3.2.6. General Procedure F: Benzyl Deprotection
To a stirred solution of benzyl ether (1 mmol) in MeOH (30 mL) was added 10% palladium on carbon (20%
w/
w) and the resultant mixture stirred under and atmosphere of hydrogen for the specified time. The reaction mixture was filtered through celite, washed with methanol (3 × 20 mL), the solvent removed in vacuo and the crude product purified by column chromatography if necessary (The
1H and
13C-NMR spectra of compounds in the
Supplemental Materials).
(E)-Ethyl 4-(3′,4′-dimethoxyphenyl)but-2-enoate (
16). To a stirred solution of NMO (7.9 g, 67.3 mmol) in H
2O/
tBuOH (1:1, 80 mL) was added 4-allyl-1,2-dimethoxybenzene
10 (3.86 mL, 22.4 mmol). A solution of OsO
4 (0.6 mL, 0.059 mmol, 2.5%
w/
v in
tBuOH) was then added dropwise and the resulting mixture stirred at room temperature for 4 days. The mixture was then quenched with saturated aqueous Na
2SO
3 (100 mL) and stirred for 1 h. The mixture was extracted with ethyl acetate (3 × 50 mL), the organic layers combined, washed with aqueous KOH (1 M, 20 mL), and dried (MgSO
4). Solvent was removed in vacuo to give
12 (4.8 g, quant.) as a white solid which was used without further purification. To a stirred solution of diol
12 (4.8 g, 22.8 mmol) in methanol/H
2O (3:1, 100 mL) was added NaIO
4 (5.9 g, 27.4 mmol) and stirred for 30 min. The reaction mixture was then quenched with addition of brine (50 mL) and extracted with ethyl acetate (3 × 40 mL). The organic extracts were combined, washed with water (2 × 20 mL), and dried (MgSO
4). Solvent was removed in vacuo to give
14 (2.68 g, 65%) as a pale-yellow oil which was used without further purification. To a stirred solution of 2-(3,4-dimethoxyphenyl)acetaldehyde
14 (2.68 g, 14.8 mmol) in CH
2Cl
2 (100 mL), under an atmosphere of nitrogen, was added (carbethoxymethylene)triphenylphosphorane (5.7 g, 16.3 mmol) and the resulting mixture stirred for 16 h. Solvent was removed in vacuo and the crude product purified by column chromatography (3:1, hexanes, ethyl acetate) to give the title compound
16 (3.13 g, 84%) as a colourless oil. R
f = 0.56 (2:1 hexanes, ethyl acetate). δ
H (400 MHz; CDCl
3) 1.27 (3H, t,
J = 7.2 Hz, 1-OCH
2C
H3), 3.45 (2H, dd,
J = 1.5, 6.7 Hz, 4-H), 3.86 (6H, s, 3′, 4′-H), 4.17 (2H, q,
J = 7.2 Hz, 1-OC
H2CH
3), 5.80 (1H, td,
J = 1.6, 15.5 Hz, 2-H), 6.67 (1H, d,
J = 1.9 Hz, 2′-H), 6.71 (1H, dd,
J = 1.9, 8.1 Hz, 6′-H), 6.81 (1H, d,
J = 8.1 Hz, 5′-H), 7.07 (1H, td,
J = 6.7, 15.5 Hz, 3-H). δ
C (100 MHz; CDCl
3) 14.3 (1-OCH
2CH
3), 38.1 (C-4), 55.9, 56.0 (3′, 4′-OCH
3), 60.3 (1-OCH
2CH
3), 111.5 (C-5′), 112.1 (C-2′), 120.8 (C-6′), 122.2 (C-2), 130.2 (C-1′), 147.6 (C-3), 147.9 (C-4′), 149.1 (C-3′), 166.6 (C-1). Values are in agreement with literature data [
42].
(E)-4-(3′,4′-Dimethoxyphenyl)but-2-en-1-ol (18). To a stirred solution of ester 16 (1.0 g, 4.0 mmol) in CH2Cl2 (20 mL), under an atmosphere of nitrogen at −78 °C, was added DIBAL (12 mL, 1 M in cyclohexane) and the resulting mixture stirred for 10 min. The reaction mixture was quenched with addition of 2 M HCl until gas evolution ceased, the organic phase separated and the aqueous phase further extracted with CH2Cl2 (3 × 10 mL). The organic layers were combined then washed with water (10 mL) and dried (MgSO4). Solvent was removed in vacuo and the crude product purified by column chromatography (1:1 hexanes, ethyl acetate) to give the title compound 18 (0.76 g, 92%) as a colourless oil. Rf = 0.18 (2:1, hexanes, ethyl acetate). δH (400 MHz; CDCl3) 3.30 (2H, d, J = 6.6 Hz, 4-H), 3.82 (3H, s, 4′-OCH3), 3.83 (3H, s, 3′-OCH3), 4.08 (2H, d, J = 5.6 Hz, 1-H), 5.64–5.69 (1H, m, 2-H), 5.78–5.83 (1H, m, 3-H), 6.68 (1H, s, 2′-H), 6.69 (1H, d, J = 8.0 Hz, 6′-H), 6.77 (1H, d, J = 8.0 Hz, 5′-H). δC (100 MHz; CDCl3) 38.2 (C-4), 55.8 and 55.9 (3′ and 4′-OCH3), 63.3 (C-1), 111.4 (C-5′), 112.0 (C-2′), 120.4 (C-6′), 130.2 (C-2), 131.6 (C-3), 132.7 (C-1′), 147.4 (C-4′), 148.9 (C-3′). IR: νMAX (film)/cm-1; 3391 (broad), 2933, 2835, 1591, 1512, 1463, 1417, 1258, 1232, 1137, 1025, 971, 852, 806, 762. HRMS (ESI+) Found [M + Na]+ 231.0995; C12H16NaO3 requires 231.0992.
(E)-4-(4-(3′,4′-Dimethoxyphenyl)but-2-en-1-yl)morpholine (9a). To a stirred solution of alcohol 18 (0.73 g, 3.5 mmol) in CH2Cl2 (20 mL), under an atmosphere of nitrogen at 0 °C, was added Et3N (1.5 mL, 10.5 mmol) and stirred for 5 min. MsCl (0.48 mL, 4.2 mmol) was added and stirred for 10 min. Morpholine (0.50 mL, 5.3 mmol) was added and the mixture brought to room temperature and stirred for 2 h. Saturated aqueous NaHCO3 (20 mL) and water (4 mL) was then added and the aqueous layer further extracted with CH2Cl2 (3 × 20 mL). The organic layers were then combined, dried (MgSO4) and the solvent removed in vacuo. The crude product was purified by column chromatography (1:1 hexanes, ethyl acetate) to give the title compound 9a (0.60 g, 62%) as a colourless oil. Rf = 0.31 (1:2 hexanes, ethyl acetate). δH (400 MHz; CDCl3) 2.41–2.44 (4H, m, O(CH2CH2)2N), 2.96 (2H, d, J = 6.8 Hz, 1-H), 3.30 (2H, d, J = 6.7 Hz, 4-H), 3.68–3.71 (4H, m, O(CH2CH2)2N), 3.83 (6H, s, 3′, 4′-OCH3), 5.52–5.57 (1H, m, 3-H), 5.71–5.78 (1H, m, 2-H), 6.67–6.70 (2H, m, 2′ and 6′-H), 6.78 (1H, d, J = 7.9 Hz, 5′-H). δC (100 MHz; CDCl3) 38.5 (C-4), 53.6 (O(CH2CH2)2N), 55.8, 56.0 (3′, 4′-OCH3), 61.1 (C-1), 67.0 (O(CH2CH2)2N), 111.4 (C-5′), 111.9 (C-2′), 120.3 (C-6′), 127.1 (C-3), 132.8 (C-1′), 133.8 (C-2), 147.5 (C-4′), 149.0 (C-3′). IR: νMAX (film)/cm−1; 2934, 2851, 1591, 1453, 1260, 1138, 1028, 976, 864, 805, 763. HRMS (ESI+) Found [M + H]+ 278.1762; C16H24NO3 requires 278.1751.
(E)-Ethyl 4-(3′,4′-methylenedioxyphenyl)but-2-enoate (
17). To a stirred solution of NMO (8.67 g, 74.0 mmol) in H
2O/
tBuOH (1:1, 80 mL) was added safrole
11 (4.0 mL, 27 mmol). A solution of OsO
4 (0.75 mL, 0.074 mmol, 2.5%
w/
v in
tBuOH) was added dropwise and the resultant mixture stirred at room temperature for 17 h. The reaction mixture was quenched with saturated aqueous Na
2SO
3 (100 mL) and stirred for 1 h. The mixture was extracted with ethyl acetate (3 × 50 mL), the organic layers were combined, washed with aqueous KOH (1 M, 20 mL) and dried (MgSO
4). Solvent was removed in vacuo to give diol
13 (5.2 g, quant.) as a white solid which was used without further purification. To a stirred solution of diol
13 (5.2 g, 27 mmol) in methanol/H
2O (3:1, 100 mL) was added NaIO
4 (6.8 g, 32 mmol) and stirred for 2 h. The mixture was then quenched with addition of brine (50 mL) and extracted with ethyl acetate (3 × 50 mL). The organic extracts were combined, washed with water (2 × 20 mL), brine (10 mL), and dried (MgSO
4). Solvent was removed in vacuo to give aldehyde
15 (4.4 g, quant.) as a yellow oil which was used without further purification. To a stirred solution of 2-(3,4-methylenedioxyphenyl)acetaldehyde
15 (4.4 g, 27 mmol) in CH
2Cl
2 (50 mL), under an atmosphere of nitrogen, was added (carbethoxymethylene)triphenylphosphorane (10.4 g, 30 mmol) and the resulting mixture stirred for 16 h. Solvent was removed in vacuo and the crude product purified by column chromatography (19:1, hexanes, ethyl acetate) to give the title compound 17 (3.54 g, 56%) as a colourless oil. R
f = 0.73 (2:1 hexanes, ethyl acetate). δ
H (400 MHz; CDCl
3) 1.27 (3H, t,
J = 7.2 Hz, 1-OCH
2C
H3), 3.42 (2H, dd,
J = 6.6, 1.6 Hz, 4-H), 4.17 (2H, q,
J = 7.2 Hz, 1-OC
H2CH
3), 5.78 (1H, dt,
J = 15.5, 1.6 Hz, 2-H), 5.93 (2H, s, OCH
2O), 6.61 (1H, dd,
J = 8.0, 2.0 Hz, 6′-H), 6.64 (1H, d,
J = 2.0 Hz, 2′-H), 6.74 (1H, d,
J = 8.0 Hz, 5′-H), 7.04 (1H, dt,
J = 15.5, 6.6 Hz, 3-H). δ
C (100 MHz; CDCl
3) 14.4 (1-OCH
2CH
3), 38.2 (C-4), 60.4 (1-O
CH
2CH
3), 101.1 (OCH
2O), 108.5 (C-5′), 109.4 (C-2′), 121.9 (C-6′), 122.4 (C-2), 131.5 (C-1′), 146.5 (C-4′), 147.5 (C-3), 148.0 (C-3′), 166.6 (C-1). Values are in agreement with literature data [
43].
(E)-4-(3′,4′-Methylenedioxyphenyl)but-2-en-1-ol (
19). To a stirred solution of ester
17 (3.2 g, 13.7 mmol) in toluene (100 mL), under an atmosphere of nitrogen at −10 °C, was added DIBAL (30 mL, 1 M in toluene) and the resultant mixture stirred for 10 min. The reaction mixture was quenched with addition of 2 M HCl until gas evolution ceased, the organic layer was separated and the aqueous phase further extracted with CH
2Cl
2 (3 × 50 mL). The organic layers were combined, washed with brine (30 mL) and dried (MgSO
4). Solvent was removed in vacuo and the crude product purified by column chromatography (3:1 hexanes, ethyl acetate) to give the title compound
19 (2.59 g, 98%) as a pale yellow oil. R
f = 0.42 (hexanes, ethyl acetate). δ
H (400 MHz; CDCl
3) 1.41 (1H, br s, 1-OH), 3.30 (2H, d,
J = 6.6 Hz, 4-H), 4.12 (2H, br d,
J = 4.5 Hz, 1-H), 5.64–5.72 (1H, m, 2-H), 5.77–5.85 (1H, m, 3-H), 5.92 (2H, s, OCH
2O), 6.63 (1H, dd,
J = 7.9, 1.9 Hz, 6′-H), 6.67 (1H, d,
J = 1.9 Hz, 2′-H), 6.73 (1H, d, 7.9 Hz, 5′-H). δ
C (100 MHz; CDCl
3) 38.4 (C-4), 63.6 (C-1), 101.0 (OCH
2O), 108.3 (C-5′), 109.2 (C-2′), 121.4 (C-6′), 130.4, 131.8 (C-2, 3), 133.9 (C-1′), 146.0, 147.8 (C-3′, 4′). Values are in agreement with literature data [
43].
(E)-4-(4-(3′,4′-Methylenedioxyphenyl)but-2-en-1-yl)morpholine (9b). To a stirred solution of alcohol 19 (1.66 g, 8.6 mmol) in CH2Cl2 (15 mL), under an atmosphere of nitrogen at 0 °C, was added Et3N (3.6 mL, 25.9 mmol) and stirred for 5 min. MsCl (1.2 mL, 10.4 mmol) was added and stirred for 10 min. Morpholine (1.3 mL, 13.8 mmol) was added and the mixture brought to room temperature and stirred for 18 h. Saturated aqueous NaHCO3 (25 mL) and water (5 mL) was added and the aqueous layer further extracted with CH2Cl2 (3 × 30 mL). The organic layers were combined, dried (MgSO4) and the solvent removed in vacuo. The crude product was purified by column chromatography (2:1 hexanes, ethyl acetate) to give the title compound 9b (1.4 g, 60%) as a pale yellow oil. Rf = 0.39 (1:2 hexanes, ethyl acetate). δH (400 MHz; CDCl3) 2.43 (4H, br t, J = 4.7 Hz, NCH2CH2O), 2.96 (2H, d, J = 6.5 Hz, 1-H), 3.28 (2H, d, J = 7.0 Hz, 4-H), 3.71 (4H, t, J = 4.7 Hz, NCH2CH2O), 5.49–5.56 (1H, m, 2-H), 5.69–5.76 (1H, m, 3-H), 5.91 (2H, d, J = 2.0 Hz, OCH2O), 6.61 (1H, dd, J = 7.5, 2.0 Hz, 6′-H), 6.65 (1H, d, J = 2.0 Hz, 2′-H), 6.72 (1H, d, J = 7.5 Hz, 5′-H). δC (100 MHz; CDCl3) 38.7 (C-4), 53.7 (NCH2CH2O), 61.2 (C-1), 67.1 (NCH2CH2O), 100.9 (OCH2O), 108.3 (C-5′), 109.1 (C-2′), 121.4 (C-6′), 127.4 (C-2), 133.7 (C-3), 134.1 (C-1′), 146.0 (C-4′), 147.8 (C-3′). IR: νMAX (film)/cm−1; 2855, 1739, 1488, 1242, 1115, 1036, 926, 864, 736. HRMS (ESI+) Found [M + H]+ 262.1428; C15H20NO3 requires 262.1438.
(E)-Ethyl-3-(3′,4′-methylenedioxyphenyl)prop-2-enoate (
23). To a stirred solution of piperonal
20 (5.0 g, 33 mmol) in CH
2Cl
2 (100 mL), under an atmosphere of nitrogen, was added (carbethoxymethylene)triphenylphosphorane (12.8 g, 37.0 mmol) and the resulting mixture stirred for 20 h. Solvent was then removed in vacuo and the crude product purified by column chromatography (3:1, hexanes, ethyl acetate) to give the title compound
23 (6.97 g, 95%) as a white solid. R
f = 0.68 (2:1 hexanes, ethyl acetate). Melting point: 62–64 °C. δ
H (400 MHz; CDCl
3) 1.32 (3H, t,
J = 7.2 Hz, 1-OCH
2C
H3), 4.25 (2H, q,
J = 7.2 Hz, 1-OC
H2CH
3), 6.00 (2H, s, -OCH
2O-), 6.25 (1H, d,
J = 15.9 Hz, 2-H), 6.80 (1H, d,
J = 8.0 Hz, 5′-H), 7.00 (1H, dd,
J = 1.4, 8.0 Hz, 6′-H), 7.02 (1H, d,
J = 1.4 Hz, 6′-H), 7.58 (1H, d,
J = 15.9 Hz, 3-H). δ
C (100 MHz; CDCl
3) 14.5 (1-OCH
2CH
3), 60.5 (1-OCH
2CH
3), 101.7 (-OCH
2O-), 106.6 (C-5′), 108.7 (C-2′), 116.4 (C-2), 124.5 (C-6′), 129.1 (C-1′), 144.4 (C-3), 148.5 (C-4′), 149.7 (C-3′), 167.3 (C-1). Values are in agreement with literature data [
44].
(E)-Ethyl-3-(3′,4′,5′-trimethoxyphenyl)prop-2-enoate (
24). To a stirred solution of 3,4,5-trimethoxybenzaldehyde
21 (3.0 g, 15.3 mmol) in CH
2Cl
2 (100 mL), under an atmosphere of nitrogen, was added (carbethoxymethylene)triphenylphosphorane (5.9 g, 16.8 mmol) and the resulting mixture stirred for 3 h. Solvent was then removed in vacuo and the crude product purified by column chromatography (3:1, hexanes, ethyl acetate) to give the title compound
24 (4.0 g, 94%) as a white solid. R
f = 0.52 (2:1 hexanes, ethyl acetate). Melting point: 64–66 °C. δ
H (400 MHz; CDCl
3) 1.34 (3H, t,
J = 7.2 Hz, 1-OCH
2C
H3), 3.87 (3H, s, 4′-OCH
3), 3.88 (6H, s, 3′-OCH
3), 4.26 (2H, q,
J = 7.2 Hz, 1-OC
H2CH
3), 6.34 (1H, d,
J = 15.9 Hz, 2-H), 6.75 (2H, s, 2′-H), 7.60 (1H, d,
J = 15.9 Hz, 3-H). δ
C (100 MHz; CDCl
3) 14.5 (1-OCH
2CH
3), 56.3 (3′-OCH
3), 60.6 (1-OCH
2CH
3), 61.1 (4′-OCH
3), 105.3 (C-2′), 117.7 (C-2), 130.1 (C-1′), 140.2 (C-4′), 144.7 (C-3), 153.6 (C-3′), 167.1 (C-1). Values are in agreement with literature data [
45].
3-(3′,4′,5′-Trimethoxyphenyl)propionic acid (31). To a stirred solution of 24 (5.4 g, 19.4 mmol) in ethyl acetate (30 mL) was added 10% palladium on activated carbon (0.54 g, 10% w/w). The solution was flushed with an atmosphere of hydrogen and stirred for 2 h. The reaction mixture was then filtered through a plug of celite and washed with ethyl acetate, solvent was then removed in vacuo to give saturated ester 27 (5.23 g, 96%) which was then used without further purification.
To a stirred solution of ester
27 (5.1 g, 17.9 mmol) in methanol (30 mL) was added aqueous NaOH (72 mL, 1 M, 4 eq.) and stirred for 20 min. The mixture was then extracted with CH
2Cl
2 (10 mL) and the aqueous layer acidified with aqueous 2 M HCl. The aqueous phase was then extracted with ethyl acetate (3 × 50 mL), dried (MgSO
4) and solvent removed in vacuo to give the title compound
31 (4.6 g, quant.) as a white solid. R
f = 0.15 (2:1 hexanes, ethyl acetate). Melting point: 104–105 °C. δ
H (400 MHz; CDCl
3) 2.68 (2H, t,
J = 7.8 Hz, 2-H), 2.90 (2H, t,
J = 7.8 Hz, 3-H), 3.82 (3H, s, 4′-OCH
3), 3.84 (6H, s, 3′-OCH
3), 6.43 (2H, s, 2′-H). δ
C (100 MHz; CDCl
3) 31.1 (C-2), 35.8 (C-3), 56.2 (3′-OCH
3), 61.0 (4′-OCH
3), 105.4 (C-2′), 136.0 (C-1′), 136.7 (C-4′), 153.4 (C-3′), 178.8 (C-1). Values are in agreement with literature data [
46].
3-(3′,4′-Methylenedioxyphenyl)propionic acid (30). To a stirred solution of 23 (6.92 g, 31.4 mmol) in ethyl acetate (30 mL) was added 10% palladium on activated carbon (0.69 g, 10% w/w). The solution was flushed with an atmosphere of hydrogen and stirred for 1 h. The reaction mixture was then filtered through a plug of celite and washed with ethyl acetate, solvent was then removed in vacuo to give saturated ester 26 (6.9 g, 99%) which was then used without further purification.
To a stirred solution of ester
26 (6.74 g, 30.0 mmol) in methanol (30 mL) was added aqueous NaOH (121 mL, 1 M, 4 eq.) and stirred for 2.5 h. The mixture was then extracted with ethyl acetate (10 mL) and the aqueous layer acidified with aqueous 2 M HCl. The aqueous phase was then extracted with ethyl acetate (3 × 50 mL), dried (MgSO
4) and solvent removed in vacuo to give the title compound
30 (5.5 g, 94%) as a white solid. R
f = 0.44 (2:1 hexanes, ethyl acetate). Melting point: 80–82°C. δ
H (400 MHz; CDCl
3) 2.64 (2H, t,
J = 7.7 Hz, 2-H), 2.88 (2H, t,
J = 7.7 Hz, 3-H), 5.93 (2H, s, -OCH
2O-), 6.66 (1H, dd,
J = 7.9, 1.4 Hz, 6′-H), 6.70 (1H, d,
J = 1.4 Hz, 2′-H), 6.74 (1H, d,
J = 7.9 Hz, 5′-H). δ
C (100 MHz; CDCl
3) 30.5 (C-2), 36.1 (C-3), 101.0 (-OCH
2O-), 108.4 (C-2′), 108.9 (C-5′), 121.2 (C-6′), 134.1 (C-1′), 146.2 (C-3′), 147.8 (C-4′), 179.1 (C-1). Values are in agreement with literature data [
47].
3-(3′-Methoxy-4′-benzyloxyphenyl)propionic acid (32). To a stirred solution of vanillin 22 (3.0 g, 19.7 mmol) in CH2Cl2 (100 mL), under an atmosphere of nitrogen, was added (carbethoxymethylene)triphenylphosphorane (7.56 g, 21.7 mmol) and the resulting mixture stirred for 18 h. Solvent was then removed in vacuo and the crude product purified by column chromatography (2:1, hexanes, ethyl acetate) to give a 2:1 mixture of E and Z isomers of unsaturated ester 25 (4.13 g, 94%) as a yellow oil which was used immediately.
To a stirred solution of unsaturated ester
25 (4.13 g, 18.6 mmol) in ethyl acetate (30 mL) was added 10% palladium on activated carbon (0.4 g, 10%
w/
w). The solution was flushed with an atmosphere of hydrogen and stirred for 2 h. The reaction mixture was then filtered through a plug of celite and washed with ethyl acetate, solvent was then removed in vacuo to give saturated ester
28 (3.9 g, 94%) as a yellow oil which was then used without further purification. To a stirred solution of phenol
28 (3.75 g, 16.7 mmol) in acetonitrile (40 mL), under an atmosphere of nitrogen, was added K
2CO
3 (6.9 g, 50.0 mmol) and stirred for 10 min. Benzyl bromide (6.0 mL, 50.0 mmol) was then added and the resulting mixture allowed to stir for 65 h. The reaction mixture was then quenched with addition of water (50 mL) and extracted with CH
2Cl
2 (3 × 30 mL). The organic phases were combined, washed with water (2 × 10 mL) and dried (MgSO
4). Solvent was then removed in vacuo and the crude product purified by column chromatography (9:1 hexanes, ethyl acetate) to give benzyl ether
29 (4.38 g, 83%) as a colourless oil which was used immediately. To a stirred solution of ester
29 (4.3 g, 13.7 mmol) in methanol (30 mL) was added aqueous NaOH (55 mL, 1 M, 4 eq.) and stirred for 2.5 h. The mixture was then acidified with aqueous 2 M HCl, extracted with ethyl acetate (3 × 50 mL), dried (MgSO
4) and solvent removed in vacuo to give the title compound
32 (3.85 g, 98%) as a white solid. R
f = 0.30 (2:1 hexanes, ethyl acetate). Melting point: 99–100°C. δ
H (400 MHz; CDCl
3) 2.66 (2H, t,
J = 7.7 Hz, 2-H), 2.90 (2H, t,
J = 7.7 Hz, 3-H), 3.88 (3H, s, 3′-OCH
3), 5.13 (2H, s, 7′-H), 6.68 (1H, dd,
J = 8.2, 2.0 Hz, 6′-H), 6.76 (1H, d,
J = 2.0 Hz, 2′-H), 6.81 (1H, d,
J = 8.2 Hz, 5′-H), 7.27–7.32 (1H, m, 11′-H), 7.34–7.39 (2H, m, 10′-H), 7.41–7.45 (2H, m, 9′-H). δ
C (100 MHz; CDCl
3) 30.4 (C-2), 35.9 (C-3), 56.1 (3′-OCH
3), 71.3 (C-7′), 112.4 (C-2′), 114.5 (C-5′), 120.3 (C-6′), 127.4 (C-9′), 127.9 (C-11′), 128.7 (C-10′), 133.5 (C-1′), 137.4 (C-8′), 146.9 (C-4′), 149.8 (C-3′), 178.8 (C-1). Values are in agreement with literature data [
48].
3-(3′,4′-Methylenedioxyphenyl)propanoyl chloride (34b). To a stirred solution of carboxylic acid 30 (0.22 g, 1.2 mmol) in CH2Cl2 (3 mL), under an atmosphere of nitrogen, was added oxalyl chloride (0.2 mL, 2.3 mmol) dropwise and the mixture stirred for 4 h. The solvent was removed in vacuo to give the title compound 34b (0.24 g, quant.) as a green oil, which was placed under nitrogen and used without further purification.
3-(3′,4′-Dimethoxyphenyl)propanoyl chloride (34a). To a stirred solution of carboxylic acid 33 (0.24 g, 1.2 mmol) in CH2Cl2 (5 mL), under an atmosphere of nitrogen, was added oxalyl chloride (0.2 mL, 2.3 mmol) dropwise and the mixture stirred for 2.5 h. The solvent was removed in vacuo to give the title compound 34a (0.26 g, quant.) as a yellow oil, which was placed under nitrogen and used without further purification.
3-(3′,4′,5′-Trimethoxyphenyl)propanoyl chloride (34c). To a stirred solution of carboxylic acid 31 (0.25 g, 1.2 mmol) in CH2Cl2 (3 mL), under an atmosphere of nitrogen, was added oxalyl chloride (0.2 mL, 2.3 mmol) dropwise and the mixture stirred for 1.5 h. The solvent removed in vacuo to give the title compound 34c (0.27 g, quant.) as a green crystalline solid, which was placed under nitrogen and used without further purification.
3-(3′,4′-Methylenedioxyphenyl)propanoyl chloride (34d). To a stirred solution of carboxylic acid 32 (0.33 g, 1.2 mmol) in CH2Cl2 (3 mL), under an atmosphere of nitrogen, was added oxalyl chloride (0.2 mL, 2.3 mmol) dropwise and the mixture stirred for 4 h. The solvent was removed in vacuo to give the title compound 34d (0.35 g, quant.) as a yellow oil, which was placed under nitrogen and used without further purification.
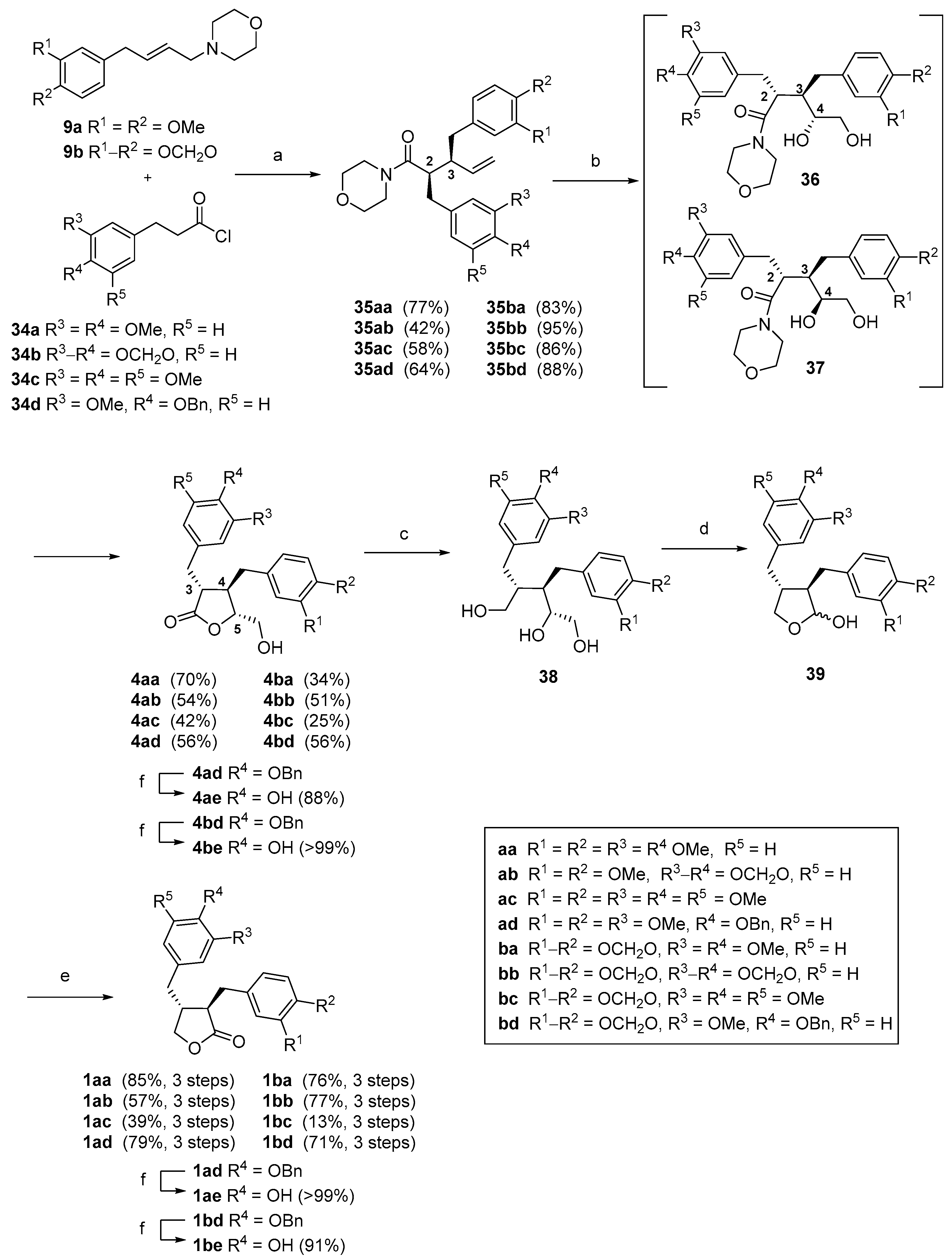
(2R*,3S*)-2-(3′,4′-Methylenedioxybenzyl)-3-(3″,4″-dimethoxybenzyl)-1-morpholinopent-4-en-1-one (35ab). Using general procedure A: Morpholine 9a (0.57 g, 2.06 mmol), acid chloride 34b (0.52 g, 2.47 mmol) and reaction time of 24 h. The crude product was purified by column chromatography (2:1 hexanes, ethyl acetate) to give the title compound 35ab (0.39 g, 42%) as a pale-yellow amorphous solid. Rf = 0.58 (1:3, hexanes, ethyl acetate). Melting point: 114–116 °C. δH (400 MHz; CDCl3) 2.57 (1H, dd, J = 13.6, 9.0 Hz, 7″-HA), 2.66–2.73 (1H, m, 3-H), 2.77–2.85 (2H, m, 7′-HA, OCHACH2N), 2.85–2.94 (4H, m, 2-H, 7′-HB, 7″-HB, OCH2CHAN), 3.06 (1H, ddd, J = 13.3, 7.9, 3.3 Hz, OCH2CHBN), 3.27–3.41 (3H, m, OCHCCHCN, OCHBCH2N), 3.53–3.60 (1H, m, OHDCH2N), 3.67–3.75 (1H, m, OCH2CHDN), 3.85 (3H, s, 4″-OCH3), 3.86 (3H, s, 3″-OCH3), 4.88 (1H, dd, J = 16.9, 1.8 Hz, 5-HA), 4.98 (1H, dd, J = 10.3, 1.8 Hz, 5-HB), 5.85 (1H, ddd, J = 16.9, 10.3, 9.5 Hz, 4-H), 5.90 (1H, d, J = 1.3 Hz, OCHAO), 5.91 (1H, d, J = 1.3 Hz, OCHBO), 6.60 (1H, dd, J = 7.8, 1.6 Hz, 6′-H), 6.64 (1H, d, J = 1.6 Hz, 2′-H), 6.65–6.68 (2H, m, 2″, 6″-H), 6.70 (1H, d, J = 7.8 Hz, 5′-H), 6.77 (1H, d, J = 8.7 Hz, 5″-H). δC (100 MHz; CDCl3) 37.4 (C-7′), 38.3 (C-7″), 42.0 (OCH2CHCDN), 46.4 (OCH2CHABN), 46.6 (C-2), 48.5 (C-3), 56.0 (3′, 4′-OCH3), 66.4 (OCHABCH2N), 67.0 (OCHCDCH2N), 101.0 (OCH2O), 108.4 (C-5′), 109.6 (C-2′), 111.1 (C-5″), 112.4 (C-2″), 116.8 (C-5), 121.3 (C-6″), 122.0 (C-6′), 132.3 (C-1″), 133.6 (C-1′), 139.3 (C-4), 146.2 (C-4′), 147.5 (C-4″), 147.7 (C-3′), 148.9 (C-3″), 172.6 (C-1). IR: νMAX (film)/cm-1; 2963, 1631, 1515, 1488, 1442, 1236, 1031, 925, 807, 730. HRMS (ESI+) Found [M + H]+ 454.2241; C26H32NO6 requires 454.2224.
(2R*,3S*)-2-(3′,4′,5′-Trimethoxybenzyl)-3-(3″,4″-dimethoxybenzyl)-1-morpholinopent-4-en-1-one (35ac). Using general procedure A: Morpholine 9a (0.47 g, 1.7 mmol), acid chloride 34c (0.53 g, 2.0 mmol) and a reaction time of 19 h. The crude product was purified by column chromatography (1:1 hexanes, ethyl acetate) to give the title compound 35ac (0.50 g, 58%) as a yellow oil. Rf = 0.38 (1:3 hexanes, ethyl acetate). δH (400 MHz; CDCl3) 2.59 (1H, dd, J = 13.6, 9.2 Hz, 7″-HA), 2.67–2.74 (1H, m, 3-H), 2.78 (1H, ddd, J = 11.4, 7.8, 3.0 Hz, NCH2CHAO), 2.82–2.96 (5H, m, 2-H, 7′-H, 7″-HB, NCHACH2O), 3.06 (1H, ddd, J = 13.2, 7.8, 3.0 Hz, NCHBCH2O), 3.25–3.40 (3H, m, NCHBCH2O, NCHCCHCO), 3.54–3.61 (1H, m, NCHDCH2O), 3.67–3.73 (1H, m, NCH2CHDO), 3.80 (3H, s, 4′-OCH3), 3.82 (6H, s, 3′-OCH3), 3.85 (3H, s, 4″-OCH3), 3.86 (3H, s, 3″-OCH3), 4.90 (1H, dd, J = 17.0, 1.8 Hz, 5-HA), 5.00 (1H, dd, J = 10.2, 1.8 Hz, 5-HB), 5.87 (1H, ddd, J = 17.0, 10.2, 9.1 Hz, 4-H), 6.37 (2H, s, 2′-H), 6.66–6.70 (2H, m, 2″, 6″-H), 6.78 (1H, d, J = 8.7 Hz, 5″-H). δC (100 MHz; CDCl3) 38.1 (C-7′), 38.3 (C-7″), 42.0 (NCHCDCH2O), 46.4 (NCHABCH2O), 46.5 (C-2), 48.7 (C-3), 56.0 (3″, 4″-OCH3), 56.3 (3′-OCH3), 61.0 (4′-OCH3), 66.4 (NCH2CHABO), 66.9 (NCH2CHCDO), 106.2 (C-2′), 111.1 (C-5″), 112.5 (C-2″), 116.8 (C-5), 121.2 (C-6″), 132.3 (C-1″), 135.6 (C-1′), 136.8 (C-4′), 139.2 (C-4), 147.5 (C-4″), 148.8 (C-3″), 153.3 (C-3′), 172.6 (C-1). IR: νMAX (film)/cm-1; 2940, 1632, 1589, 1459, 1236, 1123, 1028, 913, 735. HRMS (ESI+) Found [M + Na]+ 522.2474; C28H37NNaO7 requires 522.2462.
(2R*,3S*)-2-(3′,4′-Dimethoxybenzyl)-3-(3″,4″-dimethoxybenzyl)-1-morpholinopent-4-en-1-one (35aa). Using general procedure A: Morpholine 9a (0.53 g, 1.91 mmol), acid chloride 34a (0.52 g, 2.29 mmol) and a reaction time of 24 h. The crude product was purified by flash chromatography (1:3 hexanes, ethyl acetate) to give the title compound 35aa (0.63 g, 77% yield) as a pale-yellow amorphous solid. Rf = 0.42 (19:1 CH2Cl2, methanol). Melting point: 98–101 °C. δH (400 MHz; CDCl3) 2.55–2.63 (1H, m, 7″-HA), 2.85–2.93 (1H, m, 7″-HB), 2.67–2.85 (3H, m, 3-H, OCH2CHABN), 3.29-3.37 (4H, m, OCH2CHCDN, OCHABCH2N), 2.85–3.06 (3H, m, 2-H, 7′-H), 3.50–3.67 (2H, m, OCHCDCH2N), 3.83, 3.84, 3.85, 3.86 (12H, s, 3′, 4′, 3″, 4″-OCH3), 4.89 (1H, dd, J = 17.1, 1.7 Hz, 5-H), 4.99 (1H, dd, J = 10.3, 1.9 Hz, 5-H), 5.82–5.91 (1H, m, 4-H), 6.67–6.69 (4H, m, 2′, 6′, 2″, 6″-H), 6.75–6.78 (2H, m, 5′, 5″-H). δC (100 MHz; CDCl3) 37.2 (C-2), 38.2 (C-7″), 41.9, 46.5 (OCH2CH2N), 46.2 (C-7′), 48.5 (C-3), 55.8, 55.9 (3′, 4′, 3″, 4″-OCH3), 66.3, 66.8 (OCH2CH2N), 111.0, 111.3 (C-5′, 5″), 112.4, 112.6 (C-2′, 2″), 116.6 (C-5), 120.9, 121.2 (C-6′, 6″), 132.2, 132.3 (C-1′, 1″), 139.2 (C-4), 147.3, 147.6 (4′, 4″-OCH3), 148.7, 148.8 (3′, 3″-OCH3), 172.6 (C-1). IR: νMAX (film)/cm−1; 2935, 1628, 1591, 1462, 1260, 1155, 1027, 912, 857, 765. HRMS (ESI+) Found [M + H]+ 470.2537; C27H36NO6 requires 470.2537
(2R*,3S*)-2-(3′-Methoxy-4′-benzyloxybenzyl)-3-(3″,4″-dimethoxybenzyl)-1-morpholino-pent-4-en-1-one (35ad). Using general procedure A: Morpholine 9a (0.47 g, 1.7 mmol), acid chloride 34d (0.62 g, 2.0 mmol) and a reaction time of 22 h. The crude product was purified by column chromatography (2:1 hexanes, ethyl acetate) to give the title compound 35ad (0.59 g, 64%) as a yellow oil.
Rf = 0.58 (1:3, hexanes, ethyl acetate). δH (400 MHz; CDCl3) 2.57 (1H, dd, J = 13.5, 9.0 Hz, 7″-HA), 2.62–2.68 (1H, m, 3-H), 2.68–2.74 (1H, m, OCHACH2N), 2.75–2.82 (1H, m, OCH2CHAN), 2.83–2.92 (4H, m, 2-H, 7′-H, 7″-HB), 2.99 (1H, ddd, J = 13.3, 7.6, 3.2 Hz, OCH2CHBN), 3.20–3.32 (3H, m, OCHBCH2N, OCHCCHCN), 3.50–3.55 (1H, m, OCHDCH2N), 3.61–3.67 (1H, m, OCH2CHDN), 3.84 (3H, s, 3′-OCH3), 3.85 (3H, s, 4″-OCH3), 3.85 (3H, s, 3″-OCH3), 4.88 (1H, dd, J = 17.1, 1.9 Hz, 5-HA), 4.97 (1H, dd, J = 10.3, 1.9 Hz, 5-HB), 5.13 (1H, s, 7‴-H), 5.85 (1H, ddd, J = 17.1, 10.3, 9.0 Hz, 4-H), 6.59 (1H, dd, J = 8.1, 1.9 Hz, 6′-H), 6.65–6.68 (2H, m, 2″-H, 6″-H), 6.69 (1H, d, J = 1.9 Hz, 2′-H), 6.74 (1H, d, J = 8.1 Hz, 5′-H), 6.77 (1H, d, J = 8.5 Hz, 5″-H), 7.25–7.30 (1H, m, 4‴-H), 7.32–7.37 (2H, m, 3‴-H), 7.38–7.42 (2H, m, 2‴-H). δC (100 MHz; CDCl3) 37.4 (C-7′), 38.3 (C-7″), 41.9 (OCH2CHCDN), 46.3 (OCH2CHABN), 46.5 (C-2), 48.6 (C-3), 56.0, 56.2 (3′, 3″, 4″-OCH3), 66.4 (OCHABCH2N), 66.9 (OCHCDCH2N), 71.2 (C-7‴), 111.1 (C-5″), 112.4 (C-2″), 113.3 (C-2′), 114.6 (C-5′), 116.7 (C-5), 120.9 (C-6′), 121.3 (C-6″), 127.3 (C-2‴), 127.9 (C-4‴), 128.7 (C-3‴), 132.4 (C-1″), 133.1 (C-1′), 137.3 (C-1‴), 139.3 (C-4), 146.7 (C-4′), 147.5 (C-4″), 148.8 (C-3″), 149.7 (C-3′), 172.7 (C-1). IR: νMAX (film)/cm−1; 2936, 1736, 1633, 1513, 1454, 1261, 1140, 1028, 915, 733. HRMS (ESI+) Found [M + Na]+ 568.2671; C33H39NNaO6 requires 568.2670.
(2R*,3S*)-2-(3′,4′-Dimethoxybenzyl)-3-(3″,4″-methylenedioxybenzyl)-1-morpholinopent-4-en-1-one (35ba). Using general procedure A: Morpholine 9b (0.25 g, 0.96 mmol), acid chloride 34a (0.26 g, 1.2 mmol) and a reaction time of 21 h. The crude product was purified by column chromatography (1:1 hexanes, ethyl acetate) to give the title compound 35ba (0.36 g, 83%) as a yellow oil.
Rf = 0.50 (1:3 hexanes, ethyl acetate). δH (400 MHz; CDCl3) 2.56 (1H, dd, J = 13.4, 9.0 Hz, 7″-HA), 2.62–2.70 (1H, m, 3-H), 2.75–2.94 (6H, m, 2-H, 7′-H, 7″-HB, NCHACHAO), 3.05 (1H, ddd, J = 13.6, 7.9, 3.1 Hz, NCHBCH2O), 3.28–3.41 (3H, m, NCH2CHBO, NCHCCHCO), 3.51–3.57 (1H, m, NCH2CHDO), 3.58–3.64 (1H, m, NCHDCH2O), 3.83 (3H, s, 3′-H), 3.84 (3H, s, 4′-H), 4.89 (1H, dd, J = 17.2, 1.9 Hz, 5-HA), 4.99 (1H, dd, J = 10.2, 1.9 Hz, 5-HB), 5.86 (1H, ddd, J = 17.2, 10.2, 9.1 Hz, 4-H), 5.92 (1H, d, J = 1.4 Hz, OCHAO), 5.92 (1H, d, J = 1.4 Hz, OCHBO), 6.58 (1H, dd, J = 7.9, 1.6 Hz, 6″-H), 6.64 (1H, d, J = 1.6 Hz, 2″-H), 6.66–6.70 (2H, m, 2′, 6′-H), 6.71 (1H, d, J = 7.9 Hz, 5″-H), 6.76 (1H, d, J = 8.1 Hz, 5′-H). δC (100 MHz; CDCl3) 37.3 (C-7′), 38.5 (C-7″), 42.0 (NCHABCH2O), 46.3 (NCHCDCH2O), 46.5 (C-2), 48.8 (C-3), 56.1 (3′, 4′-OCH3), 66.4 (NCH2CHABO), 66.9 (NCH2CHCDO), 101.0 (OCH2O), 108.1 (C-5″), 109.6 (C-2″), 111.4 (C-5′), 112.7 (C-2′), 116.8 (C-5), 121.0 (C-6′), 122.1 (C-6″), 132.4 (C-1′), 133.7 (C-1″), 139.2 (C-4), 145.9 (C-4″), 147.6 (C-4′), 147.8 (C-3″), 149.0 (C-3′), 172.7 (C-1). IR: νMAX (film)/cm−1; 2908, 1740, 1630, 1515, 1441, 1237, 1029, 923, 730. HRMS (ESI+) Found [M + Na]+ 476.2042; C26H31NNaO6 requires 476.2044.
(2R*,3S*)-2-(3′,4′-Methylenedioxybenzyl)-3-(3″,4″-methylenedioxybenzyl)-1-morpholinopent-4-en-1-one (35bb). Using general procedure A: Morpholine 9b (0.5 g, 1.91 mmol), acid chloride 34b (0.49 g, 2.30 mmol) and a reaction time of 30 min. The crude product was purified by column chromatography (1:1 hexanes, ethyl acetate) to give the title compound 35bb (0.798 g, 95%) as a pale-yellow solid. Rf = 0.68 (1:3 hexanes, ethyl acetate). Melting point: 131–133 °C. δH (400 MHz; CDCl3) 2.54 (1H, dd, J = 13.5, 8.9 Hz, 7″-HA), 2.61–2.69 (1H, m, 3-H), 2.78–2.93 (6H, m, 2-H, 7′-H, 7″-HB, NCHACHAO), 3.06 (1H, ddd, J = 13.2, 7.8, 3.1 Hz, NCHBCH2O), 3.29–3.41 (3H, m, NCH2CHBO, NCHCCHCO), 3.53–3.61 (1H, m, NCH2CHDO), 3.66–3.74 (1H, m, NCHDCH2O), 4.89 (1H, dd, J = 17.0, 1.9 Hz, 5-HA), 4.99 (1H, dd, J = 10.2, 1.9 Hz, 5-HB), 5.85 (1H, ddd, J = 17.0, 10.2, 9.1 Hz, 4-H), 5.90 (1H, d, J = 1.4 Hz, 3′-OCHAO), 5.91 (1H, d, J = 1.4 Hz, 3′-OCHBO), 5.92 (1H, d, J = 1.5 Hz, 3″-OCHAO), 5.93 (1H, d, J = 1.5 Hz, 3″-OCHBO), 6.55–6.61 (2H, m, 6′, 6″-H), 6.62–6.64 (2H, m, 2′, 2″-H), 6.70, 6.71 (2 × 1H, 2 × d, J = 8.0 Hz, 5′, 5″-H). δC (100 MHz; CDCl3) 37.4 (C-7′), 38.5 (C-7″), 42.0 (NCHCDCH2O), 46.4 (NCHABCH2O), 46.5 (C-2), 48.8 (C-3), 66.4 (NCH2CHABO), 67.0 (NCH2CHCDO), 101.0 (2 × OCH2O), 108.2, 108.4 (C-5′, 5″), 109.6 (C-2′, 2″), 116.8 (C-5), 122.1 (C-6′, 6″), 133.6 (C-1′, 1″), 139.2 (C-4), 145.9, 146.2 (C-4′, 4″), 147.6, 147.7 (C-3′, 3″), 172.6 (C-1). IR: νMAX (film)/cm−1; 2897, 1630, 1487, 1440, 1244, 1036, 925, 808, 730. HRMS (ESI+) Found [M + Na]+ 460.1722; C25H27NNaO6 requires 460.1731.
(2R*,3S*)-2-(3′,4′,5′-Trimethoxybenzyl)-3-(3″,4″-methylenedioxybenzyl)-1-morpholinopent-4-en-1-one (35bc). Using general procedure A: Morpholine 9b (0.25 g, 0.96 mmol), acid chloride 34c (0.27 g, 1.2 mmol) and a reaction time of 18 h. The crude product was purified by column chromatography (1:1 hexanes, ethyl acetate) to give the title compound 35bc (0.40 g, 86%) as a pale-yellow solid. Rf = 0.55 (1:3 hexanes, ethyl acetate). Melting point: 104–106 °C. δH (400 MHz; CDCl3) 2.56 (1H, dd, J = 13.4, 9.0 Hz, 7″-HA), 2.62–2.70 (1H, m, 3-H), 2.75–2.95 (6H, m, 2-H, 7′-H, 7″-HB, NCHACHAO), 3.06 (1H, ddd, J = 13.2, 7.7, 3.0 Hz, NCHBCH2O), 3.25–3.40 (3H, m, NCH2CHBO, NCHCCHCO), 3.54–3.60 (1H, m, NCH2CHDO), 3.65–6.71 (1H, m, NCHDCH2O), 3.80 (3H, s, 4′-OCH3), 3.82 (6H, s, 3′-OCH3), 4.90 (1H, dd, J = 17.2, 1.9 Hz, 5-HA), 5.00 (1H, dd, J = 10.2, 1.9 Hz, 5-HB), 5.85 (1H, ddd, J = 17.2, 10.2, 9.0 Hz, 4-H), 5.92 (1H, d, J = 1.4 Hz, OCHAO), 5.93 (1H, d, J = 1.4 Hz, OCHBO), 6.36 (2H, s, 2′-H), 6.59 (1H, dd, J = 7.9, 1.6 Hz, 6″-H), 6.65 (1H, d, J = 1.6 Hz, 2″-H), 6.72 (1H, d, J = 7.9 Hz, 5″-H). δC (100 MHz; CDCl3) 38.1 (C-7′), 38.5 (C-7″), 42.0 (NCHCDCH2O), 46.4 (C-2, NCHABCH2O), 48.9 (C-3), 56.4 (3′-OCH3), 61.1 (4′-OCH3), 66.4 (NCH2CHABO), 67.0 (NCH2CHCDO), 101.0 (OCH2O), 106.2 (C-2′), 108.2 (C-5″), 109.6 (C-2″), 116.9 9 (C-5), 122.1 (C-6″), 133.6 (C-1″), 135.6 (C-1′), 136.9 (C-4′), 139.1 (C-4), 145.9 (C-4″), 147.7 (C-3″), 153.3 (C-3′), 172.6 (C-1). IR: νMAX (film)/cm−1; 2922, 1632, 1589, 1490, 1240, 1120, 1036, 925, 730. HRMS (ESI+) Found [M + Na]+ 506.2145; C27H33NNaO7 requires 506.2149.
(2R*,3S*)-2-(3′-Methoxy-4′-benzyloxybenzyl)-3-(3″,4″-methylenedioxybenzyl)-1-morpholinopent-4-en-1-one (35bd). Using general procedure A: Morpholine 9b (0.25 g, 0.96 mmol), acid chloride 34d (0.35 g, 1.2 mmol) and a reaction time of 18 h. The crude product was purified by column chromatography (1:1 hexanes, ethyl acetate) to give the title compound 35bd (0.45 g, 88%) as a yellow oil.
Rf = 0.67 (1:3 hexanes, ethyl acetate). δH (400 MHz; CDCl3) 2.54 (1H, dd, J = 13.5, 8.9 Hz, 7″-HA), 2.61–2.70 (2H, m, 3-H, NCH2CHAO), 2.73–2.91 (5H, m, 2-H, 7′-H, 7″-HB, NCHACH2O), 2.99 (1H, ddd, J = 13.2, 7.7, 3.0 Hz, NCHBCH2O), 3.20–3.35 (3H, m, NCH2CHBO, NCHCCHCO), 3.53 (1H, ddd, J = 11.0, 5.5, 2.5 Hz, NCH2CHDO), 3.62 (1H, ddd, J = 13.0, 5.5, 2.5 Hz, NCHDCH2O), 3.84 (3H, s, 3′-OCH3), 4.88 (1H, dd, J = 17.0, 1.9 Hz, 5-HA), 4.98 (1H, dd, J = 10.2, 1.9 Hz, 5-HB), 5.13 (2H, s, 7‴-H), 5.84 (1H, ddd, J = 17.0, 10.2, 9.1 Hz, 4-H), 5.91 (1H, d, J = 1.4 Hz, OCHAO), 5.92 (1H, d, J = 1.4 Hz, OCHBO), 6.57 (1H, dd, J = 8.0, 1.9 Hz, 6″-H), 6.59 (1H, dd, J = 8.2, 1.8 Hz, 6′-H), 6.64 (1H, d, J = 1.8 Hz, 2″-H), 6.69 (1H, d, J = 1.9 Hz, 2″-H), 6.71 (1H, d, J = 8.0 Hz, 5″-H), 6.75 (1H, d, J = 8.2 Hz, 5′-H), 7.25–7.30 (1H, m, 4‴-H), 7.32–7.37 (2H, m, 3‴-H), 7.38–7.43 (2H, m, 2‴-H). δC (100 MHz; CDCl3) 37.4 (C-7′), 38.5 (C-7″), 41.9 (NCHCDCH2O), 46.3 (NCHABCH2O), 46.4 (C-2), 48.8 (C-3), 56.2 (3′-OCH3), 66.3 (NCH2CHABO), 66.9 (NCH2CHCDO), 71.2 (C-7‴), 100.9 (OCH2O), 108.1 (C-5″), 109.6 (C-2″), 113.2 (C-2′), 114.5 (C-5′), 116.7 (C-5), 121.0 (C-6′), 122.1 (C-6″), 127.3 (C-2‴), 127.9 (C-4‴), 128.6 (C-3‴), 133.1 (C-1′) 133.6 (C-1″), 137.3 (C-1‴), 139.2 (C-4), 145.9 (C-4″), 146.7 (C-4′), 147.6 (C-3″), 149.7 (C-3′), 172.6 (C-1). IR: νMAX (film)/cm−1; 2920, 1630, 1489, 1231, 1114, 1034, 913, 729. HRMS (ESI+) Found [M + Na]+ 552.2354; C32H35NNaO6 requires 552.2357.
(3R*,4R*)-3-(3′,4′-Methylenedioxybenzyl)-4-(3″,4″-dimethoxybenzyl)-5-(hydroxymethyl)dihydrofuran-2(3H)-one (4ab). Using general procedure B: Amide 35ab (0.38 g, 0.84 mmol) in tBuOH/H2O and a reaction time of 3 days. The crude product was purified by column chromatography (1:1 hexanes, ethyl acetate) to give the title compound 4ab (180 mg, 54%) as a white foam. Rf = 0.50 (19:1 CH2Cl2, methanol). δH (400 MHz; CDCl3) 1.79 (1H, t, J = 6.4 Hz, 6-OH), 2.36–2.44 (1H, m, 4-H), 2.51 (1H, dd, J = 13.7, 7.9 Hz, 7″-HA), 2.58 (1H, dd, J = 13.7, 6.6 Hz, 7″-HB), 2.68 (1H, ddd, J = 9.3, 7.0, 5.5 Hz, 3-H), 2.85 (1H, dd, J = 14.0, 7.0 Hz, 7′-HA), 2.92 (1H, dd, J = 14.0, 5.5 Hz, 7′-HB), 3.15 (1H, ddd, J = 12.5, 6.4, 5.1 Hz, 6-HA), 3.54 (1H, ddd, J = 12.5, 6.4, 2.5 Hz, 6-HB), 3.83 (3H, s, 3″-OCH3), 3.85 (3H, s, 4″-OCH3), 4.19 (1H, ddd, J = 8.0, 5.1, 2.5 Hz, 5-H), 5.92 (1H, d, J = 1.5 Hz, OCHAHBO), 5.93 (1H, d, J = 1.5 Hz, OCHAHBO), 6.47 (1H, d, J = 2.0 Hz, 2″-H), 6.57–6.60 (2H, m, 6′ and 6″-H), 6.61 (1H, d, J = 1.5 Hz, 2′-H), 6.71 (1H, d, J = 7.8 Hz, 5′-H), 6.77 (1H, d, J = 8.1 Hz, 5″-H). δC (100 MHz; CDCl3) 35.3 (C-7′), 38.7 (C-7″), 41.6 (C-4), 47.6 (C-3), 56.0 (3″-OCH3, 4″-OCH3), 63.2 (C-6), 84.1 (C-5), 101.2 (OCH2O), 108.3 (C-5′), 109.7 (C-2′), 111.4 (C-5″), 112.0 (C-2″), 121.0 (C-6″), 122.5 (C-6′), 130.3 (C-1″), 131.6 (C-1′), 146.6 (C-4′), 148.0 (C-4″), 148.1 (C-3′), 149.3 (C-3″), 177.7 (C-2). IR: νMAX (film)/cm−1; 3496 (broad), 2936, 2254, 1760, 1515, 1489, 1442, 1239, 1025, 909, 809, 766. HRMS (ESI+) Found [M + Na]+ 423.1427; C22H24NaO7 requires 423.1414.
(3R*,4R*)-3,4-bis(3′,4′-Dimethoxybenzyl)-5-(hydroxymethyl)dihydrofuran-2(3H)-one (4aa). Using general procedure B: Amide 35aa (0.29 g, 0.61 mmol), in tBuOH/H2O and a reaction time of 6 days. The crude product was purified by flash chromatography (1:1 hexanes, ethyl acetate) to give the title compound 4aa (0.18 g, 70%) as a colourless oil. Rf = 0.32 (19:1 CH2Cl2, methanol). δH (400 MHz; CDCl3) 2.39–2.44 (1H, m, 4-H), 2.53 (1H, dd, J = 13.7, 7.3 Hz, 7″-HA), 2.58 (1H, dd, J = 13.7, 6.5 Hz, 7″-HB), 2.64 (1H, br s, 6-OH), 2.71 (1H, ddd, J = 9.3, 6.7, 5.7 Hz, 3-H), 2.88 (1H, dd, J = 14.0, 6.7 Hz, 7′-HA), 2.94 (1H, dd, J = 14.0, 5.5 Hz, 7′-HB), 3.16 (1H, dd, J = 12.6, 4.9 Hz, 6-HA), 3.53 (1H, dd, J = 12.6, 2.4 Hz, 6-HB), 3.81, 3.83, 3.84 (12H, s, 3′, 4′, 3″, 4″-OCH3), 4.15 (1H, ddd, J = 8.0, 4.9, 2.4 Hz, 5-H), 6.49 (1H, d, J = 1.9 Hz, 2″-H), 6.57 (1H, dd, J = 8.1, 1.9 Hz, 6″-H), 6.66–6.68 (2H, m, 2′, 6′-H), 6.73–6.80 (2H, m, 5′, 5″-H). δC (100 MHz; CDCl3) 35.0 (C-7′), 38.5 (C-7″), 41.6 (C-4), 47.5 (C-3), 55.8 (3′, 4′, 3″, 4″-OCH3), 62.9 (C-6), 84.0 (C-5), 111.2, 111.4 (C-5′, 5″), 112.1 (C-2″), 112.6 (C-2′), 120.9 (C-6″), 121.4 (C-6′), 130.4 (C-1′, 1″), 147.9 (C-4′, 4″), 149.0 (C-3′, 3″), 178.0 (C-2). IR: νMAX (film)/cm−1; 3505 (br), 2938, 1761, 1591, 1514, 1465, 1259, 1156, 1025, 910, 808, 766, 647. HRMS (ESI+) Found [M + H]+ 417.1909; C23H29O7 requires 417.1908.
(3R*,4R*)-3-(3′,4′,5′-Trimethoxybenzyl)-4-(3″,4″-dimethoxybenzyl)-5-(hydroxymethyl)dihydrofuran-2(3H)-one (4ac). Using general procedure B: Amide 35ac (0.45 g, 0.90 mmol) in tBuOH/H2O/THF and a reaction time of 3 days. The crude product was purified by column chromatography (1:1 hexanes, ethyl acetate) to give the title compound 4ac (0.17 g, 42%) as a pale-yellow solid.
Rf = 0.31 (19:1 CH2Cl2, methanol). Melting point: 141–142 °C. δH (400 MHz; CDCl3) 1.68 (1H, t, J = 6.5 Hz, 6-OH), 2.38–2.46 (1H, m, 4-H), 2.55 (1H, dd, J = 13.8, 8.2 Hz, 7″-HA), 2.65 (1H, dd, J = 13.8, 5.9 Hz, 7″-HB), 2.72 (1H, ddd, J = 9.7, 6.3, 5.7 Hz, 3-H), 2.90 (1H, dd, J = 14.0, 6.3 Hz, 7′-HA), 2.95 (1H, dd, J = 14.0, 5.7 Hz, 7′-HB), 3.15 (1H, ddd, J = 12.4, 5.1, 5.4 Hz, 6-HA), 3.54 (1H, ddd, J = 12.4, 6.5, 2.5 Hz, 6-HB), 3.82 (6H, s, 4′, 3″-OCH3), 3.83 (6H, s, 3′-OCH3), 3.85 (3H, s, 4″-OCH3), 4.20 (1H, ddd, J = 8.2, 5.1, 2.5 Hz, 5-H), 6.38 (2H, s, 2′-H), 6.49 (1H, d, J = 2.0 Hz, 2″-H), 6.58 (1H, dd, J = 8.1, 2.0 Hz, 6″-H), 6.76 (1H, d, J = 8.1 Hz, 5″-H). δC (100 MHz; CDCl3) 35.7 (C-7′), 38.6 (C-7″), 41.8 (C-4), 47.7 (C-3), 56.0, 56.1 (3″, 4″-OCH3), 56.3 (3′-OCH3), 61.0 (4′-OCH3), 63.2 (C-6), 83.9 (C-5), 106.5 (C-2′), 111.5 (C-5″), 112.2 (C-2″), 121.0 (C-6″), 130.3 (C-1″), 133.7 (C-1′), 137.2 (C-4′), 148.2 (C-4″), 149.3 (C-3″), 153.5 (C-3′), 177.7 (C-2). IR: νMAX (film)/cm−1; 3527 (br), 2938, 1761, 1590, 1514, 1237, 1126, 1026, 735. HRMS (ESI+) Found [M + Na]+ 469.1839; C24H30NaO8 requires 469.1833.
(3R*,4R*)-3-(3′-Methoxy-4′-benzyloxybenzyl)-4-(3″,4″-dimethoxybenzyl)-5-(hydroxymethyl)dihydrofuran-2(3H)-one (4ad). Using general procedure B: Amide 35ad (0.59 g, 1.1 mmol) in tBuOH/H2O and a reaction time of 7 days. The crude product was purified by column chromatography (1:1 hexanes, ethyl acetate) to give the title compound 4ad (0.30 g, 56%) as a cloudy oil. Rf = 0.27 (19:1 CH2Cl2, methanol). δH (400 MHz; CDCl3) 1.57 (1H, t, J = 6.5 Hz, 6-OH), 2.34–2.42 (1H, m, 4-H), 2.50 (1H, dd, J = 13.5, 8.0 Hz, 7″-HA), 2.59 (1H, dd, J = 13.5, 6.0 Hz, 7″-HB), 2.70 (1H, ddd, J = 9.7, 6.2, 5.6 Hz, 3-H), 2.90 (1H, dd, J = 14.1, 6.2 Hz, 7′-HA), 2.94 (1H, dd, J = 14.1, 5.6 Hz, 7′-HB), 3.10 (1H, ddd, J = 12.5, 6.5, 5.2 Hz, 6-HA), 3.48 (1H, ddd, J = 12.5, 6.5, 2.7 Hz, 6-HB), 3.80 (3H, s, 3″-OCH3), 3.86 (6H, s, 3′, 4″-OCH3), 4.18 (1H, ddd, J = 8.3, 5.2, 2.7 Hz, 5-H), 5.12 (2H, s, 7‴-H), 6.46 (1H, d, J = 2.0 Hz, 2″-H), 6.56 (1H, dd, J = 8.0, 2.0 Hz, 6″-H), 6.61 (1H, dd, J = 8.1, 2.0 Hz, 6′-H), 6.72 (1H, d, J = 2.0 Hz, 2′-H), 6.75 (1H, d, J = 8.0 Hz, 5″-H), 6.79 (1H, d, J = 8.1 Hz, 5′-H), 7.25–7.30 (1H, m, 4‴-H), 7.31–7.36 (2H, m, 3‴-H), 7.39–7.42 (2H, m, 2‴-H). δC (100 MHz; CDCl3) 35.1 (C-7′), 38.6 (C-7″), 41.7 (C-4), 47.6 (C-3), 56.0 (3″, 4″-OCH3), 56.2 (3′-OCH3), 63.3 (C-6), 71.3 (C-7‴), 84.0 (C-5), 111.5 (C-5″), 112.1 (C-2″), 113.3 (C-2′), 114.3 (C-5′), 121.0 (C-6″), 121.6 (C-6′), 127.4 (C-2‴), 128.0 (C-4‴), 128.7 (C-3‴), 130.3 (C-1″), 131.1 (C-1′), 137.2 (C-1‴), 147.2 (C-4′), 148.2 (C-4″), 149.3 (C-3″), 150.0 (C-3′), 177.8 (C-2). IR: νMAX (film)/cm−1; 3523 (br), 2935, 1761, 1514, 1261, 1025, 911, 730. HRMS (ESI+) Found [M + Na]+ 515.2023; C29H32NaO7 requires 515.2040.
(3R*,4R*,5S*)-4-(3″,4″-Dimethoxybenzyl)-3-(4′-hydroxy-3′-methoxybenzyl)-5-(hydroxymethyl)dihydrofuran-2(3H)-one (4ae). Using general procedure F: Benzyl ether 4ad (0.27 g, 0.55 mmol) gave the title compound 4ae (0.19 g, 88%) as a yellow solid. Rf = 0.43 (19:1 CH2Cl2, methanol). Melting point: 183–185 °C. δH (400 MHz; CDCl3) 1.63 (1H, t, J = 6.5 Hz, 6-OH), 2.34–2.43 (1H, m, 4-H), 2.53 (1H, dd, J = 13.8, 8.1 Hz, 7″-HA), 2.62 (1H, dd, J = 13.8, 6.1 Hz, 7″-HB), 2.69 (1H, dt, J = 9.5, 6.0 Hz, 3-H), 2.92 (2H, d, J = 6.0 Hz, 7′-H), 3.13 (1H, ddd, J = 12.5, 6.5, 5.3 Hz, 6-HA), 3.51 (1H, ddd, J = 12.5, 6.5, 2.5 Hz, 6-HB), 3.82 (3H, s, 3′-OCH3), 3.84 (3H, s, 3″-OCH3), 3.85 (3H, s, 4″-OCH3), 4.19 (1H, ddd, J = 8.0, 5.3, 2.5 Hz, 5-H), 5.52 (1H, s, 4′-OH), 6.46 (1H, d, J = 2.0 Hz, 2″-H), 6.57 (1H, dd, J = 8.1, 2.0 Hz, 6″-H), 6.63 (1H, dd, J = 8.0, 1.9 Hz, 6′-H), 6.66 (1H, d, J = 1.9 Hz, 2′-H), 6.76 (1H, d, J = 8.1 Hz, 5″-H), 6.83 (1H, d, J = 8.0 Hz, 5′-H). δC (100 MHz; CDCl3) 35.1 (C-7′), 38.6 (C-7″), 41.6 (C-4), 47.7 (C-3), 56.0, 56.1 (3′, 3″, 4″-OCH3), 63.4 (C-6), 84.1 (C-5), 111.5 (C-5″), 111.9 (C-2′), 112.1 (C-2″), 114.4 (C-5′), 121.0 (C-6″), 122.3 (C-6′), 129.7 (C-1′), 130.4 (C-1″), 144.7 (C-4′), 146.8 (C-3′), 148.2 (C-4″), 149.3 (C-3″), 177.8 (C-2). IR: νMAX (film)/cm−1; 3438 (br), 2937, 1755, 1514, 1236, 1155, 1025, 907, 723. HRMS (ESI+) Found [M + Na]+ 425.1564; C22H26NaO7 requires 425.1571.
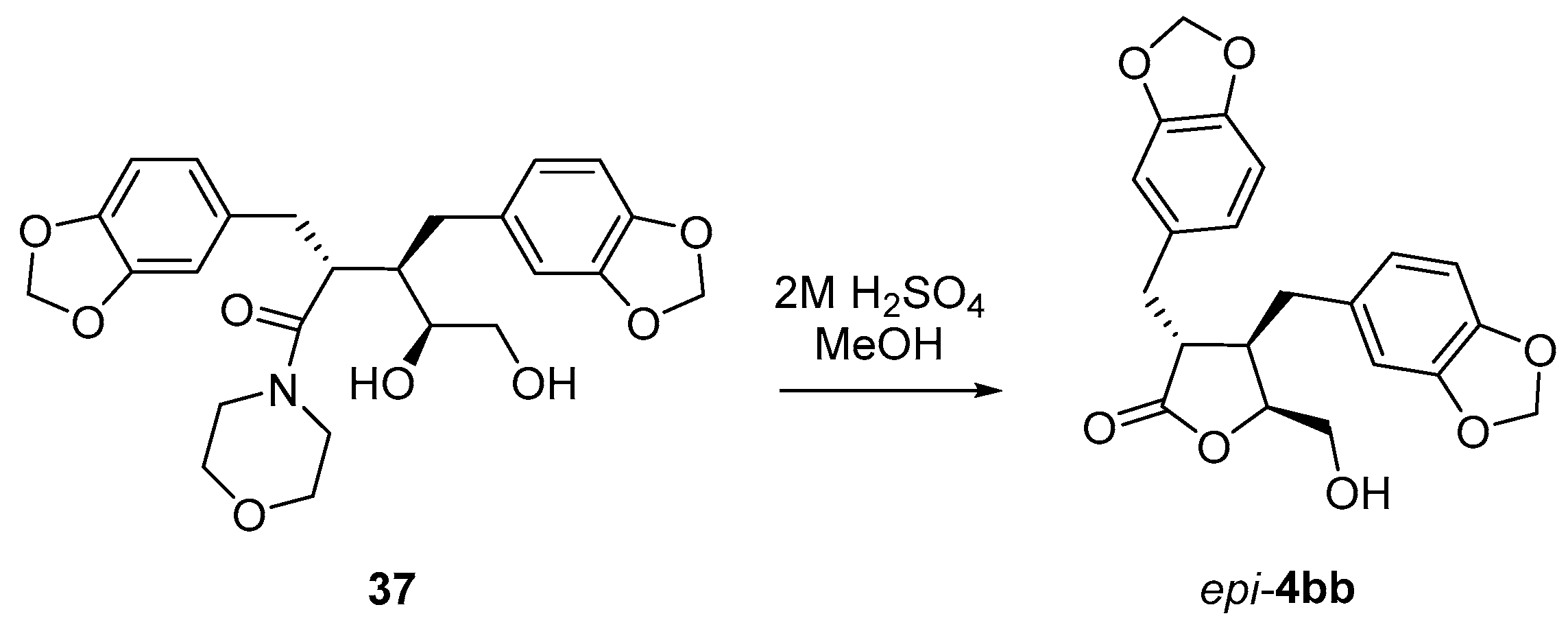
(3R*,4R*)-3,4-bis(3′,4′-Methylenedioxybenzyl)-5-(hydroxymethyl)dihydrofuran-2(3H)-one (4bb). Using general procedure B: Morpholine amide 35bb (0.322 g, 0.74 mmol) in tBuOH/H2O/THF and a reaction time of 5 days. The crude product was then purified by column chromatography (2:1 hexanes, ethyl acetate) to give the title compound 4bb (0.145 g, 51%) as a pale-yellow oil.
Rf = 0.59 (1:3 hexanes, ethyl acetate). δH (400 MHz; CDCl3) 1.72 (1H, br, 6-OH), 2.32–2.41 (1H, m, 4-H), 2.47 (1H, dd, J = 13.7, 8.1 Hz, 7″-HA), 2.56 (1H, dd, J = 13.7, 6.2 Hz, 7″-HB), 2.65 (1H, ddd, J = 9.0, 7.5, 5.3 Hz, 3-H), 2.85 (1H, dd, J = 14.0, 7.5 Hz, 7′-HA), 2.96 (1H, dd, J = 14.0, 5.3 Hz, 7′-HB), 3.15 (1H, dd, J = 12.6, 4.9 Hz, 6-HA), 3.54 (1H, dd, J = 12.6, 2.5 Hz, 6-HB), 4.18 (1H, ddd, J = 7.7, 4.9, 2.5 Hz, 5-H), 5.93–5.95 (4H, m, 2 × OCH2O), 6.45–6.49 (2H, m, 2″, 6″-H), 6.60 (1H, dd, J = 7.8, 1.7 Hz, 6′-H), 6.63 (1H, d, J = 1.7 Hz, 2′-H), 6.70 (1H, d, J = 7.8 Hz, 5″-H), 6.73 (1H, d, J = 7.8 Hz, 5′-H). δC (100 MHz; CDCl3) 35.4 (C-7′), 38.9 (C-7″), 41.8 (C-4), 47.6 (C-3), 63.3 (C-6), 83.9 (C-5), 101.1, 101.2 (2 × OCH2O), 108.4 (C-5′), 108.6 (C-5″), 109.2 (C-2″), 109.6 (C-2′), 121.9 (C-6″), 122.4 (C-6′), 131.5 (C-1′, 1″), 146.6 (C-4′, 4″), 148.0, 148.1 (C-3′, 3″), 177.6 (C-2). IR: νMAX (film)/cm−1; 3432 (br), 2922, 1760, 1503, 1490, 1444, 1247, 1038, 927, 811. HRMS (ESI+) Found [M + H]+ 385.1279; C21H21O7 requires 385.1282.
(3R*,4R*)-3-(3′,4′-Dimethoxybenzyl)-4-(3″,4″-methylenedioxybenzyl)-5-(hydroxymethyl)dihydrofuran-2(3H)-one (4ba). Using general procedure B: Morpholine amide 35ba (0.336 g, 0.74 mmol) in tBuOH/H2O/THF and a reaction time of 4 days. The crude product was then purified by column chromatography (1:3 hexanes, ethyl acetate) to give the title compound 4ba (0.103 g, 34%) as a pale yellow oil.
Rf = 0.48 (1:3 hexanes, ethyl acetate). δH (400 MHz; CDCl3) 1.68 (1H, t, J = 6.6 Hz, 6-OH), 2.33–2.42 (1H, m, 4-H), 2.48 (1H, dd, J = 13.7, 7.9 Hz, 7″-HA), 2.56 (1H, dd, J = 13.7, 6.3 Hz, 7″-HB), 2.68 (1H, ddd, J = 9.3, 6.9, 5.4 Hz, 3-H), 2.89 (1H, dd, J = 14.0, 6.9 Hz, 7′-HA), 2.96 (1H, dd, J = 14.0, 5.4 Hz, 7′-HB), 3.15 (1H, ddd, J = 12.5, 6.6, 5.2 Hz, 6-HA), 3.52 (1H, ddd, J = 12.5, 6.6, 2.6 Hz, 6-HB), 3.85 (3H, s, 3′-OCH3), 3.86 (3H, s, 4′-OCH3), 4.18 (1H, ddd, J = 7.9, 5.2, 2.6 Hz, 5-H), 5.93 (1H, d, J = 1.4 Hz, OCHAO), 5.94 (1H, d, J = 1.4 Hz, OCHBO), 6.44 (1H, d, J = 1.6 Hz, 2″-H), 6.47 (1H, dd, J = 7.8, 1.6 Hz, 6″-H), 6.67 (1H, d, J = 2.2 Hz, 2′-H), 6.68–6.72 (2H, m, 6′, 5″-H), 6.79 (1H, d, J = 8.0 Hz, 5′-H). δC (100 MHz; CDCl3) 35.2 (C-7′), 38.8 (C-7″), 41.7 (C-4), 47.6 (C-3), 56.0 (3′, 4′-OCH3), 63.4 (C-6), 83.8 (C-5), 101.3 (OCH2O), 108.5 (C-5′), 109.2 (C-2″), 111.3 (C-5″), 112.5 (C-2′), 121.6 (C-6′), 121.9 (C-6″), 130.3 (C-1′), 131.5 (C-1″), 146.7 (C-4″), 148.1 (C-4′, 3″), 149.2 (C-3′), 177.7 (C-2). IR: νMAX (film)/cm−1; 3472 (br), 2933, 1760, 1516, 1490, 1242, 1157, 1028, 925, 810, 730. HRMS (ESI+) Found [M + Na]+ 423.1423; C22H24NaO7 requires 423.1414.
(3R*,4R*)-3-(3′,4′,5′-Trimethoxybenzyl)-4-(3″,4″-methylenedioxybenzyl)-5-(hydroxymethyl)dihydrofuran-2(3H)-one (4bc). Using general procedure B: Morpholine amide 35bc (0.372 g, 0.77 mmol) in tBuOH/H2O/THF and a reaction time of 4 days. The crude product was then purified by column chromatography (1:3 hexanes, ethyl acetate) to give the title compound 4bc (0.084 g, 25%) as a pale-yellow oil.
Rf = 0.38 (1:3 hexanes, ethyl acetate). δH (400 MHz; CDCl3) 1.69 (1H, t, J = 6.6 Hz, 6-OH), 2.36–2.45 (1H, m, 4-H), 2.53 (1H, dd, J = 13.8, 7.6 Hz, 7″-HA), 2.59 (1H, dd, J = 13.8, 6.8 Hz, 7″-HB), 2.70 (1H, ddd, J = 9.5, 6.7, 5.4 Hz, 3-H), 2.87 (1H, dd, J = 14.0, 6.7 Hz, 7′-HA), 2.93 (1H, dd, J = 14.0, 5.4 Hz, 7′-HB), 3.22 (1H, ddd, J = 12.7, 6.6, 5.0 Hz, 6-HA), 3.58 (1H, ddd, J = 12.7, 6.6, 2.5 Hz, 6-HB), 3.82 (3H, s, 4′-OCH3), 3.84 (6H, s, 3′-OCH3), 3.85 (3H, s, 4″-OCH3), 4.19 (1H, ddd, J = 7.9, 5.0, 2.5 Hz, 5-H), 5.94 (1H, d, J = 1.4 Hz, OCHAO), 5.94 (1H, d, J = 1.4 Hz, OCHBO), 6.37 (2H, s, 2′-H), 6.46 (1H, d, J = 1.8 Hz, 2″-H), 6.48 (1H, dd, J = 7.9, 1.8 Hz, 6″-H), 6.70 (1H, d, J = 7.9 Hz, 5″-H). δC (100 MHz; CDCl3) 36.0 (C-7′), 38.8 (C-7″), 41.9 (C-4), 47.6 (C-3), 56.3 (3′-OCH3), 61.1 (4′-OCH3), 63.3 (C-6), 83.8 (C-5), 101.3 (OCH2O), 106.5 (C-2′), 108.5 (C-5″), 109.2 (C-2″), 121.9 (C-6″), 131.4 (C-1″), 133.6 (C-1′), 137.1 (C-4′), 146.7 (C-4″), 148.2 (C-3″), 153.5 (C-3′), 177.7 (C-2). IR: νMAX (film)/cm−1; 3475 (br), 2941, 1760, 1591, 1490, 1445, 1244, 1127, 1036, 926. HRMS (ESI+) Found [M + Na]+ 453.1519; C23H26NaO8 requires 453.1520.
(3R*,4R*)-3-(3′-Methoxy-4′-benzyloxybenzyl)-4-(3″,4″-methylenedioxybenzyl)-5-(hydroxymethyl)dihydrofuran-2(3H)-one (4bd). Using general procedure B: Morpholine amide 35bd (0.405 g, 0.77 mmol) in tBuOH/H2O/THF and a reaction time of 5 days. The crude product was then purified by column chromatography (1:3 hexanes, ethyl acetate) to give the title compound 4bd (0.205 g, 56%) as a pale-yellow oil.
Rf = 0.58 (1:3 hexanes, ethyl acetate). δH (400 MHz; CDCl3) 1.64 (1H, t, J = 6.6 Hz, 6-OH), 2.31–2.40 (1H, m, 4-H), 2.46 (1H, dd, J = 13.7, 7.9 Hz, 7″-HA), 2.53 (1H, dd, J = 13.7, 6.3 Hz, 7″-HB), 2.67 (1H, ddd, J = 9.2, 7.2, 5.3 Hz, 3-H), 2.87 (1H, dd, J = 14.0, 7.2 Hz, 7′-HA), 2.95 (1H, dd, J = 14.0, 5.3 Hz, 7′-HB), 3.13 (1H, ddd, J = 12.6, 6.6, 5.1 Hz, 6-HA), 3.48 (1H, ddd, J = 12.6, 6.6, 2.6 Hz, 6-HB), 3.86 (3H, s, 3′-OCH3), 4.17 (1H, ddd, J = 7.8, 5.1, 2.6 Hz, 5-H), 5.13 (2H, s, 7‴-H), 5.93 (1H, d, J = 1.4 Hz, OCHAO), 5.94 (1H, d, J = 1.4 Hz, OCHBO), 6.43 (1H, d, J = 1.6 Hz, 2″-H), 6.45 (1H, dd, J = 7.9, 1.6 Hz, 6″-H), 6.64 (1H, dd, J = 8.2, 2.0 Hz, 6′-H), 6.68–6.70 (2H, m, 2′, 5″-H), 6.81 (1H, d, J = 8.2 Hz, 5′-H), 7.27–7.30 (1H, m, 4‴-H), 7.32–7.36 (2H, m, 3‴-H), 7.40–7.44 (2H, m, 2‴-H). δC (100 MHz; CDCl3) 35.3 (C-7′), 38.8 (C-7″), 41.8 (C-4), 47.6 (C-3), 56.1 (3′-OCH3), 63.4 (C-6), 71.3 (C-7‴), 83.8 (C-5), 101.3 (OCH2O), 108.5 (C-5″), 109.2 (C-2″), 113.0 (C-2′), 114.4 (C-5′), 121.5 (C-6′), 121.9 (C-6″), 127.5 (C-2‴), 128.0 (C-4‴), 128.7 (C-3‴), 131.0 (C-1′), 131.5 (C-1″), 137.3 (C-1‴), 146.7 (C-4″), 147.2 (C-4′), 148.1 (C-3″), 150.0 (C-3′), 177.7 (C-2). IR: νMAX (film)/cm−1; 3471 (br), 2940, 1743, 1504, 1490, 1366, 1230, 1036, 926, 735. HRMS (ESI+) Found [M + Na]+ 499.1729; C28H28NaO7 requires 499.1727.
(3R*,4R*,5S*)-4-(3″,4″-Methylenedioxybenzyl)-3-(4′-hydroxy-3′-methoxybenzyl)-5-(hydroxymethyl)dihydrofuran-2(3H)-one (4be). Using general procedure F: Benzyl ether 4bd (0.02 g, 0.04 mmol) and a reaction time of 1 h. The crude product was then purified by column chromatography (1:3 hexanes, ethyl acetate) to give the title compound 4be (0.017 g, quant.) as a colourless oil. Rf = 0.52 (1:3 hexanes, ethyl acetate). δH (400 MHz; CDCl3) 1.74 (1H, br, 6-OH), 2.33–2.42 (1H, m, 4-H), 2.48 (1H, dd, J = 13.7, 8.0 Hz, 7″-HA), 2.57 (1H, dd, J = 13.7, 6.2 Hz, 7″-HB), 2.67 (1H, ddd, J = 9.4, 6.9, 5.5 Hz, 3-H), 2.88 (1H, dd, J = 14.0, 6.9 Hz, 7′-HA), 2.94 (1H, dd, J = 14.0, 5.5 Hz, 7′-HB), 3.15 (1H, br d, J = 12.6 Hz, 6-HA), 3.52 (1H, br d, J = 12.6 Hz, 6-HB), 3.86 (3H, s, 3′-OCH3), 4.18 (1H, ddd, J = 8.0, 5.0, 2.5 Hz, 5-H), 5.54 (1H, s, 4′-OH), 5.93 (1H, d, J = 1.4 Hz, OCHAO), 5.94 (1H, d, J = 1.4 Hz, OCHBO), 6.45 (1H, d, J = 1.9 Hz, 2″-H), 6.47 (1H, dd, J = 7.7, 1.9 Hz, 6″-H), 6.63 (1H, dd, J = 8.0, 1.9 Hz, 6′-H), 6.67 (1H, d, J = 1.9 Hz, 2′-H), 6.70 (1H, d, J = 7.7 Hz, 5″-H), 6.84 (1H, d, J = 8.0 Hz, 5′-H). δC (100 MHz; CDCl3) 35.3 (C-7′), 38.8 (C-7″), 41.7 (C-4), 47.7 (C-3), 56.1 (3′-OCH3), 63.4 (C-6), 83.9 (C-5), 101.3 (OCH2O), 108.6 (C-5″), 109.2 (C-2″), 111.8 (C-2′), 114.5 (C-5′), 121.9 (C-6″), 122.3 (C-6′), 129.6 (C-1′), 131.5 (C-1″), 144.7 (C-4′), 146.7 (C-3′), 146.8 (C-4″), 148.1 (C-3″), 177.8 (C-2). IR: νMAX (film)/cm −1; 3449 (br), 2933, 1754, 1516, 1490, 1246, 1036, 926, 812. HRMS (ESI+) Found [M + Na]+ 409.1246; C21H22NaO7 requires 409.1258.
(±)-Arcitin (
1aa). Using general procedure C: Lactone
4aa (0.16 g, 0.39 mmol) and a reaction time of 2 h to give triol
38aa (0.17 g, quant.) as a colourless oil. Then using general procedure D: Triol
38aa (0.16 g, 0.37 mmol) and a reaction time of 2.5 h to give lactol
39aa (0.14 g, 97%) which was used without further purification. Then using general procedure E: Lactol
39aa (0.054 g, 0.14 mmol) and a reaction time of 3 h. The crude product was purified by column chromatography (1:1, hexanes, ethyl acetate) to give the title compound
1aa (0.05 g, 88%) as a pale yellow amorphous solid. R
f = 0.45 (19:1, CH
2Cl
2, methanol). Melting point: 114–116 °C [lit. [
49] 113 °C]. δ
H (400 MHz; CDCl
3) 2.45–2.68 (4H, m, 8, 7′, 8′-H), 2.92 (1H, dd,
J = 14.3, 6.8 Hz, 7-H
A), 2.97 (1H, dd,
J = 14.3, 5.5 Hz, 7-H
B), 3.82 (3H, s, 3′-OCH
3), 3.83 (3H, s, 3-OCH
3), 3.85–3.90 (7H, m, 4, 4′-OCH
3, 9′-H
A), 4.13 (1H, t,
J = 7.0 Hz, 9′-H
B), 6.49 (1H, d,
J = 1.9 Hz, 2′-H), 6.55 (1H, dd,
J = 8.1, 1.9 Hz, 6′-H), 6.66 (1H, dd,
J = 8.1, 1.9 Hz, 6-H), 6.69 (1H, d,
J = 1.9 Hz, 2-H), 6.75 (1H, d,
J = 8.1 Hz, 5-H), 6.77 (1H, d,
J = 8.1 Hz, 5′-H). δ
C (100 MHz; CDCl
3) 34.5 (C-7), 38.2 (C-7′), 41.1 (C-8′), 46.6 (C-8), 55.8, 55.9 (3, 4, 3′, 4′-OCH
3), 71.2 (C-9′), 111.1 (C-5), 111.4 (C-5′), 111.9 (C-2′), 112.4 (C-2), 120.6 (C-6′), 121.4 (C-6), 130.2 (C-1), 130.5 (C-1′), 147.9 (C-4′), 148.0 (C-4), 149.1 (C-3, 3′), 178.7 (C-9). IR: ν
MAX (film)/cm
−1; 2956, 1753, 1588, 1513, 1257, 1236, 1153, 1137, 1019, 825, 764. HRMS (ESI
+) Found [M + H]
+ 387.1806; C
22H
27O
6 requires 387.1802. Values are in agreement with literature data [
50].
(±)-Bursehernin (
1a). Using general procedure C: Lactone
4ab (0.114 g, 0.28 mmol) and a reaction time of 30 min to give triol
38ab (0.111 g, 97%) as a cloudy oil. Then using general procedure D: Triol
38ab (0.111 g, 0.27 mmol) and a reaction time of 1 h to give lactol
39ab (0.093 g, 91%) which was used without further purification. Then using general procedure E: Lactol
39ab (0.093 g, 0.25 mmol) and a reaction time of 2 h. The crude product was purified by column chromatography (1:1, hexanes, ethyl acetate) to give the title compound
1ab (0.06 g, 65%) as a pale-yellow oil. R
f = 0.66 (19:1, CH
2Cl
2, methanol). δ
H (400 MHz; CDCl
3) 2.41–2.62 (4H, m, 8, 7′, 8′-H), 2.88 (1H, dd,
J = 14.0, 6.9 Hz, 7-H
A), 2.96 (1H, dd,
J = 14.0, 5.1 Hz, 7-H
B), 3.82 (3H, s, 3-OCH
3), 3.83–3.86 (4H, m, 4-OCH
3, 9′-H
A), 4.10 (1H, dd,
J = 9.1, 6.9 Hz, 9′-H
B), 5.91 (1H, d,
J = 1.4 Hz, OCH
AO), 5.92 (1H, d,
J = 1.4 Hz, OCH
BO), 6.42 (1H, d,
J = 1.5 Hz, 2′-H), 6.44 (1H, dd,
J = 7.9, 1.5 Hz, 6′-H), 6.66 (1H, d,
J = 1.9 Hz, 2-H), 6.67–6.70 (2H, m, 6, 5′-H), 6.78 (1H, d,
J = 8.0 Hz, 5-H). δ
C (100 MHz; CDCl
3) 34.7 (C-7), 38.4 (C-7′), 41.2 (C-8′), 46.6 (C-8), 55.9 (3, 4-OCH
3), 71.2 (C-9′), 101.1 (OCH
2O), 108.4 (C-5′), 108.8 (C-2′), 111.2 (C-5), 112.3 (C-2), 121.4 (C-6), 121.6 (C-6′), 130.2 (C-1), 131.7 (C-1′), 146.4 (C-4′), 148.0 (C-3′), 148.1 (C-4), 149.2 (C-3), 178.7 (C-9). IR: ν
MAX (film)/cm
−1; 2907, 1764, 1514, 1489, 1442, 1240, 1025, 923, 808, 730. HRMS (ESI
+) Found [M + Na]
+ 393.1317; C
21H
22NaO
6 requires 393.1309. Values are in agreement with literature data [
51].
(±)-4-O-Methyl traxillagenin (
1ac). Using general procedure C: Lactone
4ac (0.119 g, 0.27 mmol) and a reaction time of 45 min to give triol
38ac (0.11 g, 90%) as a cloudy oil. The using general procedure D: Triol
38ac (0.11 g, 0.24 mmol) and a reaction time of 15 min. The crude product was purified by column chromatography (1:2 hexanes, ethyl acetate) to give lactol
39ac (0.06 g, 60%) as a colourless oil. Then using general procedure E: Lactol
39ac (0.06 g, 0.15 mmol) and a reaction time of 3 h. The crude product purified by column chromatography (1:1, hexanes, ethyl acetate) to give the title compound
1ac (0.044 g, 73%) as a white solid. R
f = 0.61 (19:1, CH
2Cl
2, methanol). Melting point: 126 °C. δ
H (400 MHz; CDCl
3) 2.44–2.66 (4H, m, 8, 7′, 8′-H), 2.91 (1H, dd,
J = 14.1, 6.6 Hz, 7-H
A), 2.98 (1H, dd,
J = 14.1, 5.4 Hz, 7-H
B), 3.79 (6H, s, 3′-OCH
3), 3.80 (6H, s, 4′-OCH
3), 3.83 (3H, s, 3-OCH
3), 3.84 (3H, s, 4-OCH
3), 3.87 (1H, dd,
J = 9.2, 7.3 Hz, 9′-H
A), 4.14 (1H, dd,
J = 9.2, 7.0 Hz, 9′-H
B), 6.19 (2H, s, 2′-H), 6.63 (1H, dd,
J = 8.0, 2.0 Hz, 6-H), 6.70 (1H, d,
J = 2.0 Hz, 2-H), 6.75 (1H, d,
J = 8.0 Hz, 5-H). δ
C (100 MHz; CDCl
3) 34.6 (C-7), 39.0 (C-7′), 41.2 (C-8′), 46.7 (C-8), 56.0 (3, 4-OCH
3), 56.2 (3′-OCH
3), 60.9 (4′-OCH
3), 71.3 (C-9′), 105.7 (C-2′), 111.2 (C-5), 112.6 (C-2), 121.4 (C-6), 130.3 (C-1), 133.8 (C-1′), 137.0 (C-4′), 148.1 (C-4), 149.2 (C-3), 153.5 (C-3′), 178.7 (C-9). IR: ν
MAX (film)/cm
−1; 2938, 1764, 1590, 1509, 1460, 1237, 1123, 1014, 731. HRMS (ESI
+) Found [M + Na]
+ 439.1716; C
23H
28NaO
7 requires 439.1727. Values are in agreement with literature data [
52].
(±)-4′-O-Benzyl buplerol (1ad). Using general procedure C: Lactone 4ad (0.505 g, 1.02 mmol) and a reaction time of 3 h to give the triol 38ad (0.472 g, 93%) as a cloudy oil. Then using general procedure D: Triol 38ad (0.472 g, 0.95 mmol) and a reaction time of 30 min to give lactol 39ad (0.416 g, 94%) as a white solid which was used without further purification. Then using general procedure E: Lactol 39ad (0.416 g, 0.90 mmol) and a reaction time of 1.5 h. The crude product was purified by column chromatography (1:1, hexanes, ethyl acetate) to give the title compound 1ad (0.374 g, 90%) as a pale-yellow oil. Rf = 0.52 (1:1, hexanes, ethyl acetate). δH (400 MHz; CDCl3) 2.42–2.66 (4H, m, 8, 7′, 8′-H), 2.91 (1H, dd, J = 14.1, 6.2 Hz, 7-HA), 2.95 (1H, dd, J = 14.1, 5.7 Hz, 7-HB), 3.827, 3.829 (6H, 2 × s, 3, 3′-OCH3), 3.85 (3H, s, 4-OCH3), 3.83–3.88 (1H, m, 9′-HA), 4.11 (1H, dd, J = 8.7, 7.0 Hz, 9′-HB), 5.12 (2H, s, Ph-CH2), 6.48 (1H, dd, J = 8.0, 2.0 Hz, 6′-H), 6.51 (1H, d, J = 2.0 Hz, 2′-H), 6.64 (1H, dd, J = 8.2, 2.0 Hz, 6-H), 6.68 (1H, d, J = 2.0 Hz, 2-H), 6.76 (1H, d, J = 8.2 Hz, 5-H), 6.77 (1H, d, J = 8.0 Hz, 5′-H), 7.27–7.32 (1H, m, Ph-p-H), 7.33–7.38 (2H, m, Ph-m-H), 7.40–7.44 (2H, m, Ph-o-H). δC (100 MHz; CDCl3) 34.6 (C-7), 38.3 (C-7′), 41.2 (C-8′), 46.7 (C-8), 56.0 (3, 3′-OCH3), 56.1 (4-OCH3), 71.3, 71.4 (C-9′, Ph-CH2), 111.3 (C-5), 112.5 (C-2), 112.6 (C-5′), 114.5 (C-5′), 120.7 (C-6′), 121.5 (C-6), 127.4 (Ph-o-C), 128.0 (Ph-p-C), 128.7 (Ph-m-C), 130.3 (C-1), 131.3 (C-1′), 137.3 (Ph-i-C), 147.2 (C-4′), 148.1 (C-4), 149.2 (C-3), 149.9 (C-3′), 178.8 (C-9). IR: νMAX (film)/cm−1; 2935, 1763, 1512, 1260, 1233, 1140, 1014, 736, 697. HRMS (ESI+) Found [M + Na]+ 485.1934; C28H30NaO6 requires 485.1935.
(±)-Buplerol (
1ae). Using general procedure F: Lactone
1ad (0.336 g, 0.73 mmol) and a reaction time of 3.5 h to give the title compound
1ae (0.271 g, quant.) as a white solid. R
f = 0.33 (1:1, hexanes, ethyl acetate). Melting point: 101–103 °C. δ
H (400 MHz; CDCl
3) 2.42–2.66 (4H, m, 8, 7′, 8′-H), 2.90 (1H, dd,
J = 14.1, 6.8 Hz, 7-H
A), 2.97 (1H, dd,
J = 14.1, 5.3 Hz, 7-H
B), 3.81 (3H, s, 3-OCH
3), 3.83 (3H, s, 3′-OCH
3), 3.86 (4H, m, 4-OCH
3), 3.87 (1H, dd,
J = 8.9, 7.1 Hz, 9′-H
A), 4.13 (1H, dd,
J = 9.3, 7.1 Hz, 9′-H
B), 5.51 (1H, s, 4′-OH), 6.43 (1H, d,
J = 1.9 Hz, 2′-H), 6.52 (1H, dd,
J = 8.0, 1.9 Hz, 6′-H), 6.64–6.67 (2H, m, 2, 6-H), 6.77 (1H, d,
J = 8.6 Hz, 5-H), 6.80 (1H, d,
J = 8.0 Hz, 5′-H). δ
C (100 MHz; CDCl
3) 34.7 (C-7), 38.5 (C-7′), 41.3 (C-8′), 46.7 (C-8), 55.9, 56.0 (3, 3′, 4-OCH
3), 71.4 (C-9′), 111.1, 111.2 (C-5, 5′), 112.5 (C-2), 114.6 (C-2′), 121.5 (C-6, 6′), 129.9 (C-1′), 130.4 (C-1), 144.6 (C-4′), 146.7 (C-3′), 148.1 (C-4), 149.2 (C-3), 178.9 (C-9). IR: ν
MAX (film)/cm
−1; 3417, 2938, 1760, 1513, 1236, 1148, 1023, 812, 795. HRMS (ESI
+) Found [M + Na]
+ 395.1462; C
21H
24NaO
6 requires 395.1465. Values are in agreement with literature data [
53].
(±)-Kusunokinin (
1ba). Using general procedure C: Lactone
4ba (0.082 g, 0.20 mmol) and a reaction time of 1 h to give the triol
38ba (0.083 g, quant.) as a cloudy oil. Then using general procedure D: Triol
38ba (0.083 g, 0.20 mmol) and a reaction time of 15 min to give lactol
39ba (0.064 g, 84%) which was used without further purification. Then using general procedure E: Lactol
39ba (0.056 g, 0.15 mmol) and a reaction time of 1 h. The crude product was purified by column chromatography (2:1, hexanes, ethyl acetate) to give the title compound
1ba (0.051 g, 91%) as a colourless oil. R
f = 0.48 (1:1, hexanes, ethyl acetate). δ
H (400 MHz; CDCl
3) 2.44–2.65 (4H, m, 8, 7′, 8′-H), 2.84 (1H, dd,
J = 14.1, 7.0 Hz, 7-H
A), 2.95 (1H, dd,
J = 14.1, 5.1 Hz, 7-H
B), 3.82 (3H, s, 3′-OCH
3), 3.85 (3H, s, 4′-OCH
3), 3.87 (1H, dd,
J = 9.2, 7.2 Hz, 9′-H
A), 4.14 (1H, dd,
J = 9.2, 7.0 Hz, 9′-H
B), 5.92 (1H, d,
J = 1.4 Hz, OCH
AO), 5.93 (1H, d,
J = 1.4 Hz, OCH
BO), 6.48 (1H, d,
J = 2.0 Hz, 2′-H), 6.55–6.60 (3H, m, 2, 6, 6′-H), 6.71 (1H, d,
J = 7.7 Hz, 5-H), 6.76 (1H, d,
J = 8.2 Hz, 5′-H). δ
C (100 MHz; CDCl
3) 34.9 (C-7), 38.4 (C-7′), 41.3 (C-8′), 46.6 (C-8), 55.9 (3′-OCH
3), 56.0 (4′-OCH
3), 71.4 (C-9′), 101.1 (OCH
2O), 108.3 (C-5), 109.6 (C-2), 111.4 (C-5′), 111.8 (C-2′), 120.8 (C-6′), 122.4 (C-6), 130.6 (C-1′), 131.5 (C-1), 146.6 (C-4), 148.0 (C-3, 4′), 149.2 (C-3′), 178.6 (C-9). IR: ν
MAX (film)/cm
−1; 2908, 1764, 1515, 1489, 1442, 1242, 1024, 912, 809, 729. HRMS (ESI
+) Found [M + Na]
+ 393.1301; C
21H
22NaO
6 requires 393.1309. Values are in agreement with literature data [
50].
(±)-Hinokinin (
1bb). Using general procedure C: Lactone
4bb (0.12 g, 0.31 mmol) and a reaction time of 30 min to give the triol
38bb (0.12 g, quant.) as a cloudy oil. Then using general procedure D: Triol
38bb (0.121 g, 0.31 mmol) and a reaction time of 10 min to give lactol
39bb (0.096 g, 86%) which was used without further purification. Then using general procedure E: Lactol
39bb (0.089 g, 0.25 mmol) and a reaction time of 1 h. The crude product was purified by column chromatography (1:1, hexanes, ethyl acetate) to give the title compound
1bb (0.08 g, 90%) as a pale-yellow oil. R
f = 0.73 (1:1, hexanes, ethyl acetate). δ
H (400 MHz; CDCl
3) 2.41–2.62 (4H, m, 8, 7′, 8′-H), 2.83 (1H, dd,
J = 14.1, 7.2 Hz, 7-H
A), 2.98 (1H, dd,
J = 14.1, 5.0 Hz, 7-H
B), 3.85 (1H, dd,
J = 9.2, 7.1 Hz, 9′-H
A), 4.12 (1H, dd,
J = 9.2, 6.9 Hz, 9′-H
B), 5.91–5.94 (4H, m, 2 × OCH
2O), 6.44–6.47 (2H, m, 2′, 6′-H), 6.59 (1H, dd,
J = 7.9, 1.8 Hz, 6-H), 6.62 (1H, d,
J = 1.8 Hz, 2-H), 6.69 (1H, d,
J = 8.4 Hz, 5′-H), 6.72 (1H, d,
J = 7.9 Hz, 5-H). δ
C (100 MHz; CDCl
3) 34.9 (C-7), 38.4 (C-7′), 41.4 (C-8′), 46.6 (C-8), 71.2 (C-9′), 101.1 (2 × OCH
2O), 108.4 (C-5, 5′), 108.9 (C-2′), 109.5 (C-2), 121.6 (C-6′), 122.3 (C-6), 131.5 (C-1), 131.7 (C-1′), 146.4 (C-4), 146.6 (C-4′), 148.0 (C-3, 3′), 178.5 (C-9). IR: ν
MAX (film)/cm
−1; 2901, 1764, 1488, 1441, 1242, 1015, 924, 808, 728. HRMS (ESI
+) Found [M + Na]
+ 377.0986; C
20H
18NaO
6 requires 377.0996. Values are in agreement with literature data [
54].
(±)-Isoyatein (
1bc). Using general procedure C: Lactone
4bc (0.076 g, 0.18 mmol) and a reaction time of 1 h to give the triol
38bc (0.077 g, >99%) as a cloudy oil. Then using general procedure D: Triol
38bc (0.077 g, 0.18 mmol) and a reaction time of 1 h to give lactol
39bc (0.057 g, 80%) which was used without further purification. Then using general procedure E: Lactol
39bc (0.05 g, 0.12 mmol) and a reaction time of 3 h. The crude product was purified by column chromatography (1:1, hexanes, ethyl acetate) to give the title compound
1bc (0.8 mg, 16%) as a pale-yellow oil. R
f = 0.55 (1:1, hexanes, ethyl acetate). δ
H (400 MHz; CDCl
3) 2.46–2.64 (4H, m, 8, 7′, 8′-H), 2.86 (1H, dd,
J = 14.1, 7.0 Hz, 7-H
A), 2.98 (1H, dd,
J = 14.1, 5.1 Hz, 7-H
B), 3.81 (6H, s, 3′-OCH
3), 3.82 (3H, s, 4′-OCH
3), 3.89 (1H, dd,
J = 9.2, 7.0 Hz, 9′-H
A), 4.19 (1H, dd,
J = 9.2, 6.8 Hz, 9′-H
B), 5.93 (1H, d,
J = 1.5 Hz, OCH
AO), 5.94 (1H, d,
J = 1.5 Hz, OCH
BO), 6.20 (2H, s, 2′-H), 6.58 (1H, dd,
J = 7.9, 1.8 Hz, 6-H), 6.61 (1H, d,
J = 1.8 Hz, 2-H), 6.71 (1H, d,
J = 7.9 Hz, 5-H). δ
C (100 MHz; CDCl
3) 34.9 (C-7), 39.2 (C-7′), 41.4 (C-8′), 46.6 (C-8), 56.2 (3′-OCH
3), 61.0 (4′-OCH
3), 71.4 (C-9′), 101.2 (OCH
2O), 105.7 (C-2′), 108.3 (C-5), 109.6 (C-2), 122.4 (C-6), 131.5 (C-1), 133.8 (C-1′), 137.0 (C-4′), 146.7 (C-4), 148.1 (C-3), 153.5 (C-3′), 178.5 (C-9). IR: ν
MAX (film)/cm
−1; 2938, 1763, 1590, 1489, 1443, 1241, 1122, 1011, 927, 813, 732. HRMS (ESI
+) Found [M + Na]
+ 423.1400; C
22H
24NaO
7 requires 423.1414. Values are in agreement with literature data [
55].
(±)-4′-O-Benzyl haplomyrfolin (1bd). Using general procedure C: Lactone 4bd (0.18 g, 0.38 mmol) and a reaction time of 20 min to give the triol 38bd (0.18 g, quant.) as a cloudy oil. Then using general procedure D: Triol 38bd (0.18 g, 0.38 mmol) and a reaction time of 20 min to give lactol 39bd (0.13 g, 76%) as a white solid which was used without further purification. Then using general procedure E: Lactol 39bd (0.13 g, 0.28 mmol) and a reaction time of 2 h. The crude product was purified by column chromatography (3:1, hexanes, ethyl acetate) to give the title compound 1bd (0.12 g, 94%) as a colourless oil. Rf = 0.65 (1:1, hexanes, ethyl acetate). δH (400 MHz; CDCl3) 2.43–2.64 (4H, m, 8, 7′, 8′-H), 2.84 (1H, dd, J = 14.1, 7.0 Hz, 7-HA), 2.94 (1H, dd, J = 14.1, 5.1 Hz, 7-HB), 3.83 (3H, s, 3′-OCH3), 3.87 (1H, dd, J = 9.1, 7.2 Hz, 9′-HA), 4.14 (1H, dd, J = 9.1, 7.0 Hz, 9′-HB), 5.12 (2H, s, 7″-H), 5.91 (1H, d, J = 1.4 Hz, OCHAO), 5.93 (1H, d, J = 1.4 Hz, OCHBO), 6.49–6.52 (2H, m, 2′, 6′-H), 6.57 (1H, dd, J = 7.9, 1.8 Hz, 6-H), 6.59 (1H, d, J = 1.8 Hz, 2-H), 6.70 (1H, d, J = 7.9 Hz, 5-H), 6.78 (1H, d, J = 8.5 Hz, 5′-H), 7.27–7.32 (1H, m, 4″-H), 7.33–7.38 (2H, m, 3″-H), 7.41–7.45 (2H, m, 2″-H). δC (100 MHz; CDCl3) 34.8 (C-7), 38.4 (C-7′), 41.3 (C-8′), 46.5 (C-8), 56.0 (3′-OCH3), 71.2 (C-7″), 71.3 (C-9′), 101.1 (OCH2O), 108.3 (C-5), 109.6 (C-2), 112.4 (C-2′), 114.4 (C-5′), 120.7 (C-6′), 122.4 (C-6), 127.4 (C-2″), 128.0 (C-4″), 128.6 (C-3″), 131.2 (C-1), 131.5 (C-1′), 137.2 (C-1″), 146.6 (C-4), 147.1 (C-4′), 148.0 (C-3), 149.9 (C-3′), 178.6 (C-9). IR: νMAX (film)/cm−1; 2907, 1765, 1504, 1489, 1443, 1244, 1140, 1034, 911, 809, 730. HRMS (ESI+) Found [M + Na]+ 469.1612; C27H26NaO6 requires 469.1622.
(±)-Haplomyrfolin (
1be). Using general procedure F: Lactone
1bd (0.119 g, 0.27 mmol) and a reaction time of 1.5 h. The crude product was purified by column chromatography (1:1 hexanes, ethyl acetate) to give the title compound
1be (0.086 g, 91%) as a colourless oil. R
f = 0.47 (1:1 hexanes, ethyl acetate). δ
H (400 MHz; CDCl
3) 2.43–2.63 (4H, m, 8, 7′, 8′-H), 2.84 (1H, dd,
J = 14.1, 7.0 Hz, 7-H
A), 2.95 (1H, dd,
J = 14.1, 5.2 Hz, 7-H
B), 3.83 (3H, s, 3′-OCH
3), 3.86 (1H, dd,
J = 9.1, 7.2 Hz, 9′-H
A), 4.13 (1H, dd,
J = 9.1, 7.0 Hz, 9′-H
B), 5.63 (1H, s, 4′-OH), 5.91 (1H, d,
J = 1.4 Hz, OCH
AO), 5.92 (1H, d,
J = 1.4 Hz, OCH
BO), 6.46 (1H, d,
J = 1.9 Hz, 2′-H), 6.51 (1H, dd,
J = 8.0, 1.9 Hz, 6′-H), 6.58 (1H, dd,
J = 7.8, 1.7 Hz, 6-H), 6.60 (1H, d,
J = 1.7 Hz, 2-H), 6.70 (1H, d,
J = 7.8 Hz, 5-H), 6.80 (1H, d,
J = 8.0 Hz, 5′-H). δ
C (100 MHz; CDCl
3) 34.8 (C-7), 38.3 (C-7′), 41.4 (C-8′), 46.5 (C-8), 55.9 (3′-OCH
3), 71.3 (C-9′), 101.1 (OCH
2O), 108.3 (C-5), 109.6 (C-2), 111.2 (C-2′), 114.6 (C-5′), 121.4 (C-6′), 122.4 (C-6), 129.9 (C-1′), 131.5 (C-1), 144.5 (C-4′), 146.5 (C-4), 146.7 (C-3′), 147.9 (C-3), 178.7 (C-9). IR: ν
MAX (film)/cm
−1; 3468, 2921, 1762, 1515, 1489, 1443, 1243, 1035, 907, 725. HRMS (ESI
+) Found [M + Na]
+ 379.1151; C
20H
20NaO
6 requires 379.1152. Values are in agreement with literature data [
56].


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







