
Computational Characterization of Bidentate P-Donor Ligands: Direct Comparison to Tolman’s Electronic Parameters


1
Department of Inorganic Chemistry and MTA-PTE Research Group for Selective Chemical Syntheses, University of Pécs, H-7622 Pécs, Hungary
2
Department of Inorganic Chemistry, University of Pécs, H-7622 Pécs, Hungary
*
Author to whom correspondence should be addressed.
†
Current address: Ifjúság útja 6., H-7624 Hungary.
‡
These authors contributed equally to this work.
Molecules 2018, 23(12), 3176; https://doi.org/10.3390/molecules23123176
Submission received: 30 September 2018
/
Revised: 22 November 2018
/
Accepted: 28 November 2018
/
Published: 1 December 2018
(This article belongs to the Special Issue Organophosphorus Chemistry 2018)
Abstract
:The applicability of two types of transition-metal carbonyl complexes as appropriate candidates for computationally derived Tolman’s ligand electronic parameters were examined with density functional theory (DFT) calculations employing the B97D3 functional. Both Pd(0)L2(CO) and HRh(I)L2(CO) complexes correlated well with the experimental Tolman Electronic Parameter scale. For direct comparison of the electronic effects of diphosphines with those of monophosphines, the palladium-containing system is recommended. The trans influence of various phosphines did not show a major difference, but the decrease of the H-Rh-P angle from linear can cause a significant change.
1. Introduction
Phosphines, phosphites, and other P-donor ligands are of vital importance in the wide range of reactions catalyzed by transition-metal (TM) compounds [1,2,3]. Changing the coordinated ligands is an effective strategy for modifying the properties of transition metal containing catalysts. The chemical properties of transition-metal complexes, such as their activity in various catalytic reactions, are principally determined by the steric and electronic properties of the ligands bound to the metal center [4,5,6,7,8]. Their structural variation opens the possibility to tune catalytic activity, as well as chemo-, regio-, and enantioselectivity [9,10]. It has long been known that variation of the substituents can cause changes upon coordination in the behavior of the ligands as well as their TM complexes. Information about the nature of the transition metal–phosphorus bond and its influence on other bonds in the molecule is crucial for the characterization of catalytically active compounds and for the fine-tuning their properties in order to develop more efficient catalysts.
In the 1960s, Strohmeier et al. investigated the -donor ability and -acceptor strength of various ligand classes (nitriles, isonitriles, sulfoxides, phosphines). The phosphines were further divided into four categories designated from I to IV, classified by the frequency regions of 1927–1996, 1946–1982, 2045–2101, and 2060–2110 cm-1 for complexes C5H5Mn(CO)2L, C5H5V(CO)3L, Fe(CO)4L, and Ni(CO)3L, respectively, and sorted by wavenumber into increasing order [11]. These classes constitute the basis for Tolman’s famous formula.
In the 1970s, Tolman reported the A carbonyl-stretching frequencies of NiL(CO)3 complexes, with monodentate P-donor ligands (L = PRRR) [2,12]. The electronic effects of 70 phosphorus-containing ligands were compared. It was found that a contribution could be assigned to each substituent on phosphorus, and the sum of those contributions were equal to the entire influence of the ligand on (CO). This contribution was designated as (cm), and the most basic ligand (tri-t-butylphosphine) was chosen as reference on the Tolman electronic parameter (TEP) scale recognizing that PBu is the best -donor and the worst -acceptor ligand (Equation (1)). The NiL(CO)3 scale, that is, the TEP, can be correlated with Strohmeier’s CpMnL(CO)2 system [7,13] (Equation (2)).
Chelating ligands, such as diphosphines, however, cannot be described within the framework of the TEP scale. Crabtree et al. suggested more suitable model complexes for chelating ligands, for example, MoL2(CO)4 (L: bidentate phosphine ligand or two monodentate phosphines) [14]. They compared 11 phosphorus ligands of various types and correlated to the existing TEP scale using Equation (3). Thus, Crabtree proved Tolman’s statement that the choice of transition-metal carbonyl system is arbitrary and interchangable.
In a recent study comparing the Rh–Vaska complexes and NiL(CO)3, Otto and Roodt established a simple quadratic equation [15]. Later, the relationship between the carbonyl-stretching frequencies of the two reference complexes were revised and, by the basicities of the ligands, the (CO) range was divided into two sections [16]. The slope for the more basic phosphines showed a difference to those of less basicity.
Arsines and stibines were investigated in order to determine their electronic effects as well [16]. The contributions to the electronic effect of As and Sb, relative to P, were determined, and an additional term compared to Equation (1) was suggested addressing the effect of the donor atom (C; L = P, Sb or As). (Equation (4), C = 0)
In the past three decades several attempts have been made for invoking theoretical methods to characterize the donor and acceptor properties of phosphines and other, mainly P-donor, ligands. These methods can be divided into two categories. The first approximation deals only with the isolated ligand, focusing on its electronic and steric properties, while neglecting the influence of the metal-containing fragment. As a prominent example, the molecular electrostatic potential at the lone pair of the phosphorus atom should be mentioned, which correlates reasonably well with the TEP scale, according to Suresh and Koga [17]. Moreover, the method known as quantitative analysis of ligand effects (QALE) relies on experimental data of known ligands and provides the resolution of net donating ability into QALE parameters [18,19]. It should be noted that the pnictogen interactions, as important secondary interactions, of a wide variety of P-donor ligands were studied as well [20,21,22]. The second category uses approaches that focus on the entire transition-metal complex, thereby including the possibility to scrutinize ligand–ligand effects as well [23]. Various electronic structure methods, such as Extended Transition State theory combined with Natural Orbitals of Chemical Valence (ETS-NOCV) [24,25,26,27] and Quantum Theory of Atoms in Molecules (QTAIM) [26] have been employed as well.
Crabtree and coworkers investigated the CEP, that is, the computationally derived ligand electronic parameter using the NiL(CO)3 model complex with various neutral, cationic, and anionic ligands [28]. They found that the CEP correlated well with the Tolman electronic parameter and, for certain anionic ligands, even with the Hammett meta substituent constant.
Although the TEP scale achieved a widely accepted status, the limitations of this approximation should be mentioned as well. The coordinating properties of a ligand are governed together by its -donor and -acceptor abilities, whereas TEP (or CEP) only gives the net donor strength. Moreover, through-space ligand interactions may affect the carbonyl-stretching frequencies as reported by Sierra and coworkers for manganese half-sandwich complexes [29]. The Tolman electronic parameters failed for linear Au–carbonyl complexes [30].
The goal of this computational study is to establish a relationship between the ligand electronic effects of late transition-metal–diphosphine complexes with the Tolman’s scale, that is, the totalsymmetric carbonyl-stretching frequencies of Ni(CO)3L complexes, thereby providing a direct comparison of the electronic effects of diphosphines and monophosphines. By the selection of appropriate ligands, the set of ligands employed by Crabtree [14] and Tolman [12] has been adopted, that is, PEt3 (1), PEt2Ph (2), PMe3 (3), PMe2Ph (4), PPh3 (5), P(OMe)3 (6), P(OPh)3 (7), PCl2(OEt) (8), PCl3 (9), PF3 (10), and PF2(CF3) (11).
The secondary objective of the study is to find a dependence of the influence upon the bite angle of the diphosphines by scrutinizing the Rh–H bond distance, the rhodium–hydride stretching frequencies as well as quantum chemical descriptors within the framework of the QTAIM methodology. Those diphosphines that are involved in this study are depicted in Figure 1. As test complexes, the Pd(0)L2(CO) and the HRh(I)L2(CO) complexes were selected. The former is supposed to be sterically less strained in comparison to the Mo(CO)4L2 complexes with a natural bite angle of about 90, thereby reducing the possibility of coupling steric and electronic effects and through-space interactions, whereas the rhodium-containing species are of high importance as the potential catalysts of Rh-catalyzed hydroformylation. It should be noted that only the theoretical treatment of the selected test complexes is possible because of their high reactivity, rendering them unsuitable for the experimental spectroscopical measurements.
2. Computational Details
All the structures were optimized without symmetry constraints with tight convergence criteria using the program ORCA 4.0.1 [31] with the exchange and correlation functionals developed by Grimme [32] containing the D3 empirical dispersion correction with Becke and Johnson damping [33]. For palladium and rhodium atoms the def2-TZVP basis set [34]. was employed with the respective pseudopotentials [34]. For the other atoms, the def2-SVP basis set [34] was used. The B97D3 functional was selected for the calculations involving diphosphines as it gave slightly better linear correlations for monophosphines as compared to other GGA (BP86 [35,36], PBEPBE [37]) and meta-GGA (TPSS [38], and M06L [39]) functionals (vide infra).
For the complexes containing conformationally flexible ligands, conformational analyses have been performed in a similar manner reported earlier [26], and the lowest energy species were considered. QTAIM analyses of the wave function [40] were carried out with AIMAll software [41]. Topological analyses of the Electron Localization Function (ELF) were completed using the Topmod09 package [42]. For electronic structure calculations, the B97D3/def2-TZVP level of theory was employed on the equilibrium geometries obtained by the B97D3 calculations with the mixed (def2-TZVP/def2-SVP) basis set.
3. Results and Discussion
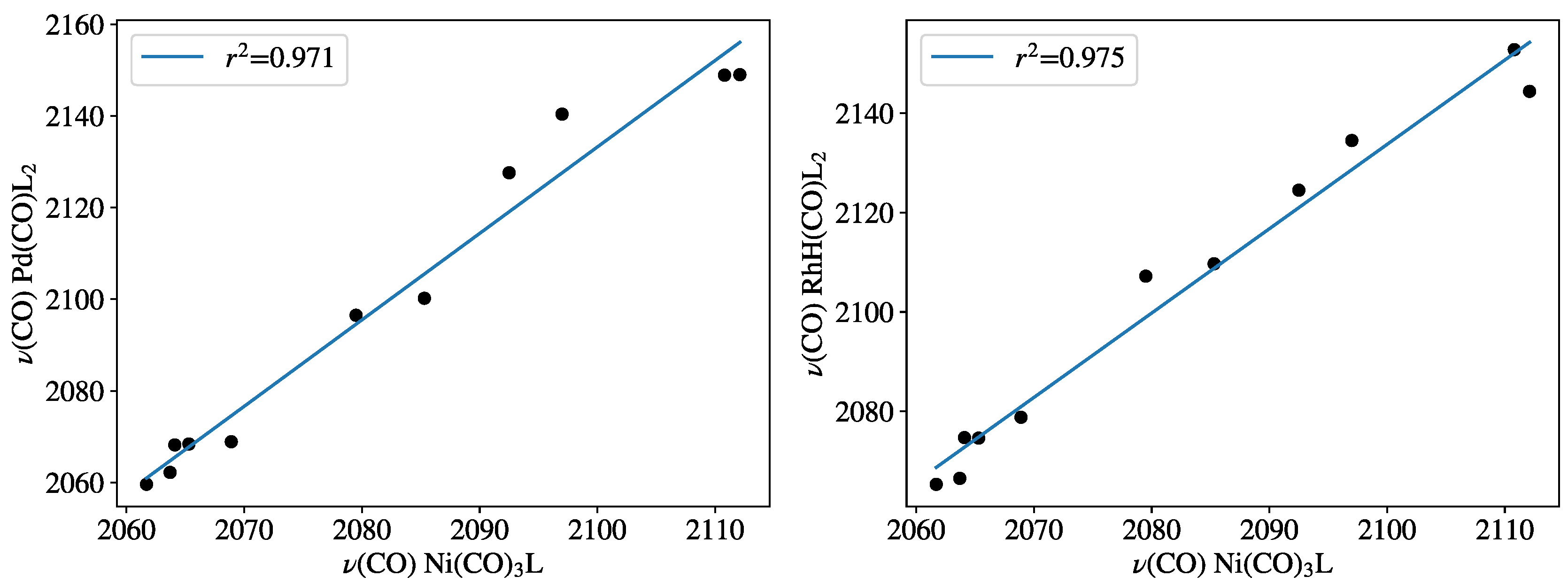
In Table 1, the experimental nickel (CO) parameters, as well as the computed palladium, rhodium (CO) parameters were collected for various phosphorus ligands, the same ligands that had also been used by Crabtree et al. In Figure 2, the 11 calculated (B97D3) carbonyl-stretching frequencies of PdL2(CO) and HRhL2(CO) training sets are plotted against the experimental (CO) frequencies for NiL(CO)3 [2,12]. The high values of the correlation coefficients r = 0.971 and r = 0.975, obtained for PdL2(CO) and HRhL2(CO), respectively, shows that the computed (CO) for these systems (CEP parameters) have reasonable linear correlation with the experimental NiL(CO)3(CO) values (TEP) according to Equations (5) and (6):
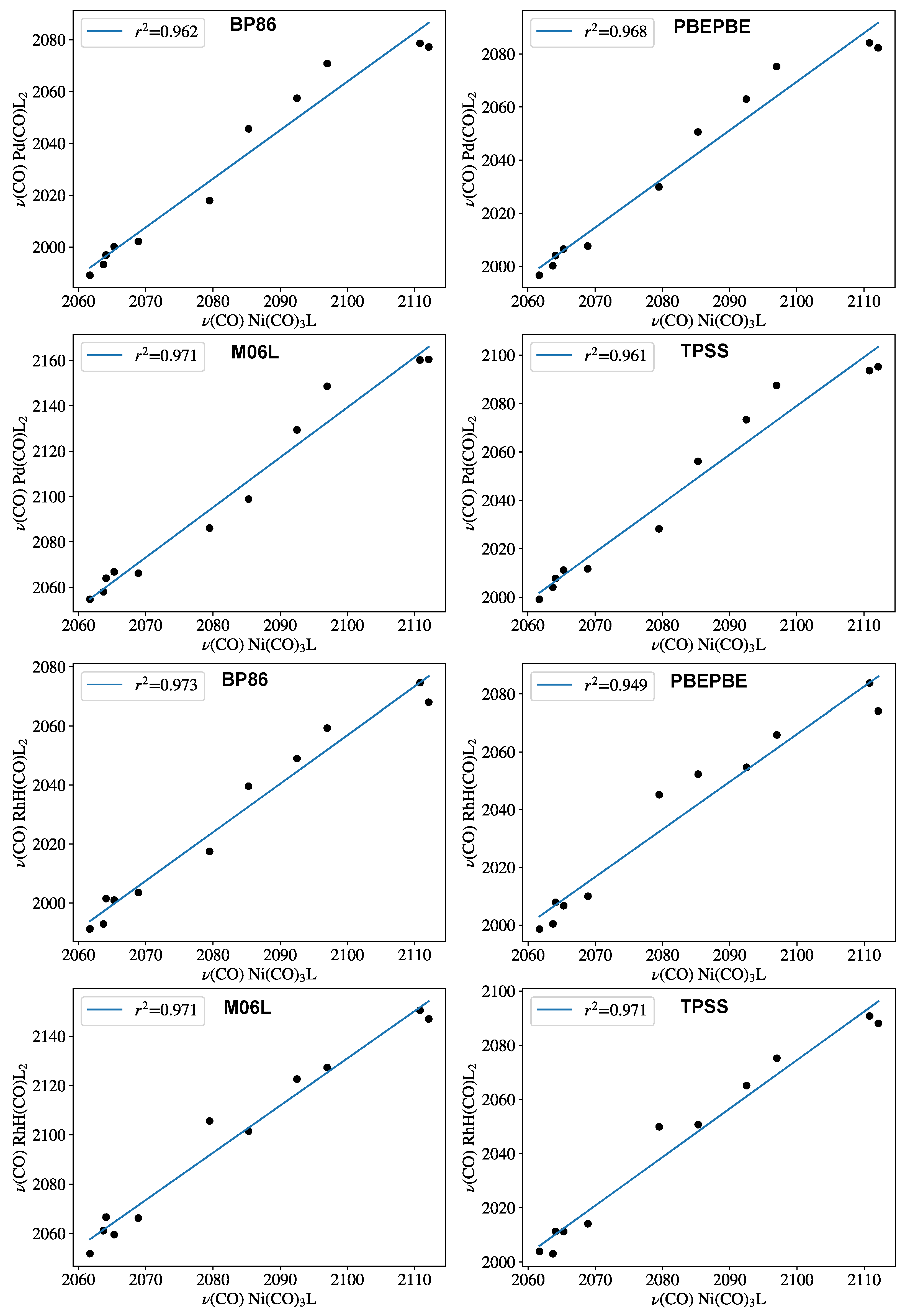
To clarify the role of the theoretical method employed for obtaining CEP parameters, the training set was recomputed with various GGA and meta-GGA functionals (Figure 3). The B97D3 results were compared with those obtained with BP86 [35,36], PBEPBE [37], TPSS [38], and M06L [39] functionals for both the palladium and the rhodium complexes. The correlation coefficients remained within quite a narrow range, as the worst correlation was found to be for the PBEPBE functional in the case of the Rh–hydride model systems. For both complexes, the B97D3 functional proved to be slightly superior; therefore, it was chosen for describing the chelating ligands as well.
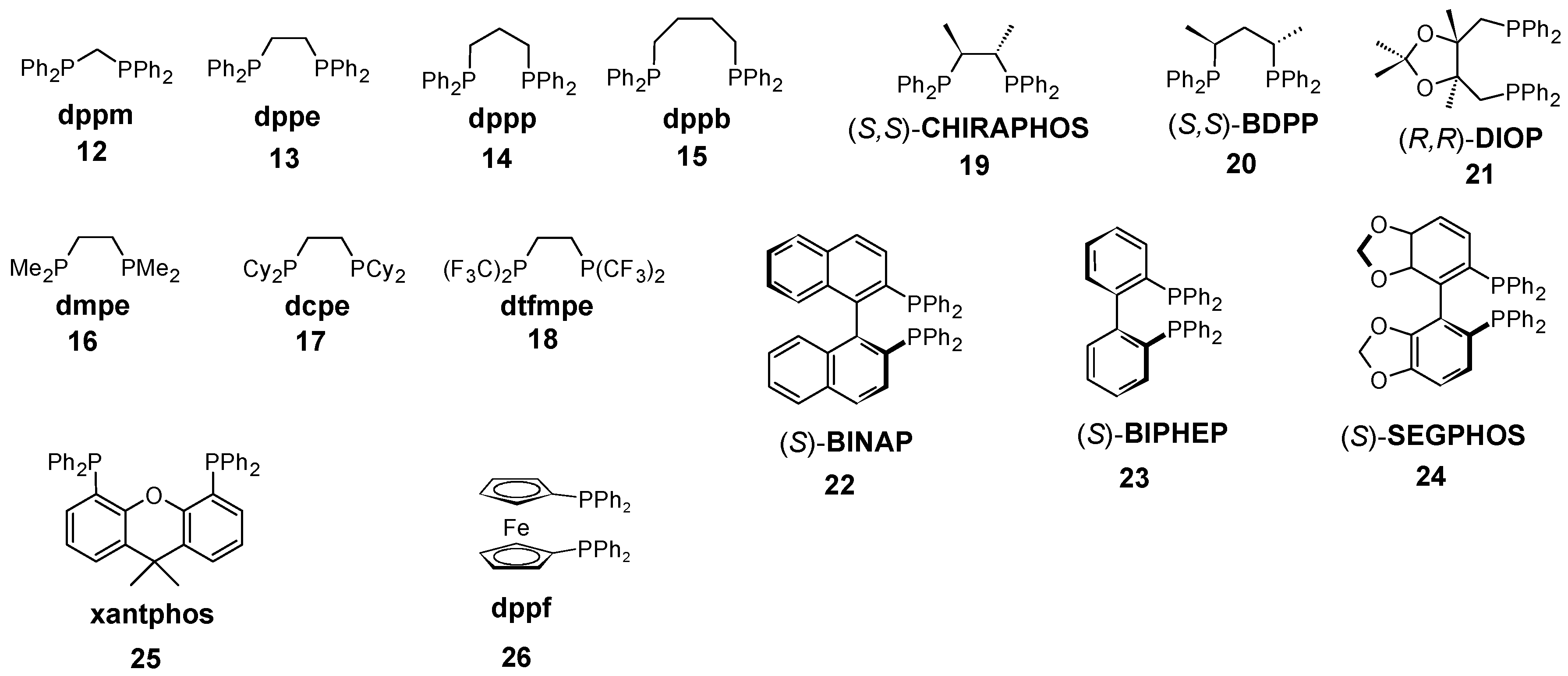
The chelating ligands shown in Table 2 were selected to address various geometrical as well as electronic parameters. Ligands 12–15 were used to illustrate the influence of the P-M-P bite angle while ligands 16–18 show the effect of the substituents on the phosphorus with ethanediphos backbone. The chiral ligands 19–21 reveal the substituent effects on the backbone. To address the structural changes of ligands with axial chirality 22–24 were selected. Finally, 25 and 26 represent ligands possessing rigid structure with wide bite angles.
The CEP determined on two different types of carbonyl complexes reveals a remarkable difference (near 10 cm) for the narrow bite-angle case 12. The increase of the bite angle by going in the direction of dppm → dppe → dppp → dppb also caused a decrease in (CO) for the palladium and an increase in (CO) for the rhodium complexes. The replacement of the phenyl substituent in dppe to the electron-withdrawing trifluoromethyl group resulted in a similar blue-shift for the two systems. On the other hand, the presence of electron-donating substituents, such as methyl or cyclohexyl, on the phosphorus results in the expected red-shift in the palladium carbonyls and a mixed effect for the Rh carbonyls as, in HRh(CO)(dmpe), virtually no change occurs in the carbonyl-stretching frequency. When the electron-donating methyl groups are introduced in the chiral backbone, such as for the ligand CHIRAPHOS, a slight blue-shift occurs for HRh(19)(CO), whereas an almost negligible red-shift takes place for Pd(19)(CO).
The change or the neglect of the condensed ring in the ligands with axial chirality has practically no effect on (CO). Moreover, the predicted CEP is almost identical for both model systems. This difference is also minor (below 2 cm) for the xantphos and dppf ligands. Not surprisingly, in the three-coordinate palladium systems, all the P-M-P bite angles were somewhat larger than in the HRh(LL)(CO) complexes, except for the dppm ligand.
The influence in the four-coordinate, distorted square-planar HRhL2(CO) complexes is formally based on a donor–acceptor interaction. Here, it is followed by the metal–hydrogen stretching frequency ((RhH)), the Rh–H distance, and two QTAIM parameters. Both the electron density at the bond critical point () and the (Rh,H) delocalization index, which provides the number of electron pairs delocalized between the atomic basins of Rh and H [43], were somewhat proportional with the bond strength. The delocalization index is, to some extent, related to formal bond orders for an equally shared pair between two atoms in a polyatomic molecule.
According to the (RhH) values in Table 3 the difference in electronic effects caused by the P-ligand to the hydride is rather subtle. As the trend in the delocalization indices is quite the opposite compared to that obtained from the geometrical parameters, it is likely that the electron sharing between atomic basins did not reflect the overall bond strengths in these cases.
The (RhH) frequencies, however, tend to follow the computed electronic parameters, as the largest red-shift occurs for the more electron donating ligands and the frequencies are increased for the electron withdrawing ones. Thus, the influence increases in the order of the substituent donor strength.
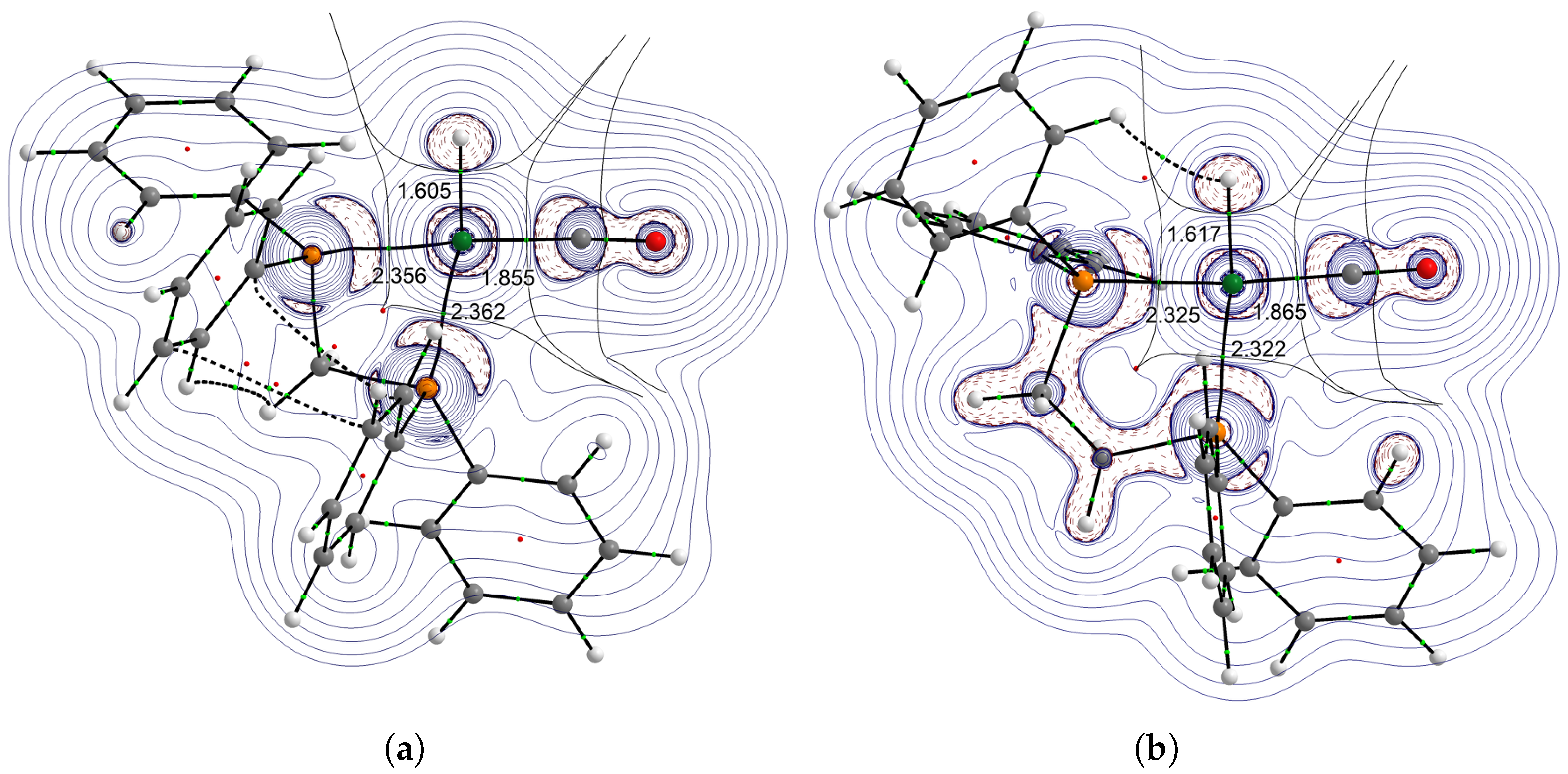
To address the effect of the P–Rh–P bite angle upon the influence, the structures and the Laplacians of the electron density were compared and are illustrated in Figure 4. The structural distortion caused by the dppmligand resulted in distortion in the electron-density distribution aswell. The stronger bond in the dppm complex, as compared to the HRh(dppe)(CO) complex, shows that even when the basicities of the phosphorus atoms of the standalone dppm and dppe ligands should be very similar, the slight decrease of the H–Rh–P angle causes a notable decrease in the influence. It should be noted that, in the dppm complex, a red-shift can be observed in (CO) in comparison to that of the dppe complex, which may be explained with the less preferred orientation to a orbital interaction.
It was found previously that, among QTAIM parameters, CO delocalization index gave good performance, whereas the electron density of the CO bond critical point gives moderate linear correlation with the computed carbonyl symmetric stretching frequencies in Ni-tricarbonyl phosphine complexes [26]. To the best of our knowledge, the relationship between the electron localization function (ELF) and the stretching frequencies in transition-metal carbonyl complexes is unexplored.
The ELF, denoted as introduced by Becke and Edgecombe [35] and reinterpreted by Savin et al. [44] using the mathematical scheme from Thom’s catastrophe theory [45] is based on the conditional pair probability function. It takes values in a range between 0 and 1, where values close to 1 are located in regions of space with strong electron localization, whereas values close to 0 correspond to electron delocalization, and = 0.5 is the value that would be expected for a homogeneous electron gas. It is worth mentioning that Putz [46,47] proposed general formulations for the ELF on the basis of the path integral formulation of Markovian open systems.
The electronic effect of the monodentate ligands was followed by the population of the V(C,O) and V(Rh,H) disynaptic basins. The population of an ELF basin is defined according to Equation (7).
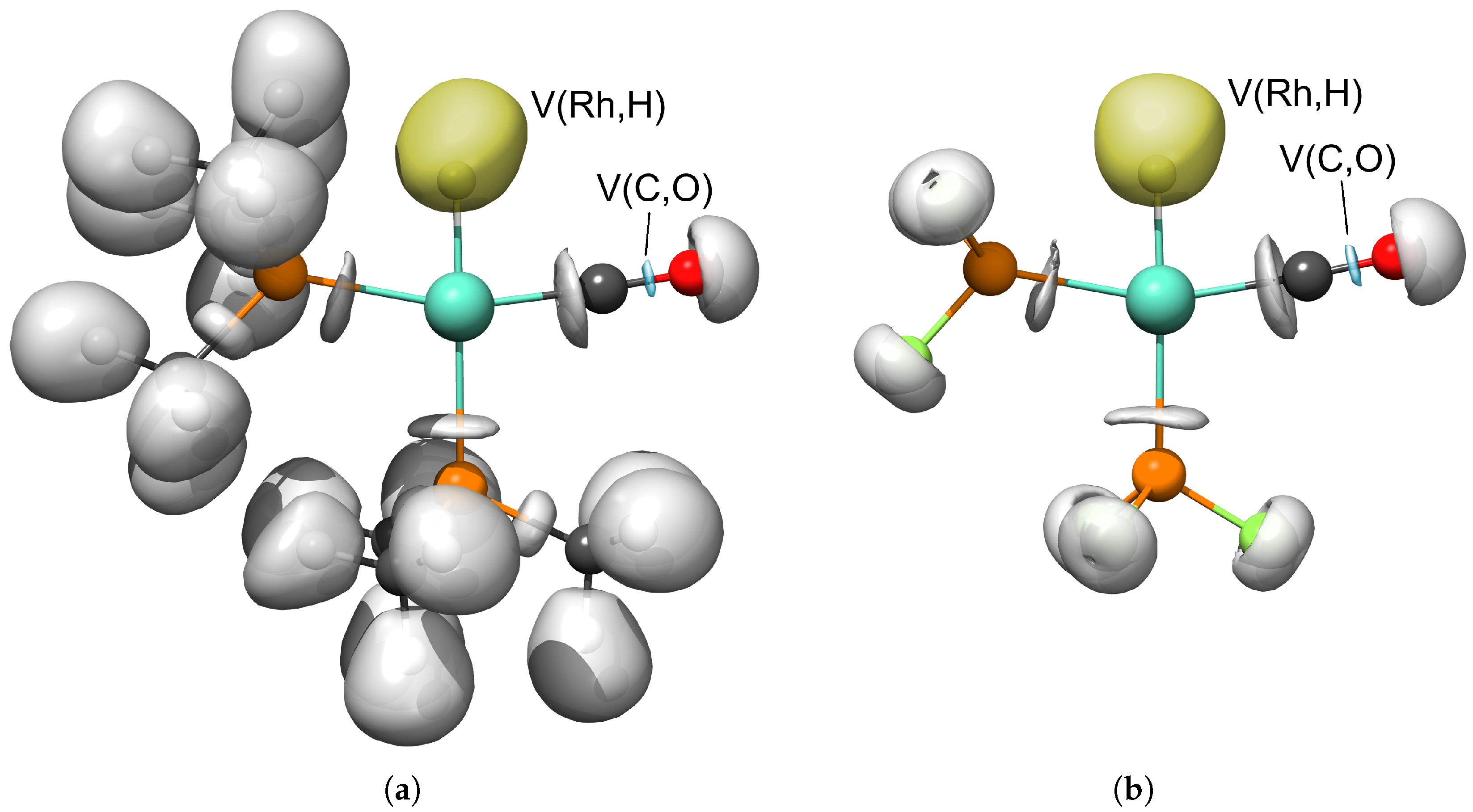
The ELF basins, which were the subject of our study, are depicted for complexes HRh(PMe3)2(CO) and HRh(PF3)2(CO) in Figure 5. The spacial distribution of the V(Rh,H) basin resembles those representing the H–C bonds, that is, it is localized around the hydrogen and not along the H–Rh bond path. On the other hand, the V(C,O) basins are situated in the middle between the carbon and oxygen of the carbonyl ligands.
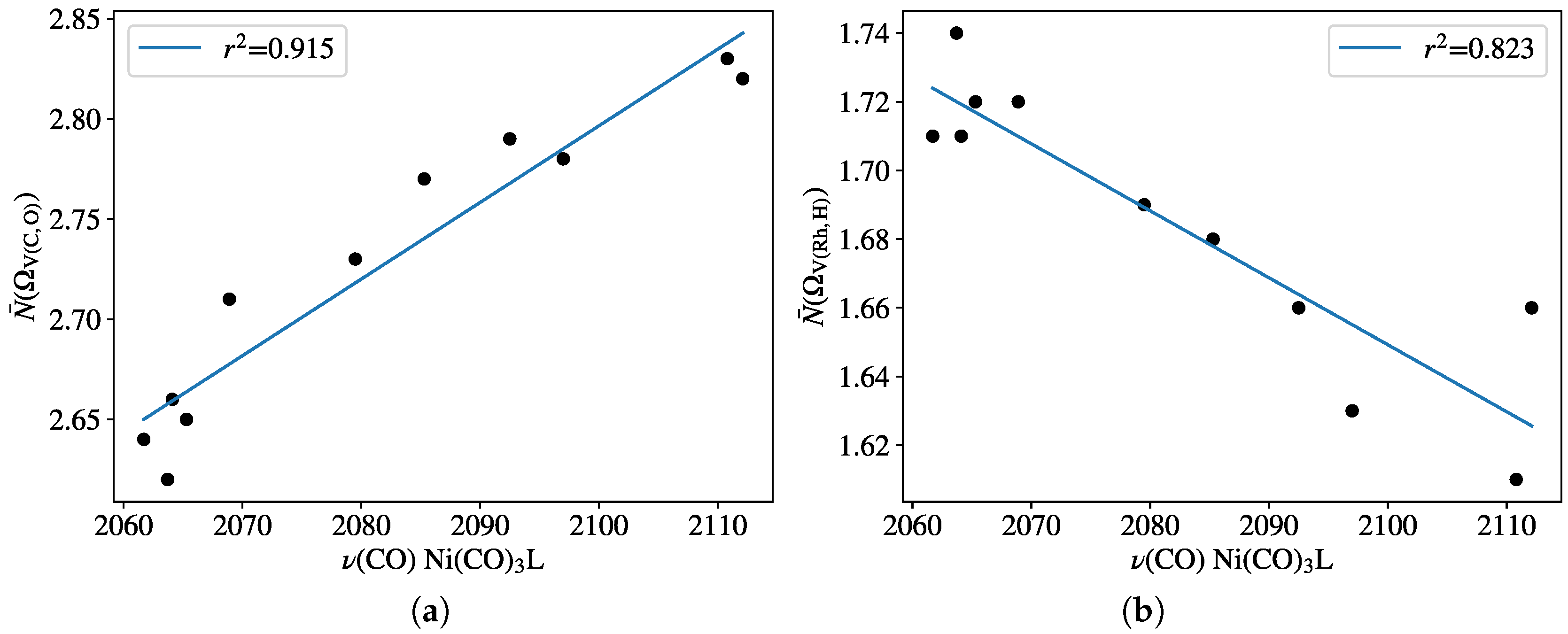
The ELF was computed for all the HRh(monophosphine)2(CO) type of complexes. The basin population associated with the C≡O bond revealed fairly good linear correlation (r2 = 0.915) with the experimental TEP parameters (Figure 6a). The extrema of the values are between 2.62 (for ligand PEt2Ph) and 2.83 (for PF3), indicating the somewhat depleted triple-bond character of the bound CO. The population change of the Rh–hydride bond, however, shows moderate linear correlation (r2 = 0.823) with TEP; only showing that the trans influence generally increases with the greater donor strength of the P-donor ligand.
4. Conclusions
In the present study, two classes of model compounds were scrutinized to check their applicability as appropriate candidates for computationally derived ligand electronic parameters (CEPs) for bidentate P-donor ligands employing the B97D3/def2-TZVP(def2-SVP) level of theory. The results of this work can be summarized as follows:
- For both model systems, no major difference in accuracy can be expected for various DFT methods in terms of the linear regression coefficient.
- For monophosphines, both Pd(0)L2(CO) and HRh(I)L2(CO) complexes showed high linearity with the experimental TEP scale.
- For monophosphines, the population of the disynaptic ELF basin V(C,O), associated with the carbonyl ligand, showed good linearity with the experimental TEP scale.
- For diphosphine-containing systems, the bite angle effect showed the opposite trend, with PdL2(CO) complexes revealing a decrease in (CO) by the increase of bite angle.
- In the case of the Pd complexes, the change of substituents on phosphorus caused a change in (CO) being consistent with the donor character of the substituent.
- For diphosphines possessing axial chirality, the nature of the condensed ring does not alter the electronic parameter of the ligand.
- The effect estimated in the HRhL2(CO) complexes is very sensitive to the H–Rh–P angle.
Supplementary Materials
The following are available online. Cartesian coordinates of palladium-diphosphine complexes occuring in this study.
Author Contributions
The research problem was conceptualized by the corresponding author (T.K.), the computational work was carried out by T.R.K. and N.P., the writing, review and editing is the joint work of all authors. All authors read and approved the final manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors thank the Hungarian Research Fund (K113177) for the financial support. This work was supported by the GINOP-2.3.2-15-2016-00049 grant. N.P. thanks Gedeon Richter’s Talentum Foundation for the scholarship. The present scientific contribution is dedicated to the 650th anniversary of the foundation of the University of Pécs, Hungary. The project has been supported by the European Union, cofinanced by the European Social Fund Grant no. EFOP-3.6.1.-16-2016-00004 entitled by Comprehensive Development for Implementing Smart Specialization Strategies at the University of Pécs. The study was also financed by the Higher Education Institutional Excellence Program of the Ministry of Human Capacities in Hungary, within the framework of the 20765-3/2018/FEKUTSTRAT thematic program of the University of Pécs.
Conflicts of Interest
The authors declare no conflict of interest.
References
- McAuliffe, C.A. Transition Metal Complexes of Phosphorus, Arsenic and Antimony Ligands; Halsted Press: New York, NY, USA, 1973. [Google Scholar]
- Tolman, C.A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Hartwig, J.F.; Collman, J.P. Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, USA, 2010. [Google Scholar]
- Brown, T.L.; Lee, K.J. Ligand steric properties. Coord. Chem. Rev. 1993, 128, 89–116. [Google Scholar] [CrossRef]
- Dias, P.B.; de Piedade, M.E.M.; Simões, J.A.M. Bonding and energetics of phosphorus (III) ligands in transition metal complexes. Coord. Chem. Rev. 1994, 135, 737–807. [Google Scholar] [CrossRef]
- Kamer, P.C.; van Leeuwen, P.W.; Reek, J.N. Wide bite angle diphosphines: Xantphos ligands in transition metal complexes and catalysis. Acc. Chem. Res. 2001, 34, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Kühl, O. Predicting the net donating ability of phosphines do we need sophisticated theoretical methods? Coord. Chem. Rev. 2005, 249, 693–704. [Google Scholar] [CrossRef]
- de Vries, J.G.; Lefort, L. The combinatorial approach to asymmetric hydrogenation: Phosphoramidite libraries, ruthenacycles, and artificial enzymes. Chem. A Eur. J. 2006, 12, 4722–4734. [Google Scholar] [CrossRef] [PubMed]
- Botteghi, C.; Paganelli, S.; Schionato, A.; Marchetti, M. The asymmetric hydroformylation in the synthesis of pharmaceuticals. Chirality 1991, 3, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Kégl, T.; Kollár, L. Chiral Phosphorous Ligands in Asymmetric Catalysis. In Comprehensive Inorganic Chemistry II (Second Edition): From Elements to Applications; Elsevier Ltd.: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Strohmeier, W.; Müller, F.J. Klassifizierung phosphorhaltiger Liganden in Metallcarbonyl-Derivaten nach der π-Acceptorstärke. Chem. Ber. 1967, 100, 2812–2821. [Google Scholar] [CrossRef]
- Tolman, C.A. Electron donor-acceptor properties of phosphorus ligands. Substituent additivity. J. Am. Chem. Soc. 1970, 92, 2953–2956. [Google Scholar] [CrossRef]
- Strohmeier, W.; Müller, F.J. π-Acceptorstärke von Phosphinen als Liganden in Cyclopentadienylmangantricarbonyl und Nickelcarbonyl. Z. Naturforschg. 1967, 22b, 451–452. [Google Scholar] [CrossRef]
- Anton, D.R.; Crabtree, R.H. Metalation-resistant ligands: Some properties of dibenzocyclooctatetraene complexes of molybdenum, rhodium and iridium. Organometallics 1983, 2, 621–627. [Google Scholar] [CrossRef]
- Roodt, A.; Otto, S.; Steyl, G. Structure and solution behaviour of rhodium(I) Vaska-type complexes for correlation of steric and electronic properties of tertiary phosphine ligands. Coord. Chem. Rev. 2003, 245, 121–137. [Google Scholar] [CrossRef]
- Otto, S.; Roodt, A. Quantifying the electronic cis effect of phosphine, arsine and stibine ligands by use of rhodium(I) Vaska-type complexes. Inorg. Chim. Acta 2004, 357, 1–10. [Google Scholar] [CrossRef]
- Suresh, C.H.; Koga, N. Quantifying the Electronic Effect of Substituted Phosphine Ligands via Molecular Electrostatic Potential. Inorg. Chem. 2002, 41, 1573–1578. [Google Scholar] [CrossRef]
- Liu, H.Y.; Eriks, K.; Prock, A.; Giering, W.P. Quantitative analysis of ligand effects (QALE). Systematic study of iron-phosphorus bond lengths and their relationship to steric thresholds. Organometallics 1990, 9, 1758–1766. [Google Scholar] [CrossRef]
- Bartholomew, J.; Fernandez, A.L.; Lorsbach, B.A.; Wilson, M.R.; Prock, A.; Giering, W.P. Comments on Coupling Graphical and Regression Analyses of Ligand Effect Data. Organometallics 1996, 15, 295–301. [Google Scholar] [CrossRef]
- Scheiner, S. The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 2012, 46, 280–288. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Intramolecular pnicogen interactions in phosphorus and arsenic analogues of proton sponges. Phys. Chem. Chem. Phys. 2014, 16, 15900–15909. [Google Scholar] [CrossRef]
- Molloy, A.D.; Sánchez-Sanz, G.; Gilheany, D.G. PP-Rotation, P-Inversion and Metathesis in Diphosphines Studied by DFT Calculations: Comments on Some Literature Conflicts. Inorganics 2016, 4, 36. [Google Scholar] [CrossRef]
- Gusev, D.G. Donor properties of a series of two-electron ligands. Organometallics 2009, 28, 763–770. [Google Scholar] [CrossRef]
- Mitoraj, M.; Michalak, A. Donor-Acceptor Properties of Ligands from the Natural Orbitals for Chemical Valence. Organometallics 2007, 26, 6576–6580. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A. σ-donor and π-acceptor properties of phosphorus ligands: An insight from the natural orbitals for chemical valence. Inorg. Chem. 2010, 49, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Kégl, T.R.; Kollár, L.; Kégl, T. Relationship of QTAIM and NOCV Descriptors with Tolman’s Electronic Parameter. Adv. Chem. 2016, 2016, 4109758. [Google Scholar] [CrossRef]
- Couzijn, E.P.; Lai, Y.Y.; Limacher, A.; Chen, P. Intuitive Quantifiers of Charge Flows in Coordinate Bonding. Organometallics 2017, 36, 3205–3214. [Google Scholar] [CrossRef]
- Perrin, L.; Clot, E.; Eisenstein, O.; Loch, J.; Crabtree, R.H. Computed ligand electronic parameters from quantum chemistry and their relation to Tolman parameters, lever parameters, and Hammett constants. Inorg. Chem. 2001, 40, 5806–5811. [Google Scholar] [CrossRef] [PubMed]
- Valyaev, D.A.; Brousses, R.; Lugan, N.; Fernández, I.; Sierra, M.A. Do ν(CO) Stretching Frequencies in Metal Carbonyl Complexes Unequivocally Correlate with the Intrinsic Electron-Donicity of Ancillary Ligands? Chem. A Eur. J. 2011, 17, 6602–6605. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Scafuri, N.; Bistoni, G.; Macchioni, A.; Tarantelli, F.; Zuccaccia, D.; Belpassi, L. When the Tolman Electronic Parameter Fails: A Comparative DFT and Charge Displacement Study of [(L)Ni(CO)3]0/− and [(L)Au(CO)]0/+. Inorg. Chem. 2014, 53, 9907–9916. [Google Scholar] [CrossRef]
- Frank, N. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101–194118. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms Moleculs—A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Keith, T.A. AIMAll (Version 15.05.18); TK Gristmill Software: Overland Park, KS, USA, 2015. [Google Scholar]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Stephens, M.E. Spatial localization of the electronic pair and number distributions in molecules. J. Am. Chem. Soc. 1975, 97, 7391–7399. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The electron localization function. Angew. Chem. Int. Ed. Engl. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- René, T. Stabilité Structurelle et Morphogénese: Essai D’une Théorie Générale des Modeles; WA Benjamin: New York, NY, USA, 1972. [Google Scholar]
- Putz, M.V. Markovian approach of the electron localization functions. Int. J. Quantum Chem. 2005, 105, 1–11. [Google Scholar] [CrossRef]
- Putz, M. Density functionals of chemical bonding. Int. J. Mol. Sci. 2008, 9, 1050–1095. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Chelating phosphines considered in this study.

Figure 2.
Carbonyl-stretching frequencies of PdL2(CO) and HRhL2(CO) against Tolman’s parameters at the B97D3 level. Frequency values are given in Table 1.
Figure 2.
Carbonyl-stretching frequencies of PdL2(CO) and HRhL2(CO) against Tolman’s parameters at the B97D3 level. Frequency values are given in Table 1.

Figure 3.
Carbonyl-stretching frequencies of PdL2(CO) (top row) and HRhL2(CO) against Tolman’s parameter computed with various DFT functionals. Frequency values are given in Table 1.
Figure 3.
Carbonyl-stretching frequencies of PdL2(CO) (top row) and HRhL2(CO) against Tolman’s parameter computed with various DFT functionals. Frequency values are given in Table 1.

Figure 4.
Laplacian () of the electron density of complexes HRh(dppm)(CO) (a) and HRh(dppe)(CO) (b). Charge concentration regions () are designated with dashed lines.
Figure 4.
Laplacian () of the electron density of complexes HRh(dppm)(CO) (a) and HRh(dppe)(CO) (b). Charge concentration regions () are designated with dashed lines.

Figure 5.
Selected electron localization function (ELF) valence basins for complexes HRh(PMe3)2(CO) (a) and HRh(PF3)2(CO) (b) at isovalue of = 0.84. Yellow and blue envelopes designate the disynaptic V(Rh,H) and V(C,O) basins, respectively.
Figure 5.
Selected electron localization function (ELF) valence basins for complexes HRh(PMe3)2(CO) (a) and HRh(PF3)2(CO) (b) at isovalue of = 0.84. Yellow and blue envelopes designate the disynaptic V(Rh,H) and V(C,O) basins, respectively.

Figure 6.
Population of ELF basins (a) V(C,O) and (b) V(Rh,H) for complexes HRhL2(CO) against Tolman’s parameters at the B97D3 level. Frequency values are given in Table 1.
Figure 6.
Population of ELF basins (a) V(C,O) and (b) V(Rh,H) for complexes HRhL2(CO) against Tolman’s parameters at the B97D3 level. Frequency values are given in Table 1.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Experimental CO-stretching frequencies (Reference [12]) for NiL(CO)3, and computed CO-stretching frequencies for PdL2(CO) and HRhL2(CO) complexes at the B97D3 level of theory. Carbonyl-stretching frequencies are given in cm.
Table 1.
Experimental CO-stretching frequencies (Reference [12]) for NiL(CO)3, and computed CO-stretching frequencies for PdL2(CO) and HRhL2(CO) complexes at the B97D3 level of theory. Carbonyl-stretching frequencies are given in cm.
| Ligand | (CO) | (CO) | (CO) |
|---|---|---|---|
| PEt3 (1) | 2061.7 | 2059.6 | 2065.3 |
| PEt2Ph (2) | 2063.7 | 2062.2 | 2066.5 |
| PMe3 (3) | 2064.1 | 2068.2 | 2074.7 |
| PMe2Ph (4) | 2065.3 | 2068.4 | 2074.6 |
| PPh3 (5) | 2068.9 | 2068.9 | 2078.8 |
| P(OMe)3 (6) | 2079.5 | 2096.5 | 2107.2 |
| P(OPh)3 (7) | 2085.3 | 2100.2 | 2109.7 |
| PCl2(OEt) (8) | 2092.5 | 2127.6 | 2124.5 |
| PCl3 (9) | 2097.0 | 2140.4 | 2134.5 |
| PF3 (10) | 2110.8 | 2148.9 | 2152.8 |
| P(CF3)F2 (11) | 2112.1 | 2149.0 | 2144.4 |
Table 2.
Computed CO-stretching frequencies and P-M-P bite angles (θPPdP and θPRhP, in degrees) for diphosphine-containing complexes PdL2(CO), HRhL2(CO), and their respective Computed Tolman Parameters (CEPs), determined according to Equations (5) and (6). Vibrational frequencies are in cm.
| Ligand | (CO) | CEP | θPPdP | (CO) | CEP | θPRhP |
|---|---|---|---|---|---|---|
| dppm (12) | 2080.8 | 2072.6 | 69.8 | 2069.8 | 2062.9 | 71.0 |
| dppe (13) | 2076.1 | 2070.1 | 85.1 | 2071.9 | 2064.1 | 84.7 |
| dppp (14) | 2068.4 | 2066.2 | 90.5 | 2074.6 | 2065.7 | 88.7 |
| dppb (15) | 2064.5 | 2064.2 | 96.2 | 2079.0 | 2068.2 | 94.6 |
| dmpe (16) | 2073.5 | 2068.8 | 86.0 | 2075.1 | 2065.9 | 85.5 |
| dcpe (17) | 2058.6 | 2061.1 | 86.2 | 2060.6 | 2057.6 | 85.3 |
| dtfmpe (18) | 2123.1 | 2094.3 | 86.3 | 2125.8 | 2095.1 | 84.9 |
| CHIRAPHOS (19) | 2074.3 | 2069.2 | 85.1 | 2076.3 | 2066.6 | 84.6 |
| BDPP (20) | 2067.7 | 2065.8 | 89.0 | 2071.2 | 2063.7 | 87.7 |
| DIOP (21) | 2069.6 | 2066.8 | 101.5 | 2077.4 | 2067.3 | 95.5 |
| BINAP (22) | 2069.0 | 2066.5 | 93.7 | 2075.9 | 2066.4 | 92.8 |
| BIPHEP (23) | 2069.6 | 2066.8 | 96.4 | 2076.8 | 2066.9 | 93.8 |
| SEGPHOS (24) | 2067.5 | 2065.7 | 94.3 | 2073.9 | 2065.2 | 92.8 |
| xantphos (25) | 2065.0 | 2064.4 | 105.9 | 2070.1 | 2063.1 | 102.8 |
| dppf (26) | 2069.3 | 2066.6 | 103.6 | 2078.6 | 2067.9 | 97.8 |
Table 3.
Computed Rh-stretching frequencies, Rh–H bond lengths (in Å), P–Rh–P bond angles (in degrees), delocalization indices, and electron densities at bond critical points for HRhL2(CO) complexes.
Table 3.
Computed Rh-stretching frequencies, Rh–H bond lengths (in Å), P–Rh–P bond angles (in degrees), delocalization indices, and electron densities at bond critical points for HRhL2(CO) complexes.
| Ligand | (RhH) | r | (Rh,H) | (RhH) | |
|---|---|---|---|---|---|
| PEt3 (1) | 1920.8 | 1.618 | 100.4 | 0.858 | 0.131 |
| PEt2Ph (2) | 1931.6 | 1.615 | 95.6 | 0.873 | 0.132 |
| PMe3 (3) | 1927.3 | 1.618 | 100.9 | 0.860 | 0.131 |
| PMe2Ph (4) | 1948.3 | 1.611 | 98.4 | 0.876 | 0.133 |
| PPh3 (5) | 1964.7 | 1.606 | 102.6 | 0.849 | 0.135 |
| P(OMe)3 (6) | 1949.0 | 1.618 | 100.7 | 0.844 | 0.132 |
| P(OPh)3 (7) | 1984.5 | 1.607 | 106.9 | 0.831 | 0.135 |
| PCl2(OEt) (8) | 2014.5 | 1.598 | 96.4 | 0.866 | 0.139 |
| PCl3 (9) | 1993.0 | 1.602 | 101.2 | 0.837 | 0.135 |
| PF3 (10) | 1970.6 | 1.614 | 99.9 | 0.841 | 0.134 |
| P(CF3)F2 (11) | 1967.5 | 1.617 | 100.2 | 0.829 | 0.133 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kégl, T.R.; Pálinkás, N.; Kollár, L.; Kégl, T. Computational Characterization of Bidentate P-Donor Ligands: Direct Comparison to Tolman’s Electronic Parameters. Molecules 2018, 23, 3176. https://doi.org/10.3390/molecules23123176
AMA Style
Kégl TR, Pálinkás N, Kollár L, Kégl T. Computational Characterization of Bidentate P-Donor Ligands: Direct Comparison to Tolman’s Electronic Parameters. Molecules. 2018; 23(12):3176. https://doi.org/10.3390/molecules23123176
Chicago/Turabian StyleKégl, Tímea R., Noémi Pálinkás, László Kollár, and Tamás Kégl. 2018. "Computational Characterization of Bidentate P-Donor Ligands: Direct Comparison to Tolman’s Electronic Parameters" Molecules 23, no. 12: 3176. https://doi.org/10.3390/molecules23123176