
Concise Synthesis of (+)-β- and γ-Apopicropodophyllins, and Dehydrodesoxypodophyllotoxin



1
University and College Key Lab of Natural Product Chemistry and Application in Xinjiang, Yili Normal University, Yining 835000, China
2
School of Life Science and Engineering, Southwest Jiaotong University, Chengdu 610031, China
3
State Key Laboratory of Applied Organic Chemistry and College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(11), 3037; https://doi.org/10.3390/molecules23113037
Submission received: 30 October 2018
/
Revised: 12 November 2018
/
Accepted: 17 November 2018
/
Published: 21 November 2018
(This article belongs to the Special Issue Lignans)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Herein, we present an expeditous synthesis of bioactive aryldihydronaphthalene lignans (+)-β- and γ-apopicropodophyllins, and arylnaphthalene lignan dehydrodesoxypodophyllotoxin. The key reaction is regiocontrolled oxidations of stereodivergent aryltetralin lactones, which were easily accessed from a nickel-catalyzed reductive cascade approach developed in our group.
1. Introduction
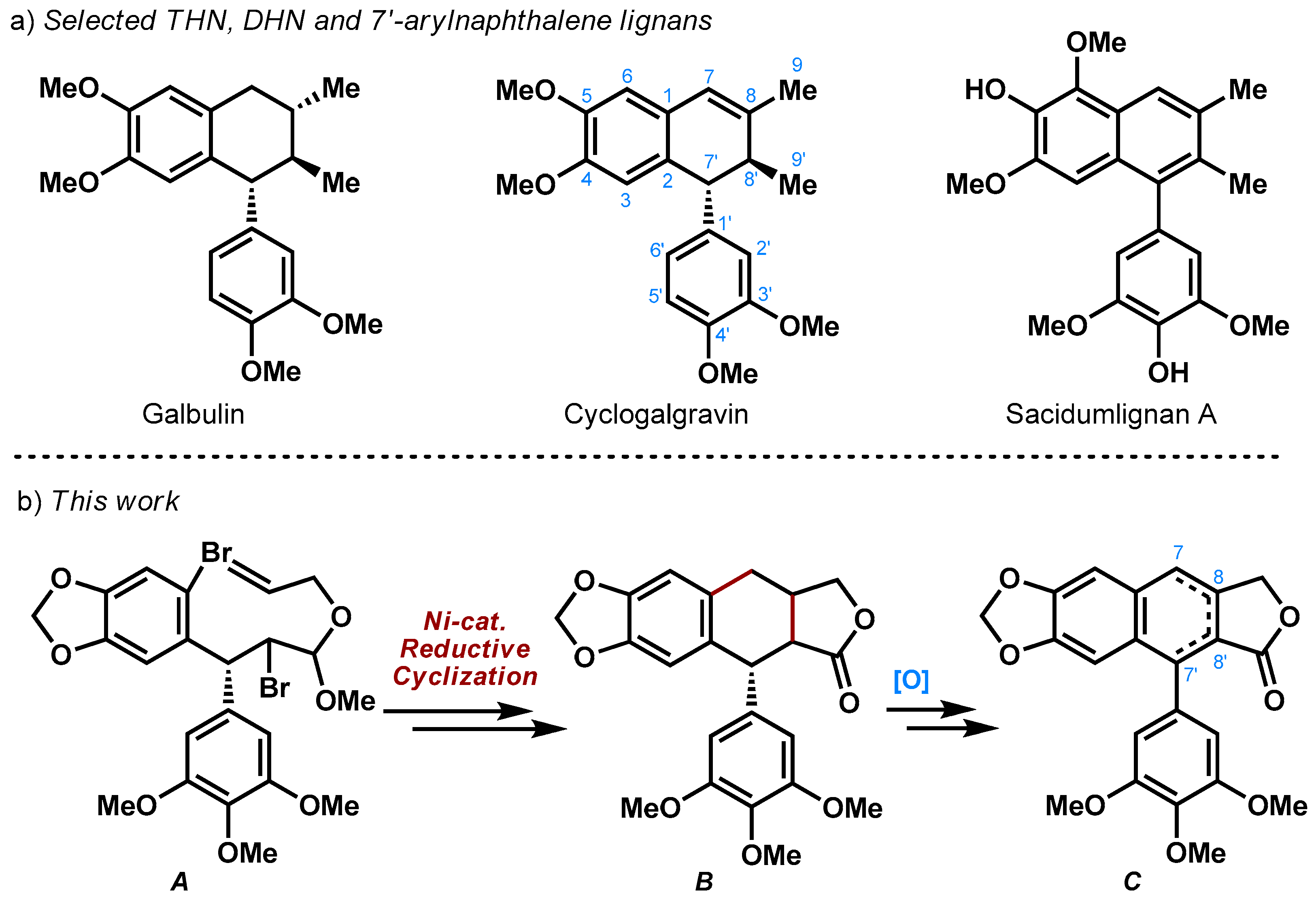
Lignans are a class of secondary metabolites in various plants, and most of them have demonstrated interesting biological properties [1,2], thus attracting the attention of the synthetic chemists [3,4]. Some of 2,7′-cyclolignans such as 7,8,8′,7′-tetrahydronaphthalene (THN), 7′,8′-dihydronaphthalene (DHN) and 7′-arylnaphthalene types are exemplified in Scheme 1a. Hong and co-workers used organocatalytic domino Michael–Michael–aldol reactions to construct THN skeleton of galbulin and realized its first enantioselective synthesis [5]. Barker and co-workers completed the first asymmetric synthesis of (−)-cyclogalgravin based on a key construction of C2–C7′ bond from in situ generated quinoid intermediate [6]. Notably, the other two structurally distinct class of lignans could also be obtained from a common precursor in their syntheses. Ramana et al. proposed a dehydrative cyclization of an aldehyde intermediate to build the DHN unit of sacidumlignan B, whose subsequent aromatization led to the synthesis of sacidumlignan A [7]. We were also involved in this fascinating field and achieved the synthesis of these three molecules through Ueno–Stork radical cyclization and Friedel–Crafts reaction [8,9]. However, almost all of the above syntheses applied stepwise strategies (i.e., a sequence of C2–C7′, C8–C8′, then C1–C7 bonds formation in our previous routes) for construction of the central core [10].
2. Results and Discussion
Recently, we completed a new synthesis of podophyllotoxin [11,12], an aryltetralin lignan used as building block for the chemotherapeutic drugs etoposide and teniposide. The key reaction is a Ni-catalyzed reductive tandem coupling [13,14,15,16,17,18,19] of dibromide A that led to the simultaneous construction of C8–C8′ and C1–C7 bonds in THN framework of B (Scheme 1b). We envision that this aryltetralin lactone could serve as an advanced intermediate for the unified synthesis of the titled arylnaphthalene, DHN and THN lignans C, by means of the regioselective late-stage oxidation. Herein, we disclosed the preliminary results.
Starting from the commercially available 6-bromopiperonal and 3,4,5-trimethoxyphenyl bromide, the chiral β-bromo acetal 1 was straightforwardly prepared as in gram-scale according to a known route [11]. Under a fully intramolecular reductive nickel-catalysis ligated by ethyl crotonate (Scheme 2), diastereodivergent (+)-deoxypicropodophyllin (2) and (+)-isodeoxypodophyllotoxin (3) were obtained in 50% overall yield after a conversion of acetal moiety to the corresponding lactone. With aryltetralin lactones 2 and 3 in hand, the designed regiocontrolled oxidation in central aliphatic ring could be executed (vide infra).
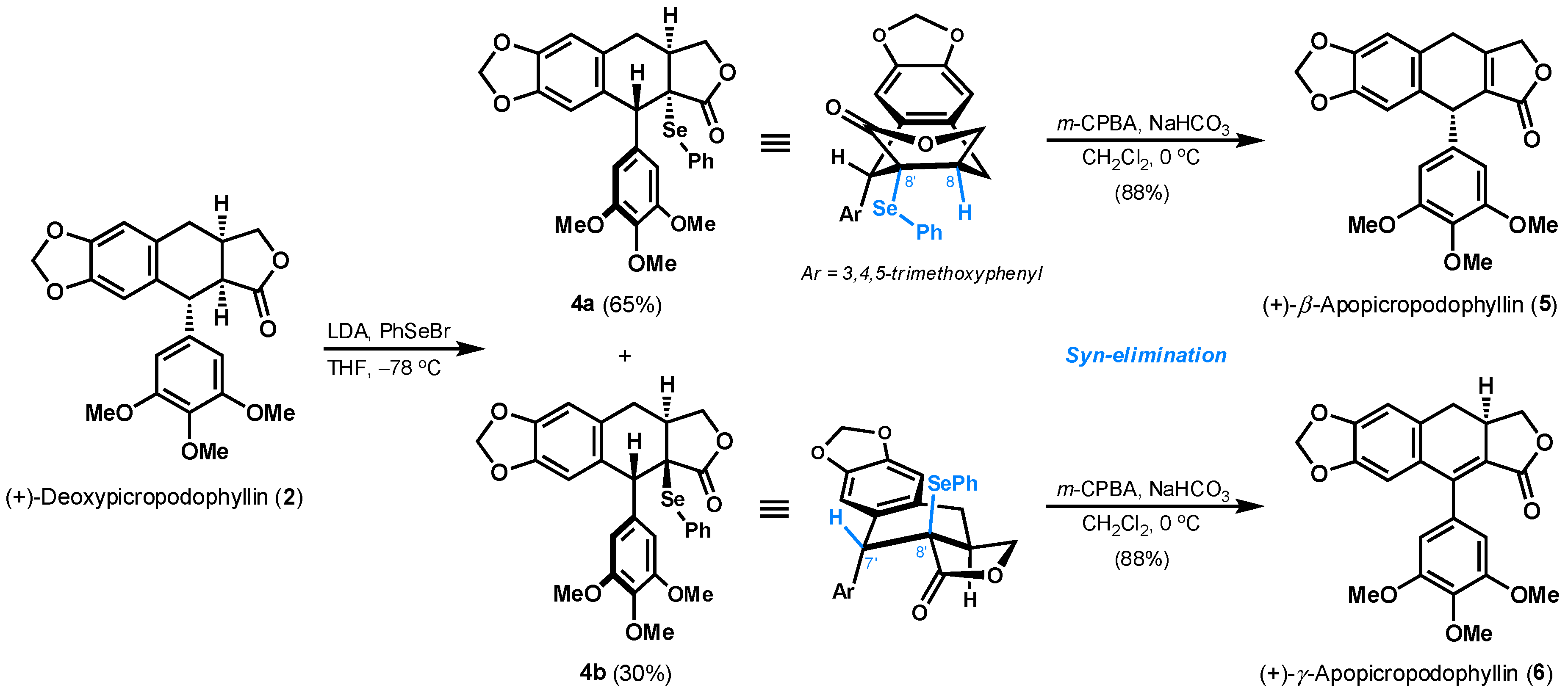
First of all, the increase of an unsaturation degree at either C8–C8′ or C7′–C8′ location was pursued in order to get (+)-β-apopicropodophyllin (5) and (+)-γ-apopicropodophyllin (6) quickly. As shown in Scheme 3, the introduction of a phenylselenyl group at C8′ position of (+)-deoxypicropodophyllin (2) was done by an initial enolization and subsequent quench with phenylselenyl bromide (PhSeBr) at −78 °C. The generated products as two diastereoisomers (4a and 4b) were separated by column chromatography on silica gel in 95% overall yield. The α-phenylselenide 4a is supposed to adopt a pseudo-boat conformation, where the hydrogen atom at C8 is arranged cis to the -SePh. The requisite syn-elimination of phenylselenoxide in situ generated from oxidation of 4a [20], eventually provided (+)-β-apopicropodophyllin (5) with in vivo insecticidal activity against the fifth-instar larvae of Brontispa longissima [21]. Its 1H NMR spectral data (Table S2) and optical rotation were in agreement with the reported data by Toste and Meyers [22,23]. The structure was later unambiguously confirmed by its single-crystal analysis (Figure 1) [24]. In contrast, the hydrogen atom at C7′ is oriented at cis-position of C8′-PhSe in the favored half-chair conformer of β-phenylselenide 4b. Thus, a double bond within C7′–C8′ was formed upon the subjection of 4b to m-CPBA, therefore affording to (+)-γ-apopicropodophyllin (6) in 88% yield. As shown in Table S3, 1H NMR spectra of the synthetic 6 was accord with the literature [25].
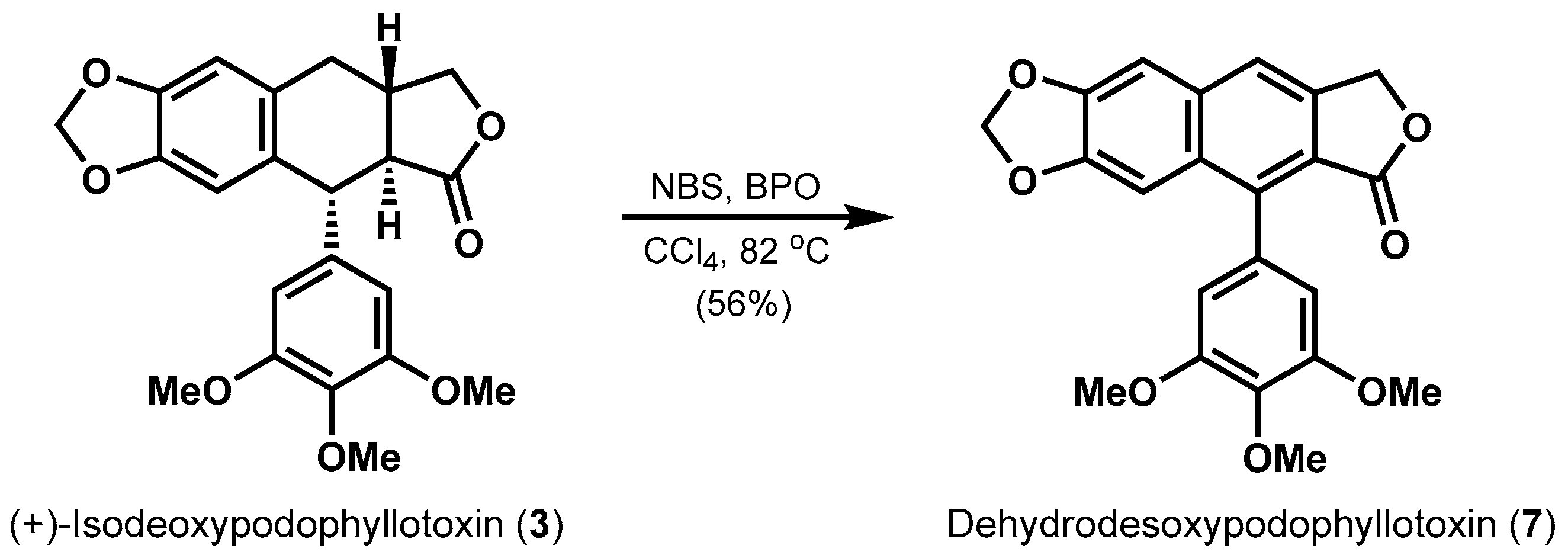
Next, the potential aromatization within tetralin lactone was investigated. As shown in Scheme 4, one-step conversion of (+)-isodeoxypodophyllotoxin (3) to dehydrodesoxypodophyllotoxin (7) was realized in 56% yield promoted by a mixture of N-bromosuccinimide (NBS) and dibenzoyl peroxide (BPO) in refluxing CCl4. The plausible mechanism of this tandem reaction would be radical bromination [26] catalyzed by BPO occurs firstly, and a fast elimination of the resulting labile benzylbromide followed by further oxidation, providing the central benzene ring in 7. 1H NMR spectra data (Table S4) of synthetic dehydrodesoxypodophyllotoxin was consistent with previous report [27].
3. Materials and Methods
3.1. General Procedure
For product purification by flash column chromatography, SiliaFlash P60 (particle size: 40–63 μm, pore size 60A) and petroleum ether (bp. 60–90 °C) were used. All solvents were purified and dried by standard techniques and distilled prior to use. All of experiments were conducted under an argon or nitrogen atmosphere in oven-dried or flame-dried glassware with magnetic stirring, unless otherwise specified. Organic extracts were dried over Na2SO4 or MgSO4, unless otherwise noted. 1H and 13C-NMR spectra were taken on a Bruker AM-400, AM-600 and Varian mercury 300 MHz spectrometer with TMS as an internal standard and CDCl3 as solvent unless otherwise noted. HRMS were determined on a Bruker Daltonics APEXII 47e FT-ICR spectrometer with ESI positive ion mode. The X-ray diffraction studies were carried out on a Bruker SMART Apex CCD area detector diffractometer equipped with graphite-monochromated Cu-Kα radiation source. Melting points were measured on Kofler hot stage and are uncorrected.
3.2. Synthesis of C9a-PhSe-Deoxypicropodophyllin (4a and 4b)
A solution of 2 [11] (100 mg, 0.25 mmol) in THF (8 mL) under argon was cooled to −78 °C, followed by the addition of freshly prepared LDA (0.5 mmol, 2.0 equiv). The stirred solution was maintained at this temperature for 20 min, and a solution of PhSeBr (118 mg, 0.5 mmol, 2.0 equiv) in THF (3 mL) was then added. The resulting mixture was stirred for 20 min at −78 °C, and then quenched by water (1 mL). The mixture was extracted with EtOAc (2 × 30 mL). The combined organic layers were washed with water (2 × 8 mL) and brine (8 mL) respectively, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash column chromatography (petroleum ether/EtOAc = 4:1 → petroleum ether/EtOAc =2:1) on silica gel to afford 4a (90 mg, 65% yield) as a white solid and 4b (42 mg, 30% yield) as a white solid. Characterization data for 4a: Rf = 0.42 (petroleum ether/EtOAc = 1:1); 1H-NMR (400 MHz, CDCl3): δ = 7.48 (d, J = 8.0 Hz, 1H), 7.47 (d, J = 8.0 Hz, 1H), 7.40 (t, J = 7.2 Hz, 1H), 7.28 (t, J = 7.2 Hz, 2H), 6.68 (s, 1H), 6.61 (s, 2H), 6.56 (s, 1H), 5.88 (d, J = 1.2 Hz, 1H), 5.87 (d, J = 1.2 Hz, 1H), 4.49 (s, 1H), 4.10 (dd, J = 9.2, 7.6 Hz, 1H), 3.85 (s, 3H), 3.84 (s, 6H), 3.75 (dd, J = 5.2, 4.0 Hz, 1H), 3.48 (dd, J = 16.4, 8.4 Hz, 1H), 3.32–3.27 (m, 1H), 2.62 (d, J = 16.4 Hz, 1H) ppm; 13C-NMR (100 MHz, CDCl3): δ = 176.7, 152.9 (2C), 147.2, 146.9, 137.7 (2C), 137.3, 134.6, 131.8, 129.9, 129.1 (2C), 126.1, 126.0, 109.3, 108.8, 106.8 (2C), 101.0, 73.3, 60.9, 56.2 (2C), 53.9, 51.3, 41.5, 35.0 ppm; HRMS (ESI): calcd. for C28H30NO7Se+ [M + NH4]+: 572.1182, found: 572.1186.
3.3. Synthesis of (+)-β-Apopicropodophyllin (5)
To a stirred solution of 4a (90 mg, 0.076 mmol) in CH2Cl2 (4 mL) was added m-CPBA (77%, 34.0 mg, 0.15 mmol, 2.0 equiv) at 0 °C followed by the addition of NaHCO3 (12.6 mg, 0.15 mmol, 2.0 equiv). After stirring for 15 min, the reaction mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (4 × 5 mL), water (5 mL) and brine (5 mL) respectively, then dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (petroleum ether/EtOAc = 3:1 → petroleum ether/EtOAc = 1:1) on silica gel to afford (+)-β-apopicropodophyllin (5) (56 mg, 88% yield) as a white solid. Rf = 0.37 (petroleum ether/EtOAc = 1:1); = +92.04 (c = 1.00, CHCl3), = +65.1 (c = 2.72, CHCl3)] [23]; m.p. 188–190 °C; 1H-NMR (300 MHz, CDCl3): δ = 6.72 (s, 1H), 6.63 (s, 1H), 6.37 (s, 2H), 5.954 (s, 1H), 5.947 (s, 1H), 4.90 (d, J = 17.4 Hz, 1H), 4.82 (d, J = 17.4 Hz, 1H), 4.81 (s, 1H), 3.86 (dd, J = 22.2, 3.9 Hz, 1H), 3.79 (s, 3H), 3.78 (s, 6H), 3.65 (dd, J = 22.2, 3.6 Hz, 1H) ppm; 13C-NMR (100 MHz, CDCl3): δ = 172.2, 157.2, 153.2 (2C), 147.3, 147.0, 138.3, 137.1, 129.7, 128.2, 123.8, 109.6, 107.7, 105.6 (2C), 101.3, 71.0, 60.8, 56.2 (2C), 42.8, 29.2 ppm.
This product (5 mg) was dissolved in EtOAc (1 mL) and hexane (2 mL). After three days, colorless single crystals were obtained by slow evaporation of solvents at room temperature.
3.4. Synthesis of (+)-γ-Apopicropodophyllin (6)
To a stirred solution of 4b (42 mg, 0.16 mmol) in CH2Cl2 (3 mL) was added m-CPBA (77%, 72.0 mg, 0.32 mmol, 2.0 equiv) at 0 °C followed by the addition of NaHCO3 (26.9 mg, 0.32 mmol, 2.0 equiv). After stirring for 15 min, the reaction mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (4 × 5 mL), water (5 mL) and brine (5 mL) respectively, then dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (petroleum ether/EtOAc = 3:1 → petroleum ether/EtOAc = 1:1) on silica gel to afford (+)-γ-apopicropodophyllin (6) (26 mg, 88% yield) as a white solid. Rf = 0.23 (petroleum ether/EtOAc = 1:1); = +27.03 (c = 1.00, CHCl3), = +25.0 (c = 1, CHCl3)] [28]; m.p. 206–208 °C; 1H-NMR (300 MHz, CDCl3): δ = 6.77 (s, 1H), 6.52 (brs, 3H), 5.97 (s, 2H), 4.70 (t, J = 8.7 Hz, 1H), 4.01 (t, J = 8.7 Hz, 1H), 3.92 (s, 3H), 3.83 (s, 6H), 3.39 (td, J = 15.9, 8.7 Hz, 1H), 2.94 (dd, J = 15.0, 6.9 Hz, 1H), 2.79 (dd, J = 15.6, 15.3 Hz, 1H) ppm; 13C-NMR (150 MHz, CDCl3): δ = 168.1, 152.7, 148.7 (2C), 147.3, 146.8, 138.1, 130.7 (2C), 129.9, 129.6, 119.9, 109.5, 108.6, 101.6 (2C), 70.9, 61.0, 56.2 (2C), 35.8, 33.3 ppm.
3.5. Synthesis of Dehydrodesoxypodophyllotoxin (7)
An oven-dried 10 mL round-bottom flask was charged with NBS (17.8 mg, 0.1 mmol, 1.0 equiv) and BPO (2.4 mg, 0.01 mmol, 0.1 equiv) at room temperature under argon, followed by the addition of a solution of 3 (40.0 mg, 0.1 mmol) in CCl4 (3 mL). The reaction mixture was stirred for 2 h at 82 °C. The reaction solvent was then evaporated in vacuo. The resulting residue was purified by flash column chromatography (petroleum ether/EtOAc = 5:1 → petroleum ether/EtOAc = 2:1) on silica gel to afford dehydrodesoxypodophyllotoxin (7) (22.2 mg, 56% yield) as a white solid. Rf = 0.45 (petroleum ether/EtOAc = 1:1); m.p. 271–273 °C; 1H-NMR (400 MHz, CDCl3): δ = 7.70 (s, 1H), 7.21 (s, 1H), 7.12 (s, 1H), 6.55 (s, 2H), 6.09 (s, 2H), 5.38 (s, 2H), 3.97 (s, 3H), 3.84 (s, 6H) ppm; 13C-NMR (150 MHz, CDCl3): δ = 169.6, 153.0 (2C), 150.0, 148.7, 140.5, 139.8, 137.8, 134.6, 130.34, 130.30, 119.1, 118.7, 107.3 (2C), 103.8, 103.6, 101.8, 68.0, 61.0, 56.1 (2C) ppm.
4. Conclusions
In summary, a two-phase strategy was developed for the unified synthesis of (+)-β-apopicropodophyllin (5), (+)-γ-apopicropodophyllin (6), and dehydrodesoxypodophyllotoxin (7). In phase I, their tetrahydronaphthalene (THN) backbone was constructed by a Ni-catalyzed reductive cascade. In phase II, regioselective oxidation of stereodivergent tetralin lactone (2 and 3) gave arylnaphthalene lignan 7 and its dihydronaphthalene (DHN) congeners (5 and 6) efficiently.
Supplementary Materials
The following are available online. Copies of 1H-, 13C-NMR, and crystallographic information files (CIFs) for 5.
Author Contributions
Y.P. conceived and designed the experiments; J.X. performed the experiments; J.X., G.N., Y.-W.W., and Y.P. analyzed the data; Y.-W.W. and Y.P. wrote the paper.
Funding
This work was supported by the Natural Science Foundation of China (nos. 21472075 and 21772078).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ayres, D.C.; Loike, J.D. Lignans, Chemical, Biological and Clinical Properties; Cambridge University Press: Cambridge, UK, 1990. [Google Scholar]
- Shi, J. Lignans Chemistry, 1st ed.; Chemical Industrial Press: Beijing, China, 2010; pp. 1–395. ISBN 978-7-122-06559-9. [Google Scholar]
- Peng, Y. Lignans, lignins, and resveratrols. In From Biosynthesis to Total Synthesis: Strategies and Tactics for Natural Products; Zografos, A.L., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 331–379. [Google Scholar]
- Sellars, J.D.; Steel, P.G. Advances in the synthesis of aryltetralin lignan lactones. Eur. J. Org. Chem. 2007, 2007, 3815–3828. [Google Scholar] [CrossRef]
- Hong, B.C.; Hsu, C.S.; Lee, G.H. Enantioselective total synthesis of (+)-galbulin via organocatalytic domino Michael-Michael-aldol condensation. Chem. Commun. 2012, 48, 2385–2387. [Google Scholar] [CrossRef] [PubMed]
- Rye, C.E.; Barker, D. Asymmetric synthesis of (+)-galbelgin, (−)-kadangustin J, (−)-cyclogalgravin and (−)-pycnanthulignenes A and B, three structurally distinct lignan classes, using a common chiral precursor. J. Org. Chem. 2011, 76, 6636–6648. [Google Scholar] [CrossRef] [PubMed]
- Route, J.K.; Ramana, C.V. Total synthesis of (−)-sacidumlignans B and D. J. Org. Chem. 2012, 77, 1566–1571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Yan, C.S.; Peng, Y.; Luo, Z.B.; Xu, X.B.; Wang, Y.W. Total synthesis of (±)-sacidumlignans D and A through Ueno-Stork radical cyclization reaction. Org. Biomol. Chem. 2013, 11, 2498–2513. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Luo, Z.B.; Zhang, J.J.; Luo, L.; Wang, Y.W. Collective synthesis of several 2,7′-cyclolignans and their correlation by chemical transformations. Org. Biomol. Chem. 2013, 11, 7574–7586. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, L.S.; Brummond, K.M. Intramolecular Dehydro-Diels–Alder Reaction Affords Selective Entry to Arylnaphthalene or Aryldihydronaphthalene Lignans. Org. Lett. 2014, 16, 4158–4161. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Cong, X.W.; Yang, G.Z.; Wang, Y.W.; Peng, Y. Divergent asymmetric syntheses of podophyllotoxin and related family members via stereoselective reductive Ni-catalysis. Org. Lett. 2018, 20, 1651–1654. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Cong, X.W.; Yang, G.Z.; Wang, Y.W.; Peng, Y. Stereoselective synthesis of podophyllum lignans core by intramolecular reductive nickel-catalysis. Chem. Commun. 2018, 54, 2040–2043. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.S.; Peng, Y.; Xu, X.B.; Wang, Y.W. Nickel-mediated inter- and intramolecular reductive cross-coupling of unactivated alkyl bromides and aryl iodides at room temperature. Chem. Eur. J. 2012, 18, 6039–6048. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.B.; Liu, J.; Zhang, J.J.; Wang, Y.W.; Peng, Y. Nickel-mediated inter- and intramolecular C-S coupling of thiols and thioacetates with aryl iodides at room temperature. Org. Lett. 2013, 15, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Luo, L.; Yan, C.S.; Zhang, J.J.; Wang, Y.W. Ni-catalyzed reductive homocoupling of unactivated alkyl bromides at room temperature and its synthetic application. J. Org. Chem. 2013, 78, 10960–10967. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Xu, X.B.; Xiao, J.; Wang, Y.W. Nickel-mediated stereocontrolled synthesis of spiroketals via tandem cyclization-coupling of β-bromo ketals and aryl iodides. Chem. Commun. 2014, 50, 472–474. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhang, J.J.; Ling, W.J.; Shao, Y.L.; Wang, Y.W.; Peng, Y. Unified synthesis of (−)-folicanthine and (−)-ditryptophenaline enabled by a nickel-mediated reductive dimerization at room temperature. Synthesis 2014, 46, 1908–1916. [Google Scholar] [CrossRef]
- Peng, Y.; Xiao, J.; Xu, X.B.; Duan, S.M.; Ren, L.; Shao, Y.L.; Wang, Y.W. Stereospecific synthesis of tetrahydronaphtho[2,3-b]furans enabled by a nickel-promoted tandem reductive cyclization. Org. Lett. 2016, 18, 5170–5173. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Wang, Y.W.; Peng, Y. Nickel-promoted reductive cyclization cascade: A short synthesis of a new aromatic strigolactone analogue. Synthesis 2017, 49, 3576–3581. [Google Scholar]
- Uchiyama, M.; Kimura, Y.; Ohta, A. Stereoselective total syntheses of (±)-arthrinone and related natural compounds. Tetrahedron Lett. 2000, 41, 10013–10017. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.Q.; Yang, L.; Feng, G. Podophyllotoxin derivatives show activity against Brontispa longissima larvae. Nat. Prod. Commun. 2010, 5, 1247–1250. [Google Scholar] [PubMed]
- Kennedy-Smith, J.J.; Young, L.A.; Toste, F.D. Rhenium-catalyzed aromatic propargylation. Org. Lett. 2004, 6, 1325–1327. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.C.; Teague, S.J.; Meyers, A.I. Asymmetric total synthesis of (−)-podophyllotoxin. J. Am. Chem. Soc. 1988, 110, 7854–7858. [Google Scholar] [CrossRef]
- CCDC-1875746 (5) Contain the Supplementary Crystallographic Data for This Paper. These Data Can Be Obtained Free of Charge. Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 28 October 2018).
- Kashima, T.; Tanoguchi, M.; Arimoto, M.; Yamaguchi, H. Studies on the constituents of the seeds of Hernandia ovigera L. VIII. Synthesis of (±)-desoxypodophyllotoxin and (±)-β-peltatin-A methyl ether. Chem. Pharm. Bull. 1991, 39, 192–194. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Arimoto, M.; Nakajima, S.; Tanoguchi, M.; Fukada, Y. Studies on the constituents of the seeds of Hernandia ovigera L.V. Syntheses of epipodophyllotoxin and podophyllotoxin from desoxypodophyllotoxin. Chem. Pharm. Bull. 1986, 34, 2056–2060. [Google Scholar] [CrossRef]
- Nishii, Y.; Yoshida, T.; Asano, H.; Wakasugi, K.; Morita, J.-I.; Aso, Y.; Yoshida, E.; Motoyoshiya, J.; Aoyama, H.; Tanabe, Y. Regiocontrolled benzannulation of diaryl(gem-dichlorocyclopropyl)methanols for the synthesis of unsymmetrically substituted α-arylnaphthalenes: Application to total synthesis of natural lignan lactones. J. Org. Chem. 2005, 70, 2667–2678. [Google Scholar] [CrossRef] [PubMed]
- Schrecker, A.W.; Hartwell, J.L. Components of podophyllin. IX. The structure of apopicropodophyllins. J. Am. Chem. Soc. 1952, 74, 5676–5683. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
Scheme 1.
(a) Several arylnaphthalene lignans and their DHN and THN derivatives; (b) Our synthetic logic.
Scheme 1.
(a) Several arylnaphthalene lignans and their DHN and THN derivatives; (b) Our synthetic logic.
Scheme 2.
Reductive tandem cyclization for tetralin lactones.

Scheme 3.
Regiodivergent oxidation of (+)-deoxypicropodophyllin (2).
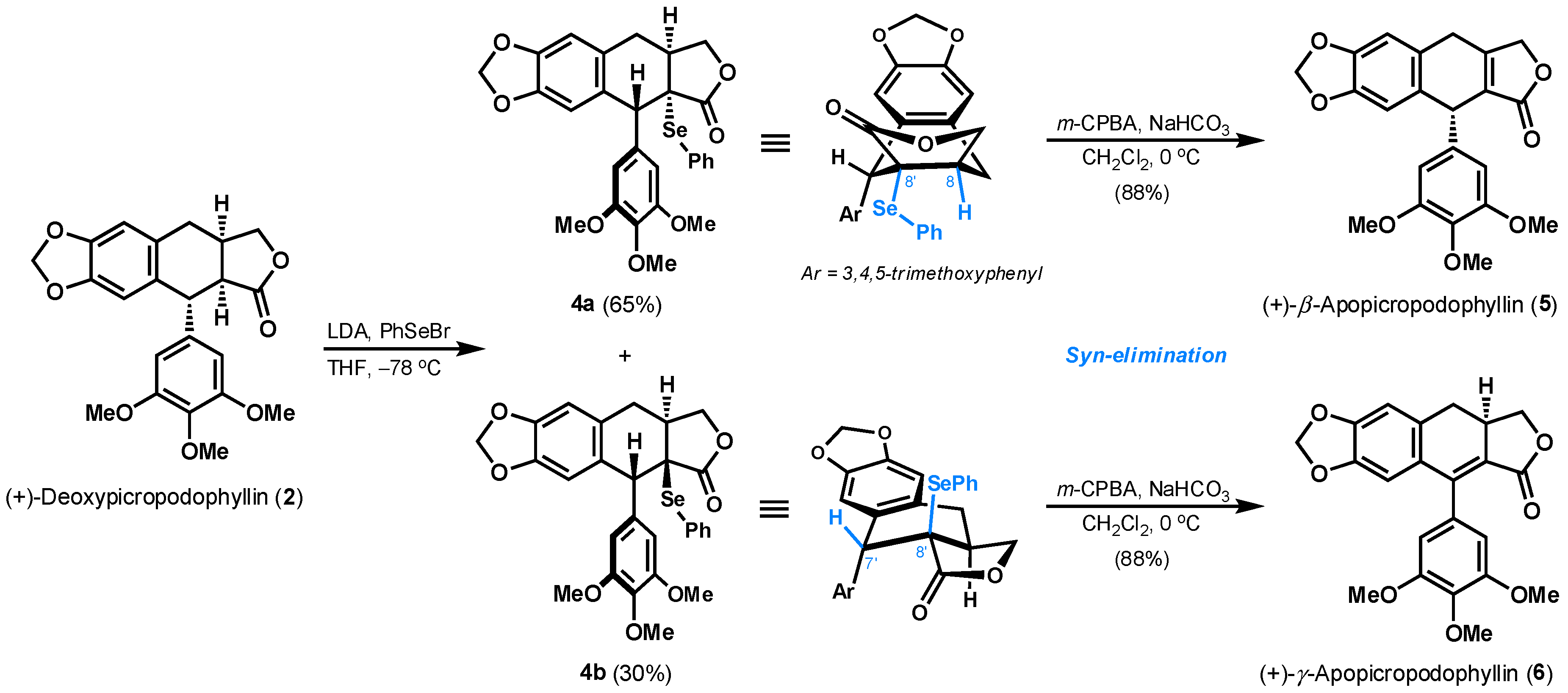
Figure 1.
X-ray crystal structure of (+)-β-apopicropodophyllin (5), selected H atoms have been omitted for clarity.
Figure 1.
X-ray crystal structure of (+)-β-apopicropodophyllin (5), selected H atoms have been omitted for clarity.

Scheme 4.
One-step conversion of tetralin to arylnaphthalene skeleton.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Xiao, J.; Nan, G.; Wang, Y.-W.; Peng, Y. Concise Synthesis of (+)-β- and γ-Apopicropodophyllins, and Dehydrodesoxypodophyllotoxin. Molecules 2018, 23, 3037. https://doi.org/10.3390/molecules23113037
AMA Style
Xiao J, Nan G, Wang Y-W, Peng Y. Concise Synthesis of (+)-β- and γ-Apopicropodophyllins, and Dehydrodesoxypodophyllotoxin. Molecules. 2018; 23(11):3037. https://doi.org/10.3390/molecules23113037
Chicago/Turabian StyleXiao, Jian, Guangming Nan, Ya-Wen Wang, and Yu Peng. 2018. "Concise Synthesis of (+)-β- and γ-Apopicropodophyllins, and Dehydrodesoxypodophyllotoxin" Molecules 23, no. 11: 3037. https://doi.org/10.3390/molecules23113037