11), 19.7 (C-6), 16.0 (C-25), 15.9 (C-26), 14.6 (C-30), 14.5 (C-27); Anal. Calcd. for C34H58N2O2: C, 77.51, H, 11.10. Found: C, 78.03, H, 11.02%. MS: m/z 527.45 [M + H]+ (calcd. for C34H58N2O2, 526.45).
3β-N-(2-Aminoethyl)-3-O-acetyl-lupane-28-amide (5), White powder, 74% yield; mp 120–122 °C (EtOH); −8° (c 0.24, CH2Cl2); IR (CHCl3) νmax 1647, 1735 (C=O), 3367 (NH) cm−1; 1H-NMR (500 MHz, CDCl3) δ: 6.14 (t, J = 5.5 Hz, 1H, CONH), 4.48–4.44 (m, 1H, H-3), 3.35–3.21 (m, 2H, H-1′), 2.81 (t, J = 6.0 Hz, 2H, H-2′), 2.43–0.97 (m, 25H, CH, CH2 in pentacyclic skeleton), 2.01 (s, 3H, CH3CO–), 0.94, 0.92, 0.85, 0.84, 0.83, 0.82 (all s, 3H each, H-23–H-27 and H-29), 0.77 (d, J = 10.0 Hz, 1H, H-5), 0.73 (d, J = 7.0 Hz, 3H, H-30); 13C-NMR (125 MHz, CDCl3) δ: 176.7 (C-28), 171.0 (COCH3), 80.9 (C-3), 56.2 (C-17), 55.4 (C-5), 50.3 (C-9), 49.5 (C-19), 44.3 (C-18), 42.6 (C-14), 41.8 (C-2′), 41.7 (C-1′), 40.8 (C-8), 38.7 (C-1), 38.4 (C-22), 37.8 (C-4), 37.6 (C-13), 37.1 (C-10), 34.4 (C-7), 33.6 (C-16), 29.9 (C-20), 29.5 (C-15), 27.9 (C-23), 27.0 (C-2), 23.7 (C-12), 23.1 (C-21, C-29), 21.3 (COCH3), 21.0 (C-11), 18.2 (C-6), 16.5 (C-25), 16.2 (C-24, C-26), 14.6 (C-30), 14.5 (C-27); Anal. Calcd. for C34H58N2O3: C, 75.23, H, 8.84. Found: C, 75.74, H, 8.79%. MS: m/z 543.40 [M + H]+ (calcd. for C34H58N2O3, 542.44).
3β-N-(4-Aminobuthyl)-3-O-acetyl-lupane-28-amide (6), White powder, 58% yield; mp 104–106 °C (EtOH); −11° (c 0.69, CHCl3); IR (CHCl3) νmax 1646, 1734 (C=O), 3367 (NH) cm−1; 1H-NMR (500 MHz, CDCl3) δ: 5.86 (t, J = 5.5 Hz, 1H, CONH), 4.48–4.45 (m, 1H, H-3), 3.30–3.17 (m, 2H, H-1′), 2.73 (t, J = 6.5 Hz, 2H, H-4′), 2.47–0.98 (m, 25H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 2.03 (s, 3H, CH3CO–), 0.95, 0.93, 0.92, 0.84, 0.83, 0.82, (all s, 3H each, H-23–H-27 and H-29), 0.78 (d, J = 10.0 Hz, 1H, H-5), 0.73 (d, J = 7.0 Hz, 3H, H-30); 13C-NMR (125 MHz, CDCl3) δ: 176.3 (C-28), 171.0 (COCH3), 80.9 (C-3), 56.0 (C-17), 55.4 (C-5), 50.3 (C-9), 49.5 (C-19), 44.3 (C-18), 42.6 (C-14), 41.6 (C-4′), 40.8 (C-8), 39.0 (C-1′), 38.7 (C-22), 38.4 (C-1), 37.8 (C-4), 37.6 (C-13), 37.1 (C-10), 34.4 (C-7), 33.6 (C-16), 30.7 (C-3′), 29.9 (C-20), 29.4 (C-15), 27.9 (C-23), 27.2 (C-2′), 27.0 (C-2), 23.7 (C-12), 23.0 (C-21, C-29), 21.3 (COCH3), 21.0 (C-11), 18.2 (C-6), 16.5 (C-25), 16.2 (C-26, C-24), 14.6 (C-30), 14.5 (C-27); Anal. Calcd. for C36H62N2O3: C, 75.74, H, 10.95. Found: C, 76.01, H, 10.89%. MS: m/z 609.43 [M + K]+ (calcd. for C36H62N2O3, 570.48).
3β-N-[2-(N,N′-bis-Aminoethyl)-aminoethyl]-3-O-acetyl-lupane-28-amide (7), White powder, 69% yield; mp 108–110 °C (EtOH); −5° (c 0.16, C2H5OH); IR (CHCl3) νmax 1648, 1735 (C=O), 3441 (NH) cm−1; 1H-NMR (400 MHz, MeOD) δ: 4.49–4.45 (m, 1H, H-3), 3.33–3.27 (m, 2H, H-1′), 2.74–2.72 (m, 4H, H-4′, H-42″), 2.60–2.58 (m, 6H, H-2′, H-3′, H-3″), 2.32–0.84 (m, 26H, CH, CH2 in pentacyclic skeleton), 2.05 (s, 3H, CH3CO–), 1.02, 1.00, 0.93, 0.90, 0.89, 0.88 (all s, 3H each, H-23–H-27 and H-29), 0.79 (d, J = 6.4 Hz, 3H, H-30); 13C-NMR (100 MHz, MeOD) δ: 179.3 (C-28), 172.8 (COCH3), 82.5 (C-3), 57.7 (C-17), 57.4 (C-5), 57.0 (C-3′, C-3″), 55.2 (C-2’), 51.9 (C-9), 51.1 (C-19), 45.5 (C-18), 43.8 (C-14), 42.2 (C-8), 40.2 (C-22), 39.9 (C-1), 39.8 (C-4′, C-4″), 39.0 (C-4), 38.8 (C-1′), 38.4 (C-13), 38.3 (C-10), 35.8 (C-7), 34.1 (C-16), 31.4 (C-20), 30.9 (C-15), 28.7 (C-23), 28.5 (C-2), 24.8 (C-12), 24.2 (C-29), 23.8 (C-21), 22.4 (COCH3), 21.4 (C-11), 19.4 (C-6), 17.2 (C-25), 17.0 (C-24, C-26), 15.3 (C-30, C-27); Anal. Calcd. for C38H68N4O3: C, 72.56, H, 10.90. Found: C, 73.04, H, 10.87%. MS: m/z 629.56 [M + H]+ (calcd. for C38H68N4O3, 628.53).
3β-N-{[3-(3-Aminopropyl)piperazinyl]propyl}-3-O-acetyl-lupane-28-amide (8a), The acid chloride of 3 was synthesized according to the general procedure for synthesis of amines 4–7. Then the N-tert-butoxycarbonyl-1,4-bis(3-aminopropyl)piperazine (0.45 g, 1.5 mmol) and Et3N (0.25 mL, 1.8 mmol) were added to acid chloride of 3 (0.50 g, 1 mmol) in dry CH2Cl2 (10 mL). The mixture was stirred at room temperature for 16 h (monitoring by TLC), then was poured into cold H2O and extracted with CH2Cl2 (2 × 15 mL). The organic phases washed brine, dried over Na2SO4 and evaporated under reduced pressure to obtain compound 8. Then the compound 8 was dissolved in CH2Cl2 (7 mL), acidified with TFA 10% (v/v) in CH2Cl2 (17 mL) for deprotection reaction and stirred for around 5 h. The reaction was quenched using saturated aqueous potassium carbonate solution (20 mL). The aqueous phase was extracted with CH2Cl2 (2 × 15 mL). The organic phases washed brine, dried over Na2SO4 and evaporated under reduced pressure. The residue was chromatographed on silica gel, using CH2Cl2/MeOH 50:1→5:1, to obtain pure compound 8a. White powder, 78% yield; mp 119–121 °C (EtOH); −7.4° (c 0.5, CHCl3); IR (CHCl3) νmax 1735 (C=O), 3361 (NH) cm−1; 1H-NMR (500 MHz, CDCl3) δ: 6.79 (t, J = 5.5 Hz, 1H, CONH), 4.47–4.44 (m, 1H, H-3), 3.31–3.25 (m, 2H, H-1′), 2.78–2.77 (m, 2H, H-1″), 2.50–2.42 (m, 12H, H-3′–H-5′, H-3″–H-5″), 2.41–0.98 (m, 25H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-2″), 2.02 (s, 3H, CH3CO–), 0.93, 0.92, 0.91, 0.83, 0.82, 0.81 (3H each, all s, H-23–H-27 and H-29), 0.77 (d, J = 10.5 Hz, 1H, H-5), 0.72 (d, J = 6.5 Hz, 3H, H-30); 13C-NMR (125 MHz, CDCl3) δ: 176.3 (C-28), 171.0 (COCH3), 80.9 (C-3), 57.8 (C-3′), 56.7 (C-3″), 55.9 (C-17), 55.4 (C-5), 53.5 (C-5′, C-5″), 53.3 (C-4′), 53.3 (C-4″), 50.3 (C-9), 49.6 (C-19), 44.1 (C-18), 42.6 (C-14), 40.9 (C-1″), 40.8 (C-8), 39.3 (C-1′), 38.8 (C-22), 38.4 (C-1), 37.8 (C-4), 37.4 (C-13), 37.1 (C-10), 34.4 (C-7), 33.7 (C-16), 29.9 (C-20), 29.8 (C-2′), 29.7 (C-15), 27.9 (C-23), 26.9 (C-2), 25.3 (C-2″), 23.7 (C-12), 23.0 (C-21, C-29), 21.3 (COCH3), 21.0 (C-11), 18.2 (C-6), 16.5 (C-25), 16.3 (C-24), 16.2 (C-26), 14.6 (C-30), 14.5 (C-27); Anal. Calcd. for C42H74N4O3: C, 73.85, H, 10.92. Found: C, 74.13, H, 10.84%. MS: m/z 683.52 [M + H]+ (calcd. for C42H74N4O3, 682.58).
3.1.2. General Procedure for the Synthesis of Compounds 15, 18–21
Acid chlorides of 3, 16, 17 was synthesized according to the general procedure for synthesis of amines 4–7. Then the acid chlorides of 3, 16, 17 (1 mmol) was dissolved in a mixture of pyridine (4 mL), CH2Cl2 (1 mL) and DMAP (0.09 g, 0.7 mmol) was added. After complete dissolution of DMAP, a solution containing of TRIS (tris(hydroxymethyl)aminomethane) (0.24 g, 2 mmol) in pyridine (0,5 mL) was added. The mixture was stirred for 10 h at room temperature and the solvent was removed rapidly under vacuum. The residue was chromatographed on silica gel, using CH2Cl2/MeOH 30:1→1:1, to obtain pure compounds 15, 18–21.
3β-[2-Amino-3-hydroxy-2-(hydroxymethyl)propyl]-3-O-acetyl-lupane-28-oate (15), White powder, 16% yield; mp 134–136 °C (EtOH); −13.1° (c 0.51, CHCl3); IR (CHCl3) νmax 1731 (C=O), 3366 (OH), 3446 (NH) cm−1; 1H-NMR (400 MHz, CDCl3) δ: 4.49–4.45 (m, 1H, H-3), 4.09, 4.03 (both d, J = 11.6 Hz, 1H each, H-1′), 3.52 (br s, 4H, H-3′, H-4′), 2.70 (br s, 4H, NH2, OH), 2.05 (s, 3H, CH3CO–), 2.24–0.81 (m, 26H, CH, CH2 in pentacyclic skeleton), 0.95, 0.91, 0.85, 0.84, 0.83, 0.82 (all s, 3H each, H-23–H-27 and H-29), 0.76 (d, J = 6.8 Hz, 3H, H-30); 13C-NMR (100 MHz, CDCl3) δ: 177.0 (C-28), 171.1 (COCH3), 80.9 (C-3), 64.9 (C-1′), 64.2 (C-3′, C-4′), 57.3 (C-17, C-2′), 55.4 (C-5), 50.2 (C-9), 48.8 (C-19), 44.2 (C-18), 42.6 (C-14), 40.7 (C-8), 38.4 (C-22), 38.1 (C-1), 37.8 (C-4), 37.5 (C-13), 37.1 (C-10), 34.3 (C-7), 32.0 (C-16), 29.7 (C-15, C-20), 27.9 (C-23), 26.9 (C-2), 23.7 (C-12), 23.0 (C-29), 22.8 (C-21), 21.3 (COCH3), 20.9 (C-11), 18.2 (C-6), 16.5 (C-25), 16.2 (C-24), 16.1 (C-26), 14.7 (C-30), 14.6 (C-27); Anal. Calcd. for C36H61NO6: C, 71.60, H, 10.18. Found: C, 72.09, H, 10.11%. MS: m/z 604.42 [M + H]+ (calcd. for C36H61NO6, 603.45).
3β-[2-Amino-3-hydroxy-2-(hydroxymethyl)propyl]-3-O-acetylurs-12-en-28-oate (18), White powder, 15% yield; mp 129–131 °C (EtOH); +40° (c 0.74, CHCl3); IR (CHCl3) νmax 1721 (C=O), 3444 (NH), 3468 (OH) cm−1; 1H-NMR (400 MHz, CDCl3) δ: 5.25 (br s, 1H, H-12), 4.51–4.47 (m, 1H, H-3), 4.02, 3.93 (both d, J = 12 Hz, 1H each, H-1′), 3.48 (br s, 4H, H-3′, H-4′), 2.68 (br s, 4H, NH2, OH), 2.19 (d, J = 11.2 Hz, 1H, H-18), 2.05 (s, 3H, CH3CO–), 1.99–0.81 (m, 22H, CH, CH2 in pentacyclic skeleton), 1.08, 0.96, 0.94, 0.88, 0.87, 0.86, 0.74 (all s, 3H each, H-23–H-27, H-29 and H-30); 13C-NMR (100 MHz, CDCl3) δ: 178.1 (C-28), 171.1 (COCH3), 138.3 (C-13), 125.6 (C-12), 80.9 (C-3), 65.7 (C-1′), 64.4 (C-3′, C-4′), 56.6 (C-2′), 55.3 (C-5), 53.0 (C-18), 48.6 (C-17), 47.4 (C-9), 42.1 (C-14), 39.5 (C-4, C-8), 38.9 (C-19), 38.3 (C-20), 37.7 (C-1), 37.1 (C-22), 36.8 (C-10), 32.9 (C-7), 30.6 (C-21), 28.1 (C-23), 27.9 (C-15), 24.3 (C-16, C-27), 23.5 (C-2, C-11), 21.3 (COCH3), 21.1 (C-30), 18.2 (C-6), 17.2 (C-29), 17.0 (C-26), 16.7 (C-24), 15.5 (C-25); Anal. Calcd. for C36H59NO6: C, 71.84, H, 9.88. Found: C, 72.13, H, 9.83%. MS: m/z 602.40 [M + H]+ (calcd. for C36H59NO6, 601.43).
3β-N-[(1′,1′,1′-tris-Hidroxymethyl)methyl]-3-O-acetyl-ursolamide (19), White powder, 24% yield; mp 203–205 °C (EtOH); +26.9° (c 0.74, CH3OH); IR (CH3OH) νmax 1734 (C=O), 2923, 2871, 2852 (OH), 3352 (NH) cm−1; 1H-NMR (500 MHz, MeOD) δ: 5.38 (br s, 1H, H-12), 4.50–4.47 (m, 1H, H-3), 3.66, 3.58 (both d, J = 10 Hz, 3H each, H-2′–H-4′), 2.12–0.88 (m, 23H, CH, CH2 in pentacyclic skeleton), 2.05 (s, 3H, CH3CO–), 1.18, 1.04, 0.99, 0.95, 0.93, 0.92, 0.91 (all s, 3H each, H-23–H-27, H-29 and H-30); 13C-NMR (125 MHz, CDCl3) δ: 178.5 (C-28), 170.7 (COCH3), 138.1 (C-13), 125.8 (C-12), 80.4 (C-3), 61.8 (C-1′), 60.8 (C-2′–C-4′), 55.1 (C-5), 53.6 (C-18), 48.3 (C-17), 47.4 (C-9), 42.3 (C-14), 39.5 (C-4, C-8), 38.9 (C-19), 38.4 (C-20), 37.7 (C-22, C-1), 36.8 (C-10), 33 (C-7), 30.9 (C-21), 28.3 (C-23), 27.8 (C-15), 24.5 (C-16), 23.7 (C-2), 23.4 (C-11, C-27), 21.5 (COCH3), 21.4 (C-30), 18.2 (C-6), 17.5 (C-29, C-26), 17.1 (C-24), 15.7 (C-25); Anal. Calcd. for C36H59NO6: C, 71.84, H, 9.88. Found: C, 72.09, H, 9.79%. MS: m/z 602.41 [M + H]+ (calcd. for C36H59NO6, 601.43).
3β-[2-Amino-3-hydroxy-2(hydroxymethyl)propyl]-3-O-acetylolean-12-en-28-oate (20), White powder, 23% yield; mp 141–143 °C (EtOH); +49° (c 0.75, CHCl3); IR (CHCl3) νmax 1618, 1727 (C=O), 3447 (NH) cm−1; 1H-NMR (500 MHz, CDCl3) δ: 5.30 (br s, 1H, H-12), 4.51–4.48 (m, 1H, H-3), 4.04, 3.99 (both d, J = 11.5 Hz, 1H each, H-1′), 3.50 (br s, 4H, H-3′, H-4′), 2.84 (d, J = 10.0 Hz, 1H, H-18), 2.68–2.60 (m, 4H, NH2, OH), 2.05 (s, 3H, CH3CO–), 2.01–0.83 (m, 22H, CH, CH2 in pentacyclic skeleton), 1.14, 0.94, 0.93, 0.92, 0.87, 0.86, 0.73 (all s, 3H each, H-23–H-27, H-29 and H-30); 13C-NMR (125 MHz, CDCl3) δ: 178.2 (C-28), 171.1 (COCH3), 143.7 (C-13), 122.6 (C-12), 80.9 (C-3), 65.7 (C-1′), 64.6 (C-3′, C-4′), 56.6 (C-2′), 55.3 (C-5), 47.5 (C-9), 47.2 (C-17), 45.7 (C-19), 41.8 (C-14), 41.5 (C-18), 39.3 (C-8), 38.1 (C-1), 37.7 (C-4), 36.9 (C-10), 33.8 (C-22), 33.0 (C-30), 32.8 (C-7), 32.7 (C-21), 30.7 (C-20), 28.0 (C-23), 27.6 (C-15), 25.8 (C-27), 23.6 (C-29), 23.5 (C-11), 23.4 (C-2), 23.1 (C-16), 21.3 (COCH3), 18.2 (C-6), 17.1 (C-26), 16.7 (C-24), 15.4 (C-25); Anal. Calcd. for C36H59NO6: C, 71.84, H, 9.88. Found: C, 72.04, H, 9.82%. MS: m/z 602.40 [M + H]+ (calcd. for C36H59NO6, 601.43).
3β-N-[(1′,1′,1′-tris-Hydroxymethyl)methyl]-3-O-acetyl-oleanolamide (21), White powder, 32% yield; mp 249–251 °C (EtOH); +33.9° (c 0.61, CH3OH); IR (CH3OH) νmax 1732(C=O), 2945, 2927 (OH), 3353 (NH) cm−1; 1H-NMR (500 MHz, d6-DMSO) δ: 6.62 (s, 1H, NH), 5.24 (br s, 1H, H-12), 4.97 (t, J = 5 Hz, 3H, –OH), 4.42–4.38 (m, 1H, H-3), 3.49–3.42 (m, 6H, H-2′–H-4′), 2.61 (d, J = 10.0 Hz, 1H, H-18), 2.00 (s, 3H, CH3CO–), 1.97–0.84 (m, 22H, CH, CH2 in pentacyclic skeleton), 1.91, 0.90, 0.88, 0.87, 0.82, 0.81, 0.78 (all s, 3H each, H-23–H-27, H-29 and H-30); 13C-NMR (125 MHz, d6-DMSO) δ: 178.3 (C-28), 170.6 (COCH3), 143.7 (C-13), 122.6 (C-12), 80.4 (C-3), 61.8 (C-1′), 60.8 (C-2′–C-4′), 55 (C-5), 47.4 (C-9), 46.8 (C-17), 46.6 (C-19), 42.0 (C-14), 41.9 (C-18), 39.5 (C-8), 38.2 (C-1), 37.7 (C-4), 36.9 (C-10), 34.1 (C-22), 33.3 (C-7), 33.2 (C-30), 32.7 (C-21), 30.8 (C-20), 28.2 (C-23), 27.4 (C-15), 25.8 (C-27), 23.8 (C-29), 23.7 (C-2), 23.4 (C-11, C-16), 21.4 (COCH3), 18.2 (C-6), 17.4 (C-26), 17.1 (C-24), 15.6 (C-25); Anal. Calcd. for C36H59NO6: C, 71.84, H, 9.88. Found: C, 72.11, H, 9.85%. MS: m/z 602.39 [M + H]+ (calcd. for C36H59NO6, 601.43).
3.1.3. General Procedure for the Guanilation of Amines 4–7, 8a, 15, 18 and 20
The amine (0.5 mmol) is added neat to a solution of 1,3-di-Boc-2-(trifluoromethyl-sulfonyl)guanidine (0.18 g, 0.45 mmol) or (0.60 g, 0.9 mmol) for amine 7 and Et3N (0.07 mL, 0.5 mmol) in CH2Cl2 (at r.t.) of compounds 4–7, 8a or in CHCl3 (at reflux) of compounds 15, 18, 20 (10 mL). The mixture was stirred for 2–12 h (TLC monitoring CH2Cl2/MeOH 20:1). After completion of the reaction, the mixture is diluted with CH2Cl2 and washed witH-NH4Cl, NaHCO3 and brine. After drying with sodium sulfate and filtering the solvent is removed under reduced pressure. The residue was chromatographed on silica gel, using hexane/EtOAc 10:1→1:1, to obtain pure compounds 9–13, 15a, 18a and 20a.
N-[4-tert-Butyloxycarbonyl buthylguanidine]-3-oxo-lupane-28-amide (9), White powder, 88% yield; mp 158–160 °C (EtOH); +3.4° (c 0.59, CH2Cl2); IR (CHCl3) νmax 1637, 1718 (C=O), 3337 (NH) cm−1; 1H-NMR (500 MHz, CDCl3) δ: 11.52 (s, 1H, NH in Boc), 8.30 (br s, 1H, NH–C=N), 5.70 (br s, 1H, CONH), 3.48–3.41 (m, 2H, H-2′), 3.37–3.20 (m, 2H, H-1′), 1.51, 1.48 (both s, 9H each, CH3 in Boc), 2.54–1.15 (m, 26H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 1.08, 1.03, 0.98, 0.97, 0.95 (all s, 3H each, H-23–H-27), 0.87 (d, J = 6.5 Hz, 3H, H-29), 0.75 (d, J = 6.5 Hz, 3H, H-30); 13C-NMR (125 MHz, CDCl3) δ: 218.1 (C-3), 176.3 (C-28), 163.6 (C=N), 156.1, 153.3 (CONH-Boc), 83.0, 79.2 (C in Boc), 56.0 (C-17), 55.0 (C-5), 49.8 (C-9), 49.5 (C-19), 47.3 (C-4), 44.1 (C-18), 42.6 (C-14), 40.7 (C-8), 40.5 (C-4′), 39.6 (C-1), 38.7 (C-22, C-1′), 37.6 (C-13), 36.9 (C-10), 34.1 (C-2), 33.8 (C-16), 33.6 (C-7), 29.9 (C-20), 29.4 (C-15), 28.4, 28.0 (CH3 in Boc), 27.7 (C-23), 27.0 (C-2′, C-3′), 26.6 (C-12), 23.0 (C-21, C-29), 21.5 (C-24), 21.0 (C-11), 19.6 (C-6), 16.0 (C-25), 15.9 (C-26), 14.6 (C-30), 14.5 (C-27); Anal. Calcd. for C45H76N4O6: C, 70.27, H, 9.96. Found: C, 70.62, H, 9.91%. MS: m/z 791.56 [M + Na]+ (calcd. for C45H76N4O6, 768.58).
3β-N-(2-tert-Butyloxycarbonylethylguanidine)-3-O-acetyl-lupane-28-amide (10), White powder, 86% yield; mp 176–177 °C (EtOH); +3.8° (c 0.56, CHCl3); IR (CHCl3) νmax 1724 (C=O), 3329 (NH) cm−1; 1H-NMR (500 MHz, CDCl3) δ: 11.53 (s, 1H, NH in Boc), 8.65 (br s, 1H, NH–C=N), 6.88 (br s, 1H, CONH), 4.50–4.47 (m, 1H, H-3), 3.69–3.54 (m, 2H, H-2′), 3.47–3.35 (m, 2H, H-1′), 2.05 (s, 3H, CH3CO–), 1.51, 1.50 (both s, 9H each, CH3 in Boc), 2.46–0.96 (m, 25H, CH, CH2 in pentacyclic skeleton), 0.94, 0.90, 0.87, 0.86, 0.85, 0.84 (all s, 3H each, H-23–H-27 and H-29), 0.79 (d, J = 9.5 Hz, 1H, H-5), 0.75 (d, J = 7.0 Hz, 3H, H-30); 13C-NMR (125 MHz, CDCl3) δ: 176.9 (C-28), 171.0 (COCH3), 163.1 (C=N), 153.5, 153.0 (CONH-Boc), 83.5 (C in Boc), 80.9 (C-3), 77.3 (C in Boc), 56.0 (C-17), 55.4 (C-5), 50.3 (C-9), 49.7 (C-19), 44.2 (C-18), 42.5 (C-14), 41.2 (C-2′), 40.7 (C-8), 39.7 (C-1′), 38.6 (C-22), 38.4 (C-1), 37.8 (C-4), 37.5 (C-13), 37.1 (C-10), 34.3 (C-7), 33.4 (C-16), 29.8 (C-20), 29.4 (C-15), 28.3, 28.0 (CH3 in Boc), 27.9 (C-23), 26.9 (C-2), 23.7 (C-12), 23.1 (C-29), 23.0 (C-21), 21.3 (COCH3), 21.0 (C-11), 18.3 (C-6), 16.5 (C-25), 16.2 (C-24), 16.1 (C-26), 14.7 (C-30), 14.6 (C-27); Anal. Calcd. for C45H76N4O7: C, 68.84, H, 9.76. Found: C, 69.23, H, 9.69%. MS: m/z 807.54 [M + Na]+ (calcd. for C45H76N4O7, 784.57).
3β-N-(4-tert-Butyloxycarbonylbutylguanidine)-3-O-acetyl-lupane-28-amide (11), White powder, 82% yield; mp 148–150 °C (EtOH); −4° (c 0.52, CHCl3); IR (CHCl3) νmax 1640, 1720 (C=O), 3288, 3337, 3410 (NH) cm−1; 1H-NMR (500 MHz, CDCl3) δ: 11.48 (s, 1H, NH in Boc), 8.30 (t, J = 5.0 Hz, 1H, NH–C=N), 5.73 (t, J = 5.5 Hz, 1H, CONH), 4.46–4.43 (m, 1H, H-3), 3.42–3.37 (m, 2H, H-2′), 3.31–3.16 (m, 2H, H-1′), 2.01 (s, 3H, CH3CO–), 1.47, 1.46 (both s, 9H each, CH3 in Boc), 2.45–0.94 (m, 25H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 0.91, 0.90, 0.83, 0.82, 0.81, 0.80 (all s, 3H each, H-23–H-27 and H-29), 0.75 (d, J = 9.5 Hz, 1H, H-5), 0.71 (d, J = 7.0 Hz, 3H, H-30); 13C-NMR (125 MHz, CDCl3) δ: 176.2 (C-28), 170.9 (COCH3), 163.6 (C=N), 156.1, 153.3 (CONH-Boc), 83.0 (C in Boc), 81.0 (C-3), 79.2 (C in Boc), 56.0 (C-17), 55.4 (C-5), 50.3 (C-9), 49.5 (C-19), 44.2 (C-18), 42.6 (C-14), 40.8 (C-8), 40.5 (C-4′), 38.7 (C-1′, C-22), 38.4 (C-1), 37.8 (C-4), 37.5 (C-13), 37.1 (C-10), 34.4 (C-7), 33.6 (C-16), 29.9 (C-3′), 29.4 (C-20), 28.3, 28.0 (CH3 in Boc), 27.8 (C-15), 27.3 (C-23), 26.9 (C-2′), 26.5 (C-2), 23.7 (C-12), 23.0 (C-21, C-29), 21.3 (COCH3), 21.0 (C-11), 18.2 (C-6), 16.5 (C-25), 16.2 (C-24, C-26), 14.6 (C-30), 14.5 (C-27); Anal. Calcd. for C47H80N4O7: C, 69.42, H, 9.92. Found: C, 69.74, H, 9.85%. MS: m/z 835.51 [M + Na]+ (calcd. for C47H80N4O7, 812.60).
3β-N-[2-(N,N′-bis-tert-Butyloxycarbonylethylgyanidine)-aminoethyl]-3-O-acetyl-lupane-28-amide (12), White powder, 87% yield; mp 188–190 °C (EtOH); +1.02° (c 0.96, CHCl3); IR (CHCl3) νmax 1641, 1722 (C=O), 3335, 3442 (NH) cm−1; 1H-NMR (500 MHz, CDCl3) δ: 11.52 (s, 2H, NH in Boc), 8.52 (br s, 2H, NH–C=N), 6.35 (t, 1H, J = 5.5 Hz, CONH), 4.49–4.46 (m, 1H, H-3), 3.53–3.43 (m, 4H, H-4′, H-4″), 3.41–3.22 (m, 2H, H-1′), 2.65–2.54 (m, 6H, H-2′, H-3′, H-3″), 2.04 (s, 3H, CH3CO–), 1.50, 1.48 (both br s, 18H each, CH3 in Boc), 2.32–0.98 (m, 25H, CH, CH2 in pentacyclic skeleton), 0.93, 0.92, 0.85, 0.84, 0.83, 0.82 (all s, 3H each, H-23–H-27 and H-29), 0.78 (d, J = 9.0 Hz, 1H, H-5), 0.73 (d, J = 6.5 Hz, 3H, H-30); 13C-NMR (125 MHz, CDCl3) δ: 176.7 (C-28), 171.0 (COCH3), 163.5 (C=N), 155.9, 153.2 (CONH-Boc), 82.9 (C in Boc), 81.0 (C-3), 79.2 (C in Boc), 55.9 (C-17), 55.5 (C-5), 54.8 (C-2′), 53.8 (C-3′, C-3″), 50.4 (C-9), 49.6 (C-19), 43.8 (C-18), 42.5 (C-14), 40.8 (C-8), 39.0 (C-4′, C-4″), 38.9 (C-22), 38.4 (C-1), 38.0 (C-13), 37.8 (C-1′), 37.2 (C-4), 37.1 (C-10), 34.4 (C-7), 33.4 (C-16), 29.8 (C-20), 29.5 (C-15), 28.3, 28.1 (CH3 in Boc), 28.0 (C-23), 27.0 (C-2), 23.7 (C-12), 23.1 (C-29), 23.0 (C-21), 21.3 (COCH3), 21.0 (C-11), 18.3 (C-6), 16.5 (C-25), 16.4 (C-24), 16.2 (C-26), 14.7 (C-30), 14.4 (C-27); Anal. Calcd. for C60H104N8O11: C, 64.72; H, 9.41. Found: C, 65.02; H, 9.34%. MS: m/z 1135.71 [M + Na]+ (calcd. for C60H104N8O11, 1112.78).
3β-N-{[3-(3-tert-Butyloxycarbonylpropylgyanidine)piperazinyl]propyl}-3-O-acetyl-lupane-28-amide (13), White powder, 60% yield; mp 131–134 °C (EtOH); −7.9° (c 0.57, CHCl3); IR (CHCl3) νmax 1640, 1722 (C=O), 3289, 3333 (NH) cm−1; 1H-NMR (500 MHz, CDCl3) δ: 11.50 (br s, 1H, NH in Boc), 8.53 (br s, 1H, NH–C=N), 6.86 (br s, 1H, CONH), 4.47–4.45 (m, 1H, H-3), 3.51–3.42 (m, 2H, H-1′′), 3.32–3.27 (m, 2H, H-1′), 2.48–2.41 (m, 14H, H-3′–H-5′, H-2″–H-5″), 2.06 (s, 3H, CH3CO–), 1.49 (br s, 18H, CH3 in Boc), 2.33–0.99 (m, 25H, CH, CH2 in pentacyclic skeleton, 2H, H-2′), 0.93, 0.92, 0.85, 0.84, 0.83, 0.81 (all s, 3H each, H-23–H-27 and H-29), 0.78 (d, J = 10.5 Hz, 1H, H-5), 0.73 (d, J = 6.5 Hz, 3H, H-30); 13C-NMR (125 MHz, CDCl3) δ: 176.4 (C-28), 171.0 (COCH3), 163.7 (C=N), 156.1, 153.0 (CONH-Boc), 82.8 (C in Boc), 80.9 (C-3), 79.2 (C in Boc), 56.4 (C-3′), 55.9 (C-17), 55.9 (C-3″), 55.4 (C-5), 53.1 (C-4′, C-4″, C-5′, C-5″), 50.3 (C-9), 49.6 (C-19), 44.1 (C-18), 42.6 (C-14), 40.8 (C-8), 39.8 (C-22, C-1″), 39.2 (C-1′), 38.8 (C-1), 38.4 (C-4), 37.8 (C-10), 37.4 (C-13), 34.5 (C-7), 33.7 (C-16), 29.8 (C-20), 29.4 (C-15), 28.3, 28.1 (CH3 in Boc), 27.9 (C-23), 26.9 (C-2), 26.9 (C-2′), 25.9 (C-2″), 23.7 (C-12), 23.1 (C-21), 23.0 (C-29), 21.3 (COCH3), 21.0 (C-11), 18.2 (C-6), 16.5 (C-25), 16.3 (C-24), 16.2 (C-26), 14.6 (C-30), 14.5 (C-27); Anal. Calcd. for C53H92N6O7: C, 68.79, H, 10.02. Found: C, 69.04, H, 9.96%. MS: m/z 947.58 [M + Na]+ (calcd. for C53H92N6O7, 924.70).
3β-[2-tert-Butyloxycarbonylguanidine-3-hydroxy-2-(hydroxymethyl)propyl]-3-O-acetyl-lupane-28-oate (15a), White powder, 63% yield; mp 106–108 °C (EtOH); −5.6° (c 0.48, CHCl3); IR (CHCl3) νmax 1655, 1714 (C=O), 3271, 3437 (NH) cm−1; 1H-NMR (400 MHz, CDCl3) δ: 11.47 (s, 1H, NH in Boc), 9.05 (s, 1H, NH–C=N), 4.49–4.45 (m, 1H, H-3), 4.25 (br s, 2H, H-1′), 3.82-3.77, 3.57–3.53 (both m, 2H each, H-3′, H-4′), 2.05 (s, 3H, CH3CO–), 1.49, 1.47 (both br s, 9H each, CH3 in Boc), 2.30–0.98 (m, 25H, CH, CH2 in pentacyclic skeleton), 0.94, 0.90, 0.88, 0.86, 0.85, 0.83 (all s, 3H each, H-23–H-27 and H-29), 0.79 (d, J = 9.6 Hz, 1H, H-5), 0.75 (d, J = 6.4 Hz, 3H, H-30); 13C-NMR (100 MHz, CDCl3) δ: 176.3 (C-28), 171.1 (COCH3), 161.8 (C=N), 155.7, 152.8 (CONH-Boc), 83.8 (C in Boc), 80.9 (C-3), 80.1 (C in Boc), 62.9 (C-1′), 62.8 (C-4′), 62.8 (C-3′), 61.9 (C-2′), 57.3 (C-17), 55.4 (C-5), 50.2 (C-9), 49.0 (C-19), 44.0 (C-18), 42.5 (C-14), 40.7 (C-8), 38.4 (C-22), 38.0 (C-1), 37.8 (C-4), 37.1 (C-13, C-10), 34.3 (C-7), 31.8 (C-16), 29.8 (C-20), 29.7 (C-15), 28.1 (CH3 in Boc), 28.0 (CH3 in Boc, C-23), 26.9 (C-2), 23.7 (C-12), 23.0 (C-29), 22.7 (C-21), 21.3 (COCH3), 20.9 (C-11), 18.2 (C-6), 16.5 (C-25), 16.2 (C-24), 15.9 (C-26), 14.7 (C-30), 14.6 (C-27); Anal. Calcd. for C47H79N3O10: C, 66.72, H, 9.41. Found: C, 67.13, H, 9.36%. MS: m/z 868.45 [M + Na]+ (calcd. for C47H79N3O10, 845.58).
3β-[2-tert-Butyloxycarbonylguanidine-3-hydroxy-2-(hydroxymethyl)propyl]-3-O-acetyl-urs-12-en-28-oate (18a), White powder, 75% yield; mp 118–120 °C (EtOH); +31.3° (c 0.53, CHCl3); IR (CHCl3) νmax 1653, 1729 (C=O), 3271, 3443 (NH) cm−1; 1H-NMR (500 MHz, CDCl3) δ: 11.50 (s, 1H, NH in Boc), 9.06 (s, 1H, NH–C=N), 5.26 (br s, 1H, H-12), 4.51–4.48 (m, 1H, H-3), 4.13 (br s, 2H each, H-1′), 3.82–3.76 (m, 2H, H-3′), 3.54–3.52 (m, 2H, H-4′), 2.26 (d, J = 11.0 Hz, 1H, H-18), 2.04 (s, 3H, CH3CO–), 1.99–0.81 (m, 22H, CH, CH2 in pentacyclic skeleton), 1.50, 1.47 (both br s, 9H each, CH3 in Boc), 1.07, 0.94, 0.93, 0.86, 0.85, 0.83, 0.75 (all s, 3H each, H-23–H-27, H-29 and H-30); 13C-NMR (125 MHz, CDCl3) δ: 177.4 (C-28), 171.0 (COCH3), 161.8 (C=N), 155.6, 152.8 (CONH-Boc), 138.1 (C-13), 125.8 (C-12), 83.7 (C in Boc), 80.9 (C-3), 79.9 (C in Boc), 63.4 (C-1′), 62.9 (C-3′), 62.6 (C-4′), 61.6 (C-2′), 55.3 (C-5), 52.8 (C-18), 48.5 (C-17), 47.5 (C-9), 42.0 (C-14), 39.5 (C-8), 39.0 (C-4), 38.7 (C-19), 38.3 (C-20), 37.7 (C-1), 36.8 (C-22), 36.5 (C-10), 32.9 (C-7), 30.6 (C-21), 28.1 (CH3 in Boc), 28.0 (C-15), 28 (C-23), 24.1 (C-16), 23.6 (C-2), 23.5 (C-27), 23.3 (C-11), 21.3 (C-30), 21.0 (COCH3), 18.2 (C-6), 17.0 (C-29), 16.9 (C-26), 16.7 (C-24), 15.5 (C-25); Anal. Calcd. for C47H77N3O10: C, 66.87, H, 9.19. Found: C, 67.27, H, 9.14%. MS: m/z 866.43 [M + Na]+ (calcd. for C47H77N3O10, 843.56).
3β-[2-tert-Butyloxycarbonylguanidine-3-hydroxy-2-(hydroxymethyl)propyl]-3-O-acetyl-olean-12-en-28-oate (20a), White powder, 79% yield; mp 140–142 °C (EtOH); +30.4° (c 0.56, CHCl3); IR (CHCl3) νmax 1618, 1727 (C=O), 3434 (NH) cm−1; 1H-NMR (400 MHz, CDCl3) δ: 11.49 (s, 1H, NH in Boc), 9.03 (s, 1H, NH–C=N), 5.30 (br s, 1H, H-12), 5.10 (br s, 1H, OH), 4.49–4.45 (m, 1H, H-3), 4.26, 4.15 (both d, J = 11.6 Hz, 1H each, H-1′), 3.77, 3.54 (both d, J = 12.0 Hz, J = 11.6 Hz, 2H each, H-3′, H-4′), 2.85 (d, J = 10.0 Hz, 1H, H-18), 2.03 (s, 3H, CH3CO–), 1.99–0.80 (m, 22H, CH, CH2 in pentacyclic skeleton), 1.51, 1.48 (both br s, 9H each, CH3 in Boc), 1.12, 0.91, 0.90, 0.89, 0.85, 0.84, 0.71 (3H each, all s, H-23–H-27, H-29 and H-30); 13C-NMR (100 MHz, CDCl3) δ: 177.4 (C-28), 171.0 (COCH3), 161.8 (C=N), 155.6, 152.8 (CONH-Boc), 138.1 (C-13), 125.8 (C-12), 83.7 (C in Boc), 80.9 (C-3), 79.9 (C in Boc), 63.4 (C-1′), 62.9 (C-3′), 62.6 (C-4′), 61.6 (C-2′), 55.3 (C-5), 52.8 (C-18), 48.5 (C-17), 47.5 (C-9), 42.0 (C-14), 39.5 (C-8), 39.0 (C-4), 38.7 (C-19), 38.3 (C-20), 37.7 (C-1), 36.8 (C-22), 36.5 (C-10), 32.9 (C-7), 30.6 (C-21), 28.1 (CH3 in Boc), 28.0 (C-15, C-23), 24.1 (C-16), 23.6 (C-2), 23.5 (C-27), 23.3 (C-11), 21.3 (C-30), 21.0 (COCH3), 18.2 (C-6), 17.0 (C-29), 16.9 (C-26), 16.7 (C-24), 15.5 (C-25); Anal. Calcd. for C47H77N3O10: C, 66.87, H, 9.19. Found: C, 67.24, H, 9.14%. MS: m/z 866.47 [M + Na]+ (calcd. for C47H77N3O10, 843.56).
3.1.4. General Procedure for the Synthesis of Compounds 9a–13a, 15b, 18b and 20b
Compounds 9–13 and 15a, 18a, 20a (0.2 mmol) in 1 mL of dry CH2Cl2 were treated with TFA (1 mL) and the mixture was stirred for 4–6 h at room temperature (TLC control, hexane:EtOAc, 1:1, v/v). The solution was evaporated to dryness to obtain pure compounds 9a–13a, 15b, 18b and 20b.
N-(4-Butylgyanidine)-3-oxolupane-28-amide trifluoroacetate (9a), White powder, 96% yield; mp 142–144 °C (EtOH); −0.6° (c 0.34, CHCl3); IR (CHCl3) νmax 1669 (C=O), 3206, 3437 (NH) cm−1; 19F-NMR (376.50 MHz, CDCl3) δ: −75.91; 1H-NMR (400 MHz, CDCl3) δ: 11.13 (br s, 1H, NH in Boc), 7.50 (br s, 1H, NH–C=N), 6.82 (br s, 2H, NH2), 6.39 (br s, 1H, CONH), 3.26 (m, 4H, H-1′, H-4′), 2.45–1.19 (m, 26H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 1.07, 1.01, 0.96, 0.92, 0.90 (all s, 3H each, H-23–H-27), 0.85 (d, J = 6.0 Hz, 3H, H-29), 0.75 (d, J = 6.0 Hz, 3H, H-30); 13C-NMR (100 MHz, CDCl3) δ: 220.4 (C-3), 178.8 (C-28), 161.4 (q, JC,F = 37 Hz), 157.2 (C=N), 117.5 (q, JC,F = 288 Hz), 56.4 (C-17), 54.8 (C-5), 49.6 (C-9), 49.2 (C-19), 47.4 (C-4), 44.4 (C-18), 42.7 (C-14), 40.9 (C-1, C-4′), 40.7 (C-8), 38.8 (C-22), 38.1 (C-1′), 38.0 (C-13), 36.8 (C-10), 34.2 (C-2), 33.6 (C-16), 33.3 (C-7), 29.9 (C-20), 29.7 (C-3′), 29.4 (C-15), 27.0 (C-2′), 26.7 (C-23), 25.5 (C-12), 23.0 (C-21), 22.9 (C-29), 20.9 (C-11, C-24), 19.6 (C-6), 15.8 (C-25), 15.6 (C-26), 14.5 (C-30), 14.4 (C-27); Anal. Calcd. for C37H61F3N4O4: C, 65.08, H, 9.00. Found: C, 65.52, H, 8.94%. MS: m/z 569.43 [M + H]+ (calcd. for C35H60N4O2, 568.47).
3β-N-(2-Ethylgyanidine)-3-O-acetyl-lupane-28-amide trifluoroacetate (10a), White powder, 93% yield; mp 132–134 °C (EtOH); −5.6° (c 0.31, CHCl3); IR (CHCl3) νmax 1671 (C=O), 3351 (NH) cm−1; 19F-NMR (470.59 MHz, CDCl3) δ: −75.80; 1H-NMR (500 MHz, CDCl3) δ: 10.63 (br s, 1H, NH in Boc), 8.05 (br s, 1H, NH–C=N), 7.00 (br s, 2H, NH2), 6.79 (br s, 1H, CONH), 4.48–4.45 (m, 1H, H-3), 3.39–3.28 (m, 4H, H-1′, H-2′), 2.09 (s, 3H, CH3CO–), 2.31–1.01 (m, 25H, CH, CH2 in pentacyclic skeleton), 0.95, 0.92, 0.89, 0.86, 0.85, 0.83 (all s, 3H each, H-23–H-27 and H-29), 0.79 (d, J = 11.0 Hz, 1H, H-5), 0.75 (d, J = 6.0 Hz, 3H, H-30); 13C-NMR (125 MHz, CDCl3) δ: 179.6 (C-28), 171.6 (COCH3), 161.2 (q, JC,F = 37 Hz), 157.9 (C=N), 117.2 (q, JC,F = 287 Hz), 81.2 (C-3), 56.4 (C-17), 55.4 (C-5), 50.2 (C-9), 49.3 (C-19), 44.3 (C-18), 42.6 (C-14), 40.7 (C-1, C-2′), 40.6 (C-8), 38.6 (C-1′), 38.4 (C-22), 37.8 (C-4, C-13), 37.0 (C-10), 34.2 (C-7), 33.1 (C-16), 29.9 (C-20), 29.3 (C-15), 27.9 (C-23), 26.9 (C-2), 23.6 (C-12), 23.0 (C-21, C-29), 21.3 (COCH3), 21.0 (C-11), 18.1 (C-6), 16.4 (C-25), 16.0 (C-24), 15.9 (C-26), 14.5 (C-27, C-30); Anal. Calcd. for C37H61F3N4O5: C, 63.59, H, 8.80. Found: C, 63.88, H, 8.73%. MS: m/z 585.62 [M + H]+ (calcd. for C35H60N4O3, 584.47).
3β-N-(4-Butylgyanidine)-3-O-acetyl-lupane-28-amide trifluoroacetate (11a), White powder, 95% yield; mp 148–150 °C (EtOH); −12° (c 0.49, CHCl3); IR (CHCl3) νmax 1672 (C=O), 3207, 3354 (NH) cm−1; 19F-NMR (470.59 MHz, CDCl3) δ: −77.19; 1H-NMR (500 MHz, MeOD) δ: 4.48–4.45 (m, 1H, H-3), 3.28–3.15 (m, 4H, H-1′, H-4′), 2.04 (s, 3H, CH3CO–), 2.61–1.04 (m, 25H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 1.02, 0.98, 0.92, 0.90, 0.89, 0.88 (3H each, all s, H-23–H-27 and H-29), 0.84 (d, J = 11.0 Hz, 1H, H-5), 0.79 (d, J = 7.0 Hz, 3H, H-30); 13C-NMR (125 MHz, MeOD) δ: 179.6 (C-28), 172.9 (COCH3), 161.2 (q, JC,F = 37 Hz), 158.8 (C=N), 117.2 (q, JC,F = 287 Hz), 82.6 (C-3), 57.5 (C-17), 57.0 (C-5), 51.9 (C-9), 51.1 (C-19), 45.5 (C-18), 43.8 (C-14), 42.2 (C-8, C-4′), 40.0 (C-22), 39.8 (C-1), 39.4 (C-1′), 39.0 (C-4), 38.9 (C-13), 38.4 (C-10), 35.8 (C-7), 34.1 (C-16), 31.3 (C-20), 30.7 (C-3′), 28.7 (C-15), 28.5 (C-23), 28.1 (C-2′), 27.4 (C-2), 24.8 (C-12), 24.2 (C-29), 23.7 (C-21), 22.5 (C-11), 21.3 (COCH3), 19.4 (C-6), 17.2 (C-25), 17.0 (C-24, C-26), 15.3 (C-30), 15.2 (C-27); Anal. Calcd. for C39H65F3N4O5: C, 64.44, H, 9.01. Found: C, 64.87, H, 8.94%. MS: m/z 613.48 [M + H]+ (calcd. for C37H64N4O3, 612.50).
3β-N-[2-(N,N′-bis-Ethylgyanidine)-aminoethyl]-3-O-acetyl-lupane-28-amide trifluoroacetate (12a), White powder, 96% yield; mp 116–118 °C (EtOH); −10.5° (c 0.2, C2H5OH); IR (CHCl3) νmax 1681 (C=O), 3199, 3362 (NH) cm−1; 19F-NMR (470.59 MHz, MeOD) δ: −76.98; 1H-NMR (500 MHz, MeOD) δ: 4.48–4.44 (m, 1H, H-3), 3.73–3.70 (m, 4H, H-4′, H-4″), 3.56–3.53 (m, 2H, H-1′), 3.46 (t, J = 6.5 Hz, 4H, H-3′, H-3″), 3.29–3.26 (m, 2H, H-2′), 2.04 (s, 3H, CH3CO–), 2.52–1.06 (m, 25H, CH, CH2 in pentacyclic skeleton), 1.02, 0.97, 0.92, 0.90, 0.88, 0.87 (all s, 3H each, H-23–H-27 and H-29), 0.85 (d, J = 11.5 Hz, 1H, H-5), 0.79 (d, J = 6.5 Hz, 3H, H-30); 13C-NMR (125 MHz, MeOD) δ: 181.2 (C-28), 173.0 (COCH3), 161.7 (q, JC,F = 37 Hz), 159.1 (C=N), 117.5 (q, JC,F = 288 Hz), 35.6 (C-4″), 53.4 (C-3″), 82.6 (C-3), 57.6 (C-17), 56.9 (C-5), 53.9 (C-2′), 53.4 (C-3′), 51.8 (C-9), 51.0 (C-19), 45.5 (C-18), 43.8 (C-14), 42.2 (C-8), 39.7 (C-1), 39.7 (C-22), 39.0 (C-13, C-4), 38.4 (C-1′), 37.8 (C-10), 35.6 (C-4′, C-7), 33.8 (C-16), 31.3 (C-20), 30.7 (C-15), 28.6 (C-23), 28.4 (C-2), 24.8 (C-12), 24.1 (C-21), 23.5 (C-29), 22.3 (C-11), 21.3 (COCH3), 19.4 (C-6), 17.1 (C-25), 17.0 (C-24), 16.9 (C-26), 15.2 (C-30), 15.1 (C-27); Anal. Calcd. for C44H74F6N8O7: C, 56.16, H, 7.93. Found: C, 56.63, H, 7.86%. MS: m/z 713.59 [M + H]+ (calcd. for C40H72N8O3, 712.57).
3β-N-{[3-(3-Propylguanidine)piperazinyl]propyl}-3-O-acetyl-lupane-28-amide trifluoroacetate (13a), White powder, 96% yield; mp 92–94 °C (EtOH); +8° (c 0.54, CHCl3); IR (CHCl3) νmax 1673, 1773 (C=O), 3190, 3367 (NH) cm−1; 19F-NMR (470.59 MHz, MeOD) δ: −77.25; 1H-NMR (500 MHz, MeOD) δ: 4.48–4.45 (m, 1H, H-3), 3.49–3.40 (m, 4H, H-2″, H-3″), 3.31–3.22 (m, 8H, H-4′, H-4″, H-5′, H-5″), 3.13–3.10 (m, 2H, H-1″), 3.00–2.98 (m, 2H, H-1′), 2.58–0.84 (m, 26H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 2.08 (s, 3H, CH3CO–), 1.02, 0.99, 0.92, 0.89, 0.86, 0.84 (all s, 3H each, H-23–H-27 and H-29), 0.80 (d, J = 7.0 Hz, 3H, H-30); 13C-NMR (125 MHz, MeOD) δ: 180.2 (C-28), 173.0 (COCH3), 161.7 (q, JC,F = 37 Hz), 158.9 (C=N), 117.5 (q, JC,F = 288 Hz), 82.6 (C-3), 57.6 (C-17), 57.0 (C-5), 56.0 (C-3″), 55.2 (C-3′), 51.9 (C-9), 51.0 (C-19), 50.1 (C-4′, C-4″, C-5′, C-5″), 45.5 (C-18), 43.8 (C-14), 42.2 (C-8), 39.9 (C-1′), 39.8 (C-1″), 39.6 (C-1, C-22), 39.0 (C-4), 38.9 (C-13), 38.4 (C-10), 35.7 (C-7), 34.0 (C-16), 31.3 (C-20), 30.7 (C-15), 28.6 (C-23), 28.5 (C-2″), 25.6 (C-2′), 24.9 (C-2), 24.2 (C-12), 23.6 (C-21, C-29), 22.4 (C-11), 21.3 (COCH3), 19.4 (C-6), 17.1 (C-25), 17.0 (C-24), 16.9 (C-26), 15.2 (C-30), 15.1 (C-27); Anal. Calcd. for C45H77F3N6O5: C, 64.41, H, 9.25. Found: C, 64.88, H, 9.19. MS: m/z 747.58 [M + Na]+ (calcd. for C43H76N6O3, 724.60).
3β-[2-Guanidine-3-hydroxy-2-(hydroxymethyl)propyl]-3-O-acetyl-lupane-28-oate trifluoroacetate (15b), White powder, 95% yield; mp 126–129 °C (EtOH); −9.5° (c 0.34, CHCl3); IR (CHCl3) νmax 1620, 1698 (C=O), 3437 (NH) cm−1; 19F-NMR (470.59 MHz, CDCl3) δ: −75.97; 1H-NMR (500 MHz, MeOD) δ: 4.50–4.47 (m, 1H, H-3), 4.31 (2H, m, H-1′), 3.75 (m, 4H, H-3′, H-4′), 2.18–0.99 (m, 26H, CH, CH2 in pentacyclic skeleton), 2.08 (s, 3H, CH3CO–), 0.98, 0.97, 0.96, 0.87, 0.86 (all s, 3H each, H-23–H-27), 0.89 (d, J = 6.0 Hz, 3H, H-29), 0.76 (d, J = 6.0 Hz, 3H, H-30); 13C-NMR (125 MHz, MeOD) δ: 177.4 (C-28), 172.3 (COCH3), 161.4 (q, JC,F = 37 Hz), 157.5 (C=N), 117.5 (q, JC,F = 288 Hz), 81.7 (C-3), 63.1 (C-3′, C-4′), 62.0 (C-2′), 60.2 (C-1′), 57.5 (C-17), 55.4 (C-5), 50.2 (C-9), 48.8 (C-19), 44.1 (C-18), 42.6 (C-14), 40.7 (C-8), 38.4 (C-22), 38.2 (C-1), 37.8 (C-4, C-13), 37.0 (C-10), 34.2 (C-7), 31.9 (C-16), 29.7 (C-20), 29.0 (C-15), 27.9 (C-23), 26.8 (C-2), 23.6 (C-12), 22.9 (C-29, C-21), 21.3 (COCH3), 20.9 (C-11), 18.1 (C-6), 16.4 (C-25), 16.1 (C-24), 15.8 (C-26), 14.6 (C-27, C-30); Anal. Calcd. for C39H64F3N3O8: C, 61.64; H, 8.49. Found: C, 62.34; H, 8.43%. MS: m/z 646.39 [M + H]+ (calcd. for C37H63N3O6, 645.47).
3β-[2-Guanidine-3-hydroxy-2-(hydroxymethyl)propyl]-3-O-acetyl-urs-12-en-28-oate trifluoroacetate (18b), White powder, 98% yield; mp 124–126 °C (EtOH); +30.0° (c 0.72, CH2Cl2); IR (CHCl3) νmax 1619, 1682 (C=O), 3438 (NH) cm−1; 19F-NMR (470.59 MHz, MeOD) δ: −77.17; 1H-NMR (500 MHz, MeOD) δ: 5.29 (br s, 1H, H-12), 4.49–4.46 (m, 1H, H-3), 4.31, 4.17 (both d, J = 11.5 Hz, 1H each, H-1′), 3.75 (m, 4H, H-3′, H-4′), 2.24 (d, J = 11.0 Hz, 1H, H-18), 2.04 (s, 3H, CH3CO–), 2.13–0.87 (m, 22H, CH, CH2 in pentacyclic skeleton), 1.15, 1.01, 0.97, 0.98, 0.92, 0.90, 0.81 (3H each, all s, H-23–H-27, H-29 and H-30); 13C-NMR (125 MHz, MeOD) δ: 179.2 (C-28), 173.0 (COCH3), 161.4 (q, JC,F = 37 Hz), 159.2 (C=N), 140.1 (C-13), 127.2 (C-12), 117.5 (q, JC,F = 288 Hz), 82.5 (C-3), 64.3 (C-3′), 64.2 (C-4′), 62.7 (C-2′), 62.2 (C-1′), 56.8 (C-5), 54.4 (C-18), 49.5 (C-17), 48.6 (C-9), 43.3 (C-14), 40.9 (C-8), 40.4 (C-4, C-19), 39.6 (C-20), 38.8 (C-1), 38.1 (C-22), 38.0 (C-10), 34.2 (C-7), 31.7 (C-21), 29.2 (C-15), 28.7 (C-23), 25.5 (C-16), 24.6 (C-2), 24.5 (C-27), 24.4 (C-11), 21.6 (C-30), 21.3 (COCH3), 19.4 (C-6), 17.9 (C-29), 17.8 (C-26), 17.3 (C-24), 16.2 (C-25); Anal. Calcd. for C39H62F3N3O8: C, 61.80, H, 8.25. Found: C, 62.33, H, 8.19%. MS: m/z 644.47 [M + H]+ (calcd. for C37H61N3O6, 643.46).
3β-[2-Guanidine-3-hydroxy-2-(hydroxymethyl)propyl]-3-O-acetyl-olean-12-en-28-oate trifluoroacetate (20b), White powder, 97% yield; mp 130–132 °C (EtOH); +26.6° (c 0.53, C2H5OH); IR (CHCl3) νmax 1619, 1682 (C=O), 3438 (NH) cm−1; 19F-NMR (470.59 MHz, CDCl3) δ: −77.24; 1H-NMR (500 MHz, MeOD) δ: 5.31 (br s, 1H, H-12), 4.49–4.46 (m, 1H, H-3), 4.33, 4.20 (d, J = 11.5 Hz, 1H each, H-1′), 3.75 (m, 4H, H-3′, H-4′), 2.89 (d, 1H, J = 9.5 Hz, H-18), 2.09–0.88 (m, 22H, CH, CH2 in pentacyclic skeleton), 2.04 (s, 3H, CH3CO–), 1.19, 1.00, 0.97, 0.95, 0.90, 0.89, 0.79 (all s, 3H each, H-23–H-27, H-29 and H-30); 13C-NMR (125 MHz, MeOD) δ: 178.9 (C-28), 173.0 (COCH3), 161.4 (q, JC,F = 37 Hz), 159.2 (C=N), 145.0 (C-13), 124.1 (C-12), 117.5 (q, JC,F = 288 Hz), 82.6 (C-3), 64.3 (C-4′), 64.2 (C-3′), 62.7 (C-2′), 62.3 (C-1′), 56.9 (C-5), 48.5 (C-9, C-17), 47.1 (C-19), 43.0 (C-14), 42.9 (C-18), 40.8 (C-8), 39.5 (C-1), 38.9 (C-4), 38.3 (C-10), 34.9 (C-22), 33.9 (C-30), 33.7 (C-7), 33.6 (C-21), 31.7 (C-20), 28.9 (C-15), 28.7 (C-23), 26.6 (C-27), 24.7 (C-11, C-29), 24.3 (C-2), 24.2 (C-16), 21.3 (COCH3), 19.5 (C-6), 17.9 (C-26), 17.3 (C-24), 16.1 (C-25); Anal. Calcd. for C39H62F3N3O8: C, 61.80, F, 7.52, H, 8.25. Found: C, 62.12, H, 8.21%. MS: m/z 644.57 [M + H]+ (calcd. for C37H61N3O6, 643.46).
3.1.5. General Procedure for the Synthesis of Compounds 9b–12b, 15c, 18c and 20c
The compounds 9a–12a, 15b, 18b, 20b (0.2 g) were dissolved in 2 mL MeOH and 5M HCl was added dropwise until the precipiate formed. The solution was evaporated to dryness and this procedure was repeated three times. The precipitate which formed was filtered off and washed with water to pH = 7. The salts 9b–12b and 15c, 18c, 20c were obtained as white solids with a quantitative yield.
N-(4-Butylgyanidine)-3-oxolupane-28-amide dihydrochloride (9b), White powder, 87% yield; mp 176–178 °C (EtOH); −11° (c 0.29, C2H5OH); IR (CHCl3) νmax 1721 (C=O), 3342 (NH) cm−1; 1H-NMR (500 MHz, d6-DMSO) δ: 7.71 (br s, 1H, NH), 7.59 (br s, 1H, CONH), 3.10–3.01 (m, 4H, H-1′, H-4′), 2.61–1.04 (m, 26H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 0.99, 0.93, 0.91, 0.87, 0.86 (3H each, all s, H-23–H-27), 0.81 (d, J = 6.5 Hz, 3H, H-29), 0.71 (d, J = 7.0 Hz, 3H, H-30); 13C-NMR (125 MHz, d6-DMSO) δ: 217.1 (C-3), 176.1 (C-28), 157.4 (C=N), 55.7 (C-17), 54.3 (C-5), 49.6 (C-9), 49.5 (C-19), 47.0 (C-4), 43.8 (C-18), 42.6 (C-14), 40.9 (C-4′), 40.3 (C-8), 39.7 (C-1), 38.1 (C-22, C-1′), 37.0 (C-13), 36.8 (C-10), 34.1 (C-2), 33.8 (C-16), 32.7 (C-7), 30.0 (C-20), 29.4 (C-15), 27.2 (C-3′), 27.0 (C-2′), 26.9 (C-23), 26.4 (C-12), 23.6 (C-29), 23.1 (C-21), 21.7 (C-11), 21.2 (C-24), 19.7 (C-6), 16.2 (C-25), 16.1 (C-26), 15.0 (C-30), 14.6 (C-27); Anal. Calcd. for C35H62Cl2N4O2: C, 65.50, Cl, 11.05, H, 9.74. Found: C, 65.97, Cl, 11.78, H, 9.68%. MS: m/z 569.49 [M + H]+ (calcd. for C35H60N4O2, 568.47).
3β-N-(2-Ethylgyanidine)-3-O-acetyl-lupane-28-amide hydrochloride (10b), White powder, 82% yield; mp 192–194 °C (EtOH); −16° (c 0.23, C2H5OH); IR (CHCl3) νmax 1652, 1716 (C=O), 3155, 3327 (NH) cm−1; 1H-NMR (500 MHz, MeOD) δ: 4.48–4.45 (m, 1H, H-3), 3.43–3.16 (m, 4H, H-1′, H-2′), 2.04 (s, 3H, CH3CO–), 2.60–0.81 (m, 25H, CH, CH2 in pentacyclic skeleton), 1.02, 0.98, 0.92, 0.90, 0.88, 0.87 (all s, 3H each, H-23–H-27 and H-29), 0.80 (d, J = 6.5 Hz, 3H, H-30); 13C-NMR (125 MHz, MeOD) δ: 180.8 (C-28), 173.0 (COCH3), 159.0 (C=N), 82.6 (C-3), 57.6 (C-17), 57.0 (C-5), 51.1 (C-9, C-19), 45.5 (C-18), 42.7 (C-14, C-1′), 42.2 (C-8), 39.8 (C-1), 39.7 (C-2′), 39.4 (C-22), 39.0 (C-4), 38.9 (C-13), 38.4 (C-10), 35.7 (C-7), 34.0 (C-16), 31.4 (C-20), 30.7 (C-15), 28.6 (C-23), 28.5 (C-2), 24.8 (C-12), 24.1 (C-21), 23.6 (C-29), 22.4 (C-11), 21.3 (COCH3), 19.4 (C-6), 17.1 (C-25), 16.9 (C-24, C-26), 15.2 (C-30), 15.1 (C-27); Anal. Calcd. for C35H61ClN4O3: C, 67.66, Cl, 5.71, H, 9.90. Found: C, 67.99, Cl, 5.40, H, 9.84%. MS: m/z 585.54 [M + H]+ (calcd. for C35H60N4O3, 584.47).
3β-N-(4-Butylgyanidine)-3-O-acetyl-lupane-28-amide hydrochloride (11b), White powder, 79% yield; mp 156–158 °C (EtOH); −14.5° (c 0.53, C2H5OH); IR (CHCl3) νmax 1645, 1716 (C=O), 3168, 3338 (NH) cm−1; 1H-NMR (500 MHz, MeOD) δ: 7.45 (br s, 1H, NH), 4.48–4.45 (m, 1H, H-3), 3.23–3.18 (m, 4H, H-1′, H-4′), 2.04 (s, 3H, CH3CO–), 2.61–0.84 (m, 26H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 1.01, 0.98, 0.92, 0.89, 0.88, 0.86 (all s, 3H each, H-23–H-27 and H-29), 0.79 (d, J = 7.0 Hz, 3H, H-30); 13C-NMR (125 MHz, MeOD) δ: 179.6 (C-28), 172.9 (COCH3), 158.8 (C=N), 82.6 (C-3), 57.5 (C-17), 57.0 (C-5), 51.9 (C-9), 51.1 (C-19), 45.5 (C-18), 43.8 (C-14), 42.4 (C-8), 42.2 (C-4′), 40.0 (C-22, C-1′), 39.8 (C-1), 39.0 (C-4), 38.9 (C-13), 38.4 (C-10), 35.8 (C-7), 34.1 (C-16), 31.4 (C-20), 30.8 (C-3′), 28.6 (C-15), 28.2 (C-23, C-2′), 27.4 (C-2), 24.8 (C-12), 24.4 (C-21), 24.2 (C-29), 22.5 (C-11), 21.3 (COCH3), 19.4 (C-6), 17.1 (C-25), 17 (C-24), 16.9 (C-26), 15.2 (C-30), 15.1 (C-27); Anal. Calcd. for C37H65ClN4O3: C, 68.43, Cl, 5.46, H, 10.09. Found: C, 68.88; Cl, 5.12; H, 10.02%. MS: m/z 614.51 [M + 2H]+ (calcd. for C37H64N4O3, 612.50).
3β-N-[2-(N,N′-bis-Ethylgyanidine)-aminoethyl]-3-O-acetyl-lupane-28-amide dihydrochloride (12b), White powder, 88% yield; mp 198–200 °C (EtOH); −16° (c 0.29, C2H5OH); IR (CHCl3) νmax 1637, 1672, 1734 (C=O), 3162, 3271, 3324 (NH) cm−1; 1H-NMR (500 MHz, MeOD) δ: 4.48–4.45 (m, 1H, H-3), 3.90–3.83 (m, 4H, H-4′, H-4″), 3.66–3.59 (m, 6H, H-2′, H-3′, H-3″), 3.38–3.33 (m, 2H, H-1′), 2.04 (s, 3H, CH3CO–), 2.51–0.85 (m, 26H, CH, CH2 in pentacyclic skeleton), 1.03, 0.99, 0.93, 0.90, 0.88, 0.87 (all s, 3H each, H-23–H-27 and H-29), 0.80 (d, J = 6.5 Hz, 3H, H-30); 13C-NMR (125 MHz, MeOD) δ: 181.0 (C-28), 172.9 (COCH3), 158.9 (C=N), 82.5 (C-3), 57.7 (C-17), 56.9 (C-5), 53.7 (C-3′, C-3″), 53.6 (C-2′), 51.8 (C-9), 51.0 (C-19), 45.5 (C-18), 43.8 (C-14), 42.2 (C-8), 39.7 (C-22), 39.0 (C-1, C-13), 38.4 (C-4), 37.7 (C-10, C-1′), 35.7 (C-7), 35.0 (C-4′, C-4″), 33.9 (C-16), 31.3 (C-20), 30.8 (C-15), 28.6 (C-23), 28.4 (C-2), 24.8 (C-12), 24.2 (C-21), 23.6 (C-29), 22.4 (C-11), 21.3 (COCH3), 19.4 (C-6), 17.1 (C-25, C-24), 16.9 (C-26), 15.2 (C-30), 15.1 (C-27); Anal. Calcd. for C40H74Cl2N8O3: C, 61.13, Cl, 9.02, H, 9.49. Found: C, 61.55, Cl, 11.40, H, 9.42%. MS: m/z 713.51 [M + H]+ (calcd. for C40H72N8O3, 712.57).
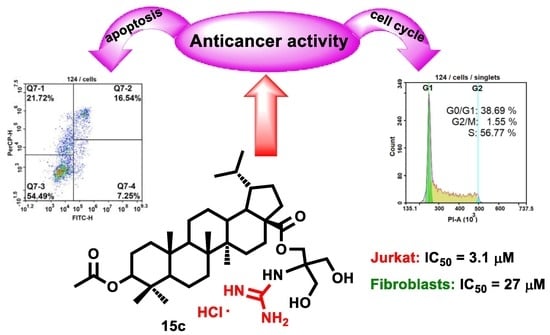
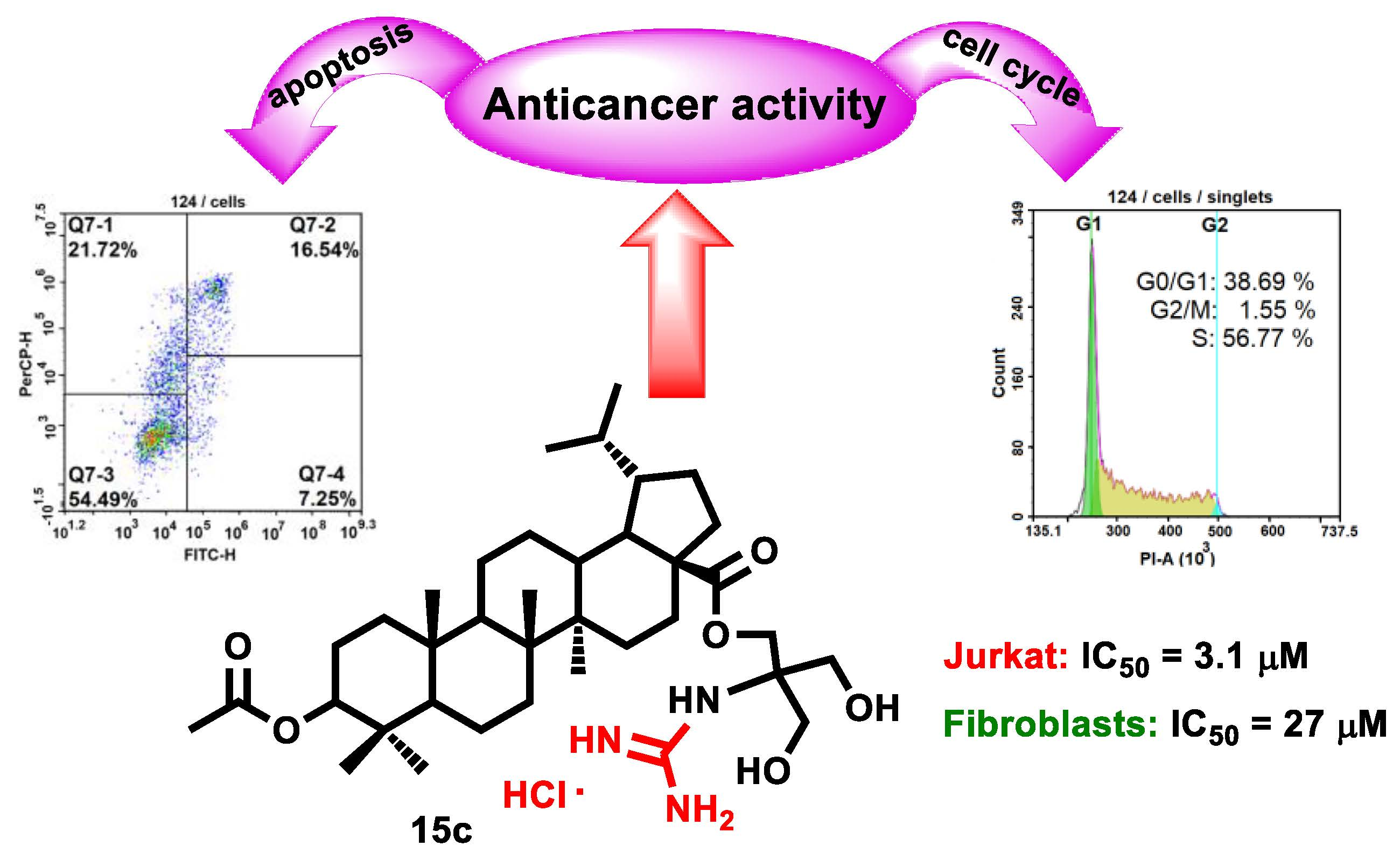
3β-[2-Guanidine-3-hydroxy-2-(hydroxymethyl)propyl]-3-O-acetyl-lupane-28-oate hydrochloride (15c), White powder, 68% yield; mp 136–138 °C (EtOH); −14.5° (c 0.53, C2H5OH); IR (CHCl3) νmax 1673, 1733 (C=O), 3325 (NH) cm−1; 1H-NMR (500 MHz, MeOD) δ: 4.48–4.45 (m, 1H, H-3), 4.31 (2H, m, H-1′), 3.75 (m, 4H, H-3′, H-4′), 2.04 (s, 3H, CH3CO–), 2.29–0.85 (m, 26H, CH, CH2 in pentacyclic skeleton), 1.03, 1.00, 0.98, 0.91, 0.88, 0.87 (all s, 3H each, H-23–H-27 and H-29), 0.81 (d, J = 6.5 Hz, 3H, H-30); 13C-NMR (125 MHz, MeOD) δ: 177.1 (C-28, COCH3), 159.1 (C=N), 82.6 (C-3), 64.1 (C-3′), 64.1 (C-4′), 62.8 (C-2′), 61.8 (C-1′), 58.7 (C-17), 56.9 (C-5), 51.8 (C-9), 50.4 (C-19), 45.8 (C-18), 43.9 (C-14), 42.1 (C-8), 39.7 (C-22), 39.6 (C-1), 39.0 (C-4), 38.4 (C-10, C-13), 35.6 (C-7), 33.2 (C-16), 31.2 (C-20), 30.9 (C-15), 28.6 (C-23), 28.4 (C-2), 24.8 (C-12), 23.9 (C-21), 23.5 (C-29), 22.3 (C-11), 21.3 (COCH3), 19.4 (C-6), 17.1 (C-25), 16.9 (C-24), 16.8 (C-26), 15.2 (C-30), 15.1 (C-27); Anal. Calcd. for C37H64ClN3O6: C, 65.13, Cl, 5.20, H, 9.45. Found: C, 65.6 4, Cl, 4.50; H, 9.39%. MS: m/z 647.47 [M + 2H]+ (calcd. for C37H63N3O6, 645.47).
3β-[2-Guanidine-3-hydroxy-2-(hydroxymethyl)propyl]-3-O-acetyl-urs-12-en-28-oate hydrochloride (18c), White powder, 62% yield; mp 142–144 °C (EtOH); +37° (c 0.51, C2H5OH); IR (CHCl3) νmax 1675, 1733 (C=O), 3186, 3335 (NH) cm−1; 1H-NMR (500 MHz, MeOD) δ: 7.07 (s, 1H, NH), 5.30 (br s, 1H, H-12), 4.50–4.47 (m, 1H, H-3), 4.29, 4.23 (both d, J = 11.5 Hz, 1H each, H-1′), 3.75 (m, 4H, H-3′, H-4′), 2.25 (d, J = 11.5 Hz, 1H, H-18), 2.05 (s, 3H, CH3CO–), 2.14–0.88 (m, 22H, CH, CH2 in pentacyclic skeleton), 1.16, 1.02, 1.00, 0.92, 0.91, 0.90, 0.81 (all s, 3H each, H-23–H-27, H-29 and H-30); 13C-NMR (125 MHz, MeOD) δ: 178.6 (C-28), 173.0 (COCH3), 159.2 (C=N), 139.6 (C-13), 127.3 (C-12), 82.6 (C-3), 64.2 (C-3′, C-4′), 62.9 (C-2′), 62.3 (C-1′), 56.8 (C-5), 54.5 (C-18), 49.3 (C-17), 49.0 (C-9), 43.4 (C-14), 41.0 (C-8), 40.5 (C-4, C-19), 39.6 (C-20), 38.9 (C-1), 38.2 (C-22), 38.0 (C-10), 34.2 (C-7), 31.8 (C-21), 29.3 (C-15), 28.8 (C-23), 25.5 (C-16), 24.7 (C-2), 24.5 (C-27), 24.4 (C-11), 21.7 (C-30), 21.3 (COCH3), 19.5 (C-6), 17.9 (C-29), 17.8 (C-26), 17.4 (C-24), 16.2 (C-25); Anal. Calcd. for C37H62ClN3O6: C, 65.32, Cl, 5.21, H, 9.19. Found: C, 65.74, Cl, 4.51, H, 9.14%. MS: m/z 644.34 [M + H]+ (calcd. for C37H61N3O6, 643.46).
3β-[2-Guanidine-3-hydroxy-2-(hydroxymethyl)propyl]-3-O-acetyl-olean-12-en-28-oate hydrochloride (20c), White powder, 53% yield; mp 124–128 °C (EtOH); +34° (c 0.34, C2H5OH); IR (CHCl3) νmax 1662, 1733 (C=O), 3344 (NH) cm−1; 1H-NMR (500 MHz, MeOD) δ: 5.32 (br s, 1H, H-12), 4.49–4.46 (m, 1H, H-3), 4.33, 4.20 (both d, J = 11.5 Hz, 1H each, H-1′), 3.76 (m, 4H, H-3′, H-4′), 2.89 (d, J = 9.5 Hz, 1H, H-18), 2.13–0.90 (m, 22H, CH, CH2 in pentacyclic skeleton), 2.05 (s, 3H, CH3CO–), 1.20, 1.00, 0.98, 0.96, 0.94, 0.91, 0.88 (all s, 3H each, H-23–H-27, H-29 and H-30); 13C-NMR (125 MHz, MeOD) δ: 178.8 (C-28), 172.9 (COCH3), 159.1 (C=N), 145.0 (C-13), 124.1 (C-12), 82.6 (C-3), 64.2 (C-4′), 64.1 (C-3′), 62.8 (C-2′), 62.3 (C-1′), 56.8 (C-5), 49.0 (C-9), 48.5 (C-17), 47.1 (C-19), 43.0 (C-14), 42.9 (C-18), 40.7 (C-8), 39.5 (C-1), 38.9 (C-4), 38.2 (C-10), 34.9 (C-22), 33.9 (C-30), 33.7 (C-7, C-21), 31.7 (C-20), 28.9 (C-15), 28.7 (C-23), 26.6 (C-27), 24.7 (C-11, C-29), 24.3 (C-2), 24.2 (C-16), 21.3 (COCH3), 19.5 (C-6), 17.9 (C-26), 17.3 (C-24), 15.6 (C-25); Anal. Calcd. for C37H62ClN3O6: C, 65.32, Cl, 5.21, H, 9.19. Found: C, 65.74, Cl, 4.80, H, 9.12%. MS: m/z 644.44 [M + H]+ (calcd. for C37H61N3O6, 643.46).
3β-N-(4-Butylgyanidine)-3-hydroxy-lupane-28-amide (14), To a solution of the compound 11b (0.33 g, 0.5 mmol) in MeOH (4 mL) and THF (4 mL) was added 4 N NaOH (4 mL). The reaction mixture was stirred at room temperature for 24 h (monitoring by TLC) and then neutralized with 20% HCl. The solution was dried under vacuum and reconstituted with CH2Cl2. The organic layer was washed with brine and dried over anhydrous MgSO4 and concentrated under reduced pressure to obtain pure compound 14. White powder, 79% yield; mp 260–262 °C (EtOH); −10° (c 0.24, DMSO); IR (CHCl3) νmax 1731 (C=O), 3366 (NH) cm−1; 1H-NMR (500 MHz, DMSO-d5) δ: 7.73 (br s, 1H, NH), 7.58 (br s, 1H, CONH), 4.29 (br s, 1H, OH), 3.09–2.98 (m, 5H, H-3, H-1′, H-4′), 2.19–1.02 (m, 26H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 0.89, 0.87, 0.84, 0.78, 0.66 (all s, 3H each, H-23–H-27), 0.81 (d, J = 6.5 Hz, 3H, H-29), 0.72 (d, J = 6.5 Hz, 3H, H-30); 13C-NMR (125 MHz, DMSO-d5) δ: 176.1 (C-28), 157.4 (C=N), 77.2 (C-3), 55.7 (C-17), 55.4 (C-5), 50.3 (C-9), 49.6 (C-19), 43.8 (C-18), 42.5 (C-14), 40.9 (C-8), 40.8 (C-4′), 39.0 (C-22), 38.8 (C-1), 38.6 (C-4), 38.1 (C-1′), 37.2 (C-13), 36.9 (C-10), 34.6 (C-7), 32.8 (C-16), 30.0 (C-20), 29.4 (C-15), 28.6 (C-23), 27.4 (C-2), 27.2 (C-3′), 27.0 (C-2′), 26.4 (C-12), 23.6 (C-29), 23.2 (C-21), 21.1 (C-11), 18.5 (C-6), 16.4 (C-25), 16.3 (C-24, C-26), 15.0 (C-30), 14.7 (C-27); Anal. Calcd. for C35H62N4O2: C, 73.63, H, 10.95. Found: C, 73.74, H, 10.88%. MS: m/z 593.31 [M + Na]+ (calcd. for C35H62N4O2, 570.49).


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}