
The Triple-Decker Complex [Cp*Fe(µ,η5:η5-P5)Mo(CO)3] as a Building Block in Coordination Chemistry


Abstract
:
1. Introduction
2. Results and Discussion
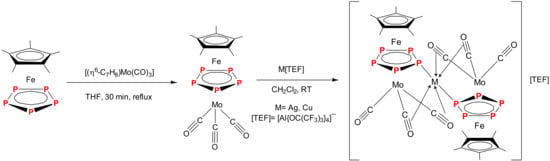
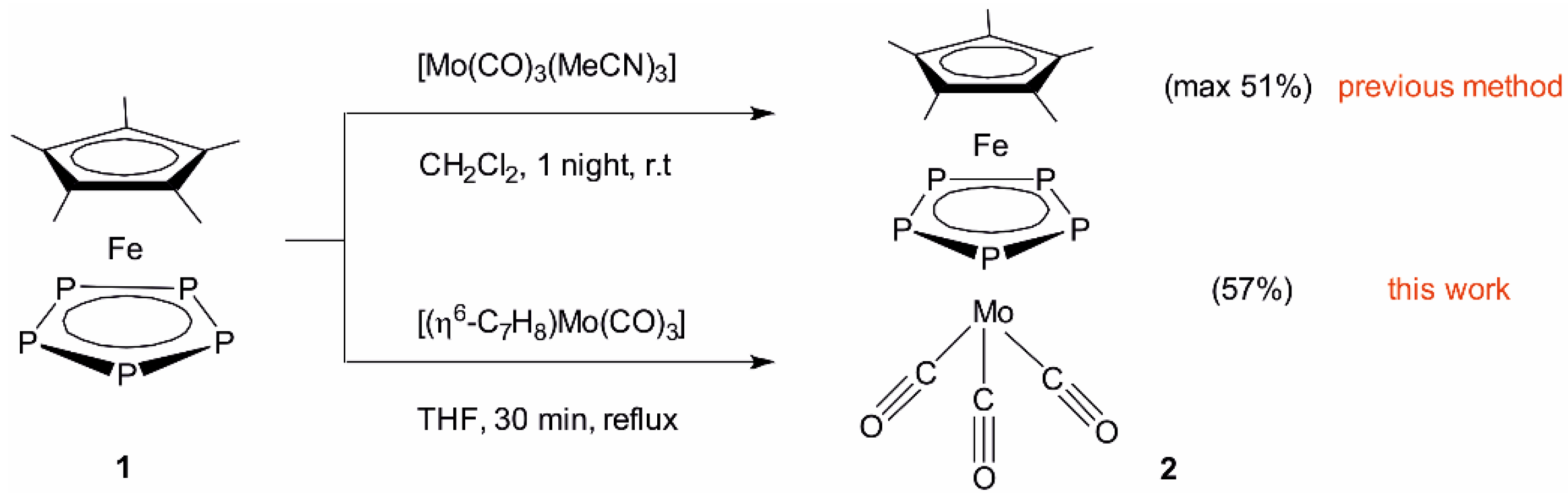
2.1. Synthesis and X-Ray Structure of [Cp*Fe(µ,η5:η5-P5)Mo(CO)3] (2)
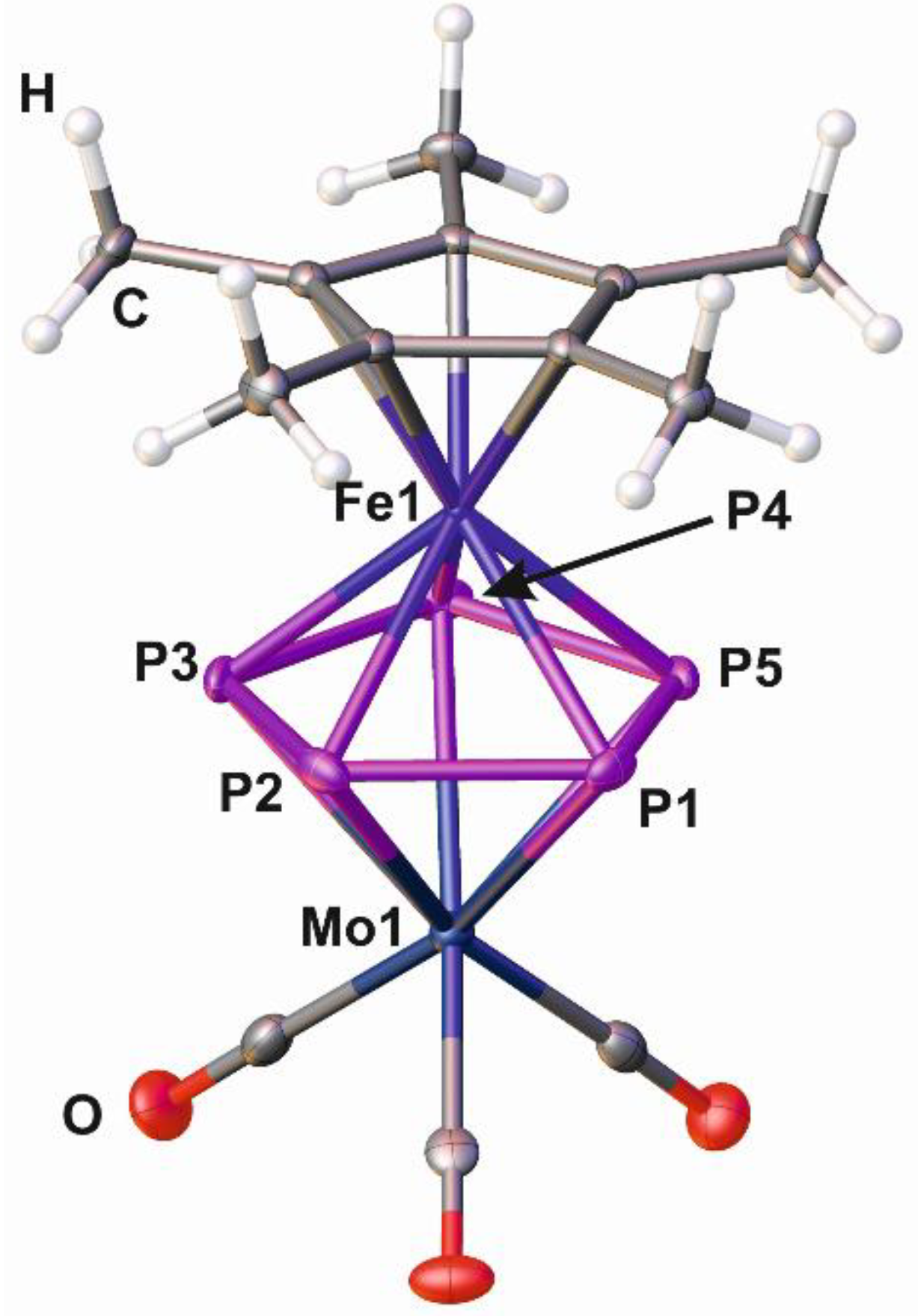
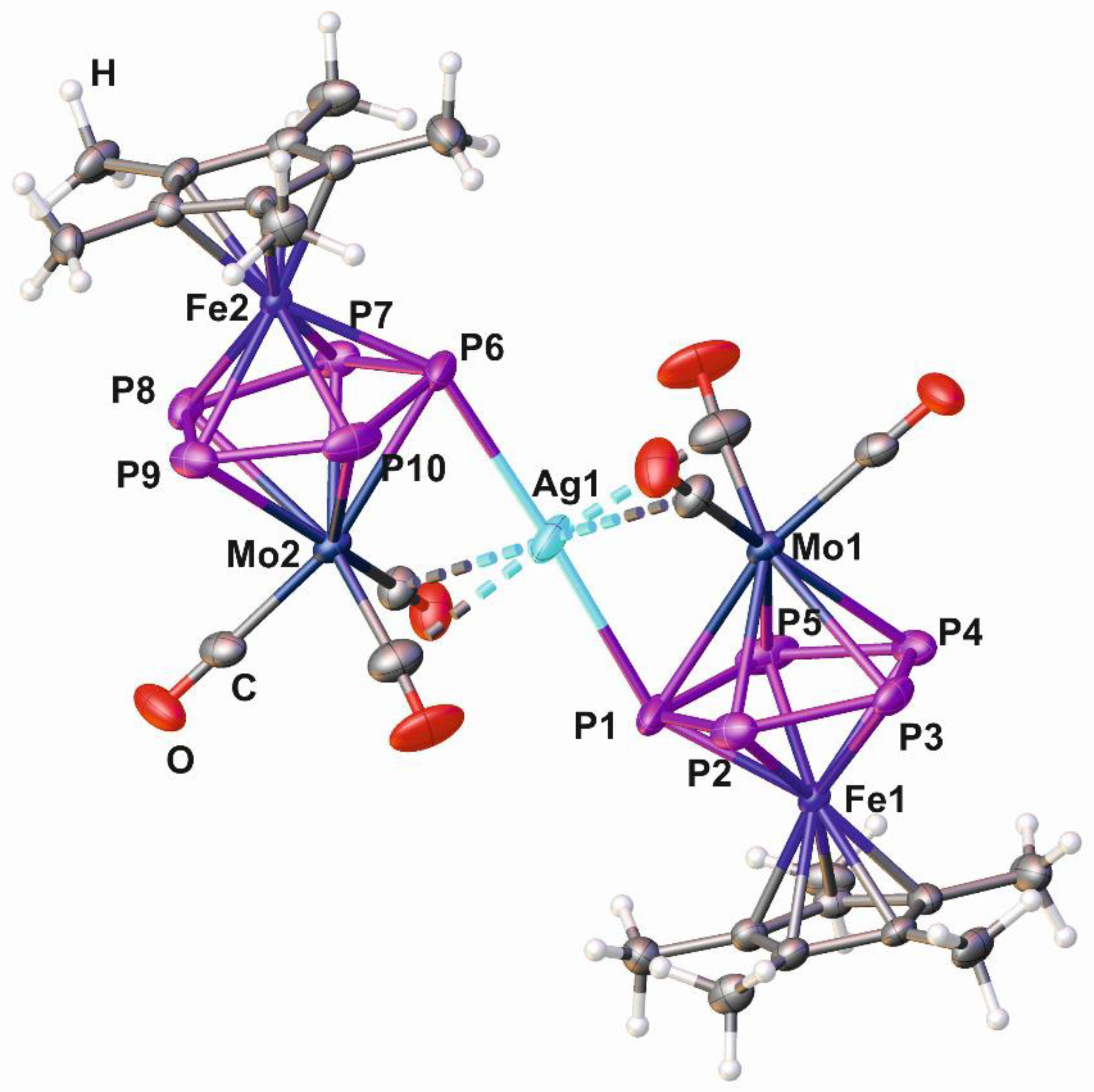
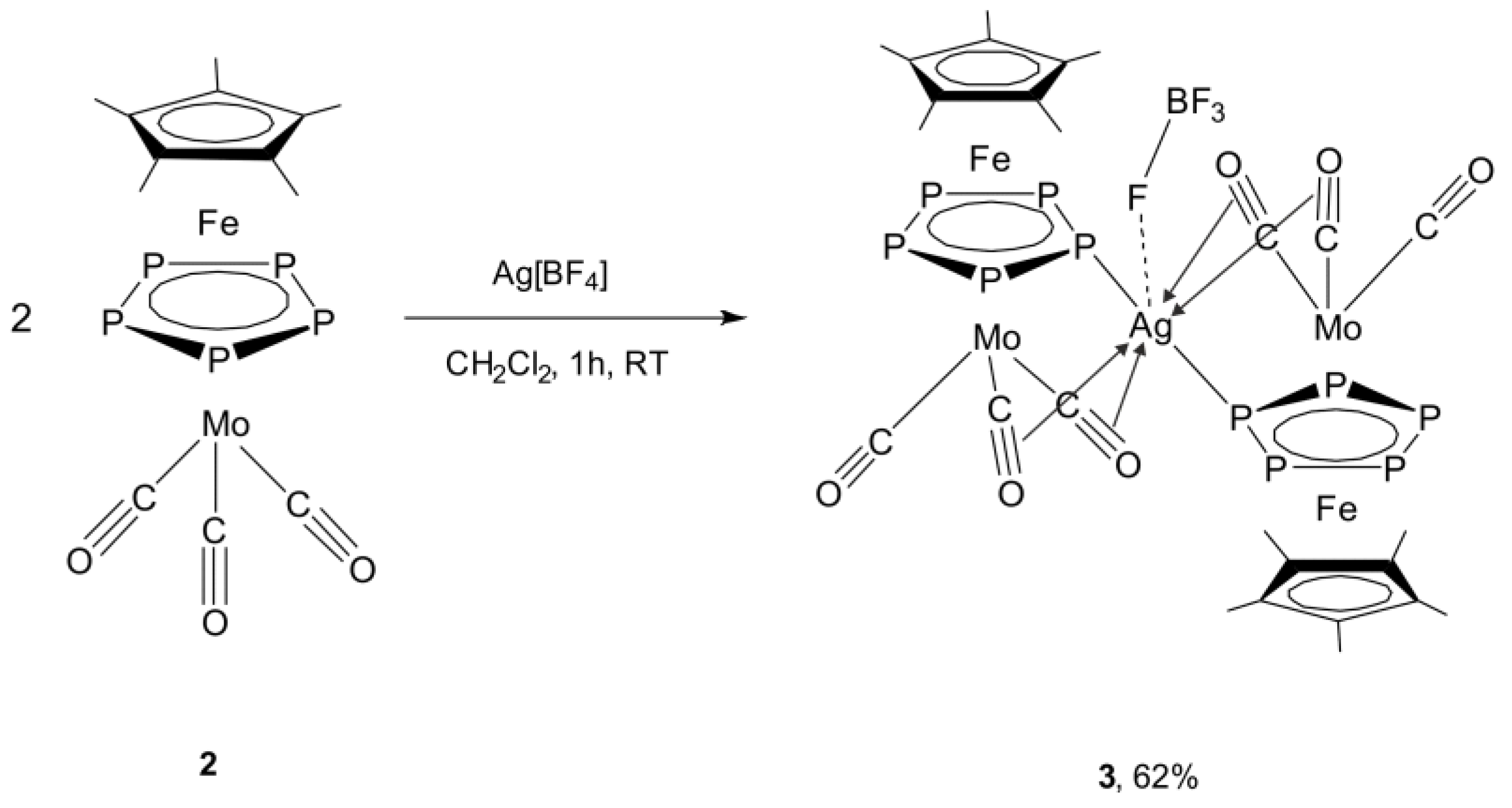
2.2. Synthesis and X-Ray Structure of the Coordination Compound [Ag{Cp*Fe(µ,η5:η5-P5)Mo(CO)3}2][BF4] (3)
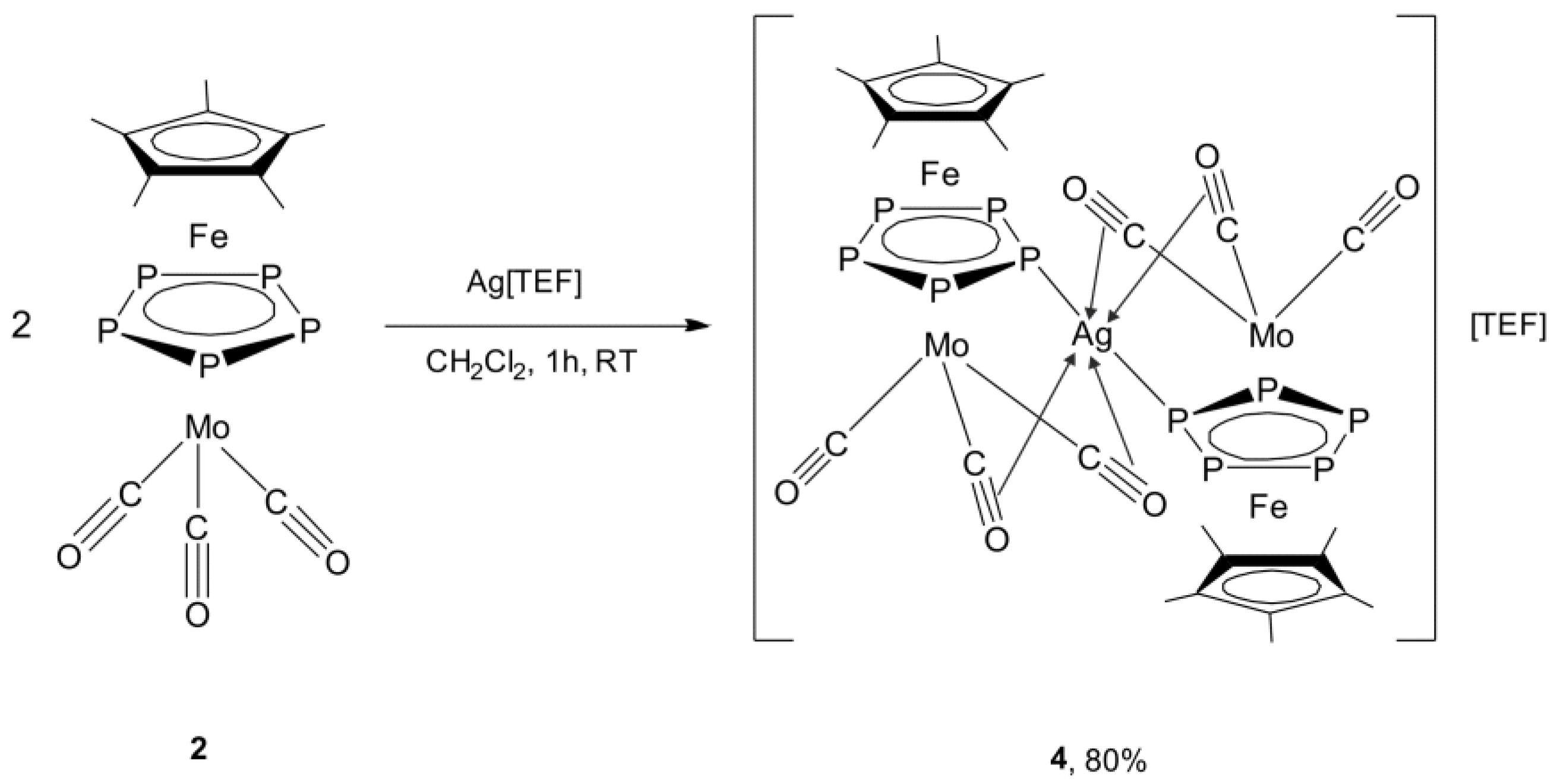
2.3. Synthesis and X-Ray Structure of the Coordination Compound [Ag{Cp*Fe(µ,η5:η5-P5)Mo(CO)3}2][TEF] (4)
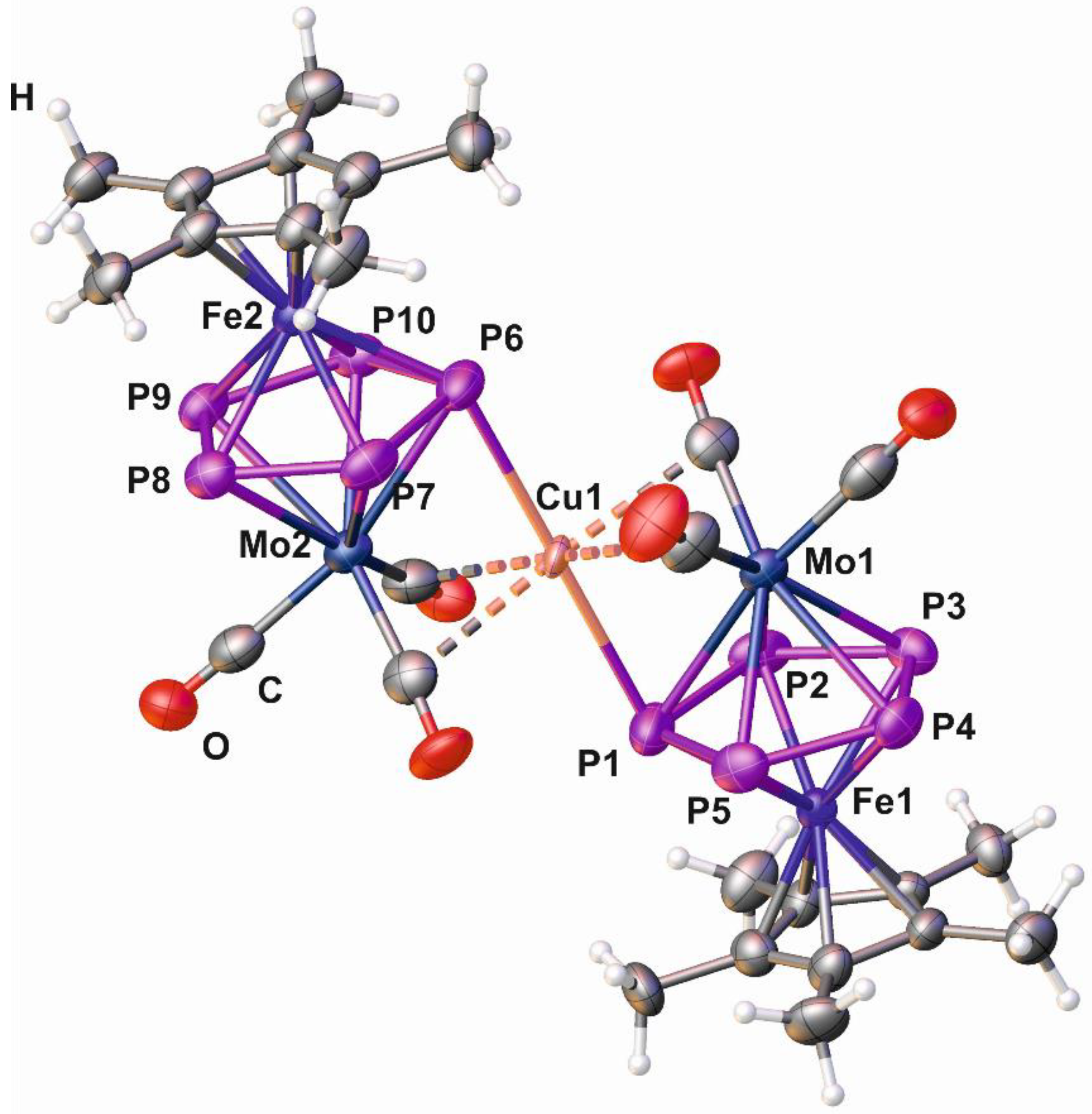
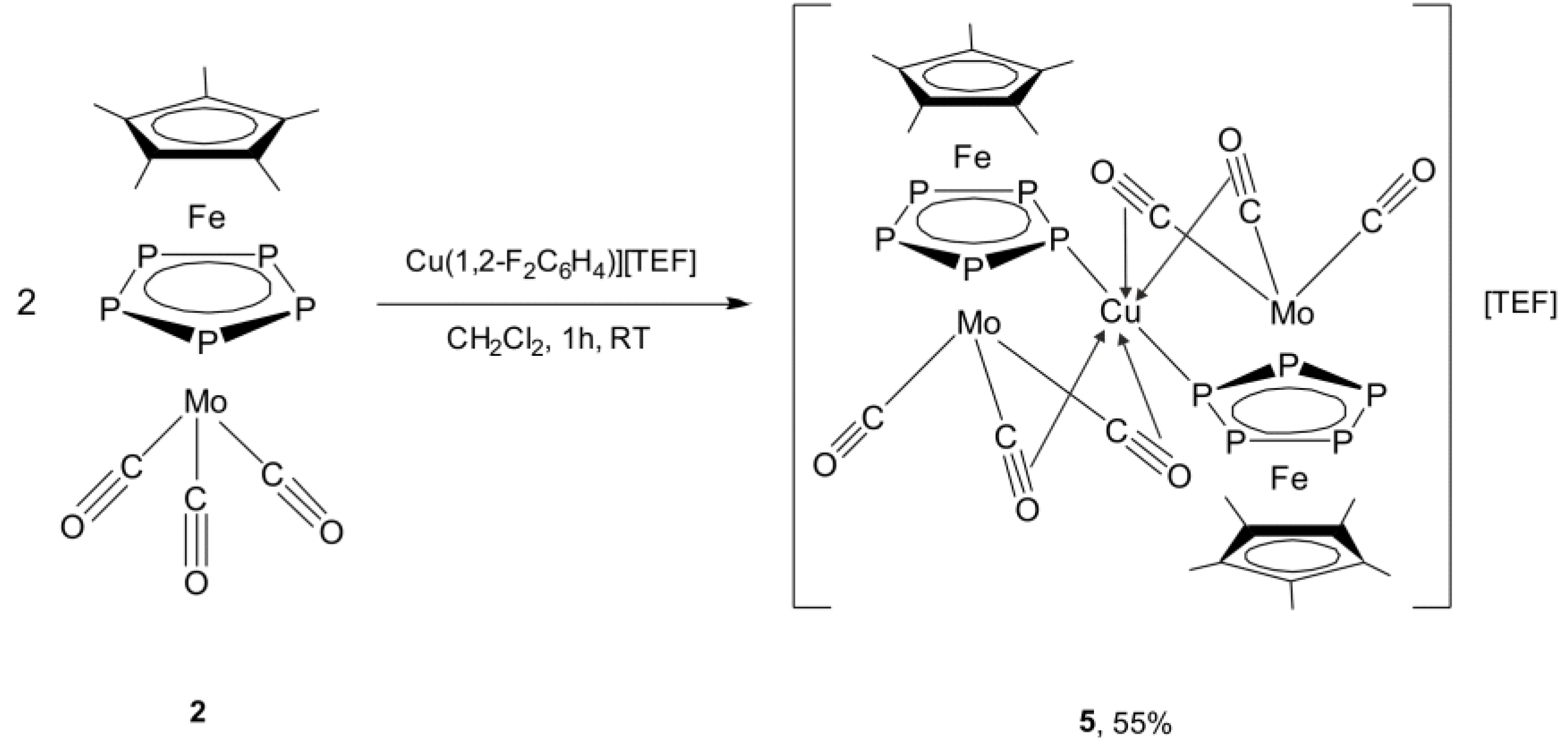
2.4. Synthesis and X-Ray Structure of the Complex [Cu{Cp*Fe(µ,η5:η5-P5)Mo(CO)3}2][TEF] (5)
3. Materials and Methods
3.1. General Information
3.2. Synthesis and Characterization of the Compounds 2 and 3–5
3.2.1. Synthesis and Characterization of [Cp*Fe(µ,η5:η5-P5)Mo(CO)3] (2)
3.2.2. Synthesis and Characterization of [Ag{Cp*Fe(µ,η5:η5-P5)Mo(CO)3}2][BF4] (3)
3.2.3. Synthesis and Characterization of [Ag{Cp*Fe(µ,η5:η5-P5)Mo(CO)3}2][TEF] (4)
3.2.4. Synthesis and Characterization of [Cu{Cp*Fe(µ,η5:η5-P5)Mo(CO)3}2][TEF] (5)
3.3. Crystallography
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Whitmire, K.H. Transition metal complexes of the naked pnictide elements. Coord. Chem. Rev. 2018, 376, 114–195. [Google Scholar] [CrossRef]
- Scheer, M. The coordination chemistry of group 15 element ligand complexes-a developing area. Dalton Trans. 2008, 4372–4386. [Google Scholar] [CrossRef] [PubMed]
- Scherer, O.J.; Brück, T. [(η5-P5)Fe(η5-C5Me5)], a pentaphosphaferrocene derivative. Angew. Chem. Int. Ed. 1987, 99, 59. [Google Scholar] [CrossRef]
- Fleischmann, M.; Welsch, S.; Peresypkina, E.V.; Virovets, A.V.; Scheer, M. Highly Dynamic Coordination Behavior of Pn Ligand Complexes towards “Naked” Cu+ Cations. Chem. Eur. J. 2015, 21, 14332–14336. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, M.; Welsch, S.; Krauss, H.; Schmidt, M.; Bodensteiner, M.; Peresypkina, E.V.; Sierka, M.; Gröger, C.; Scheer, M. Complexes of Monocationic Group 13 Elements with Pentaphospha- and Pentaarsaferrocene. Chem. Eur. J. 2014, 20, 3759–3768. [Google Scholar] [CrossRef] [PubMed]
- Scheer, M.; Gregoriades, L.J.; Merkle, R.; Johnson, B.P.; Dielmann, F. Formation of Spherical Giant Molecules and Dynamic Behaviour of Supramolecular Assemblies Based on Pn-Ligand Complexes. Phosphorus Sulfur Silicon Relat. Elem. 2008, 182, 504–508. [Google Scholar] [CrossRef]
- Welsch, S.; Gregoriades, L.J.; Sierka, M.; Zabel, M.; Virovets, A.V.; Scheer, M. Unusual Coordination Behavior of Pn-Ligand Complexes with Tl+. Angew. Chem. Int. Ed. 2007, 46, 9323–9326. [Google Scholar] [CrossRef] [PubMed]
- Scheer, M.; Gregoriades, L.J.; Virovets, A.V.; Kunz, W.; Neueder, R.; Krossing, I. Reversible Formation of Polymeric Chains by Coordination of Pentaphosphaferrocene with Silver(I) Cations. Angew. Chem. Int. Ed. 2006, 45, 5689–5693. [Google Scholar] [CrossRef] [PubMed]
- Dielmann, F.; Schindler, A.; Scheuermayer, S.; Bai, J.; Merkle, R.; Zabel, M.; Virovets, A.V.; Peresypkina, E.V.; Brunklaus, G.; Eckert, H.; et al. Coordination Polymers Based on [Cp*Fe(η5-P5)]: Solid-State Structure and MAS NMR Studies. Chem. Eur. J. 2012, 18, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Virovets, A.V.; Scheer, M. Pentaphosphaferrocene as a Linking Unit for the Formation of one- and Two-Dimensional Polymers. Angew. Chem. Int. Ed. 2002, 41, 1737–1740. [Google Scholar] [CrossRef]
- Schindler, A.; Heindl, C.; Balázs, G.; Gröger, C.; Virovets, A.V.; Peresypkina, E.V.; Scheer, M. Size-Determining Dependencies in Supramolecular Organometallic Host-Guest Chemistry. Chem. Eur. J. 2012, 18, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Scheer, M.; Schindler, A.; Bai, J.; Johnson, B.P.; Merkle, R.; Winter, R.; Virovets, A.V.; Peresypkina, E.V.; Blatov, V.A.; Sierka, M.; et al. Structures and Properties of Spherical 90-Vertex Fullerene-Like Nanoballs. Chem. Eur. J. 2010, 16, 2092–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheer, M.; Schindler, A.; Gröger, C.; Virovets, A.V.; Peresypkina, E.V. A Spherical Molecule with a Carbon-Free Ih-C80 Topological Frameworks. Angew. Chem. Int. Ed. 2009, 48, 5046–5049. [Google Scholar] [CrossRef] [PubMed]
- Scheer, M.; Schindler, A.; Merkle, R.; Johnson, B.P.; Linseis, M.; Winter, R.; Anson, C.E.; Virovets, A.V. Fullerene C60 as an Endohedral Molecule within an Inorganic Supramolecule. J. Am. Chem. Soc. 2007, 129, 13386–13387. [Google Scholar] [CrossRef] [PubMed]
- Scheer, M.; Bai, J.; Johnson, B.P.; Merkle, R.; Virovets, A.V.; Christopher, E.A. Fullerene-Like Nanoballs Formed by Pentaphosphaferrocene and CuBr. Eur. J. Inorg. Chem. 2005, 2005, 4023–4026. [Google Scholar] [CrossRef]
- Bai, J.; Virovets, A.V.; Scheer, M. Synthesis of Inorganic Fullerene-Like Molecules. Science 2003, 300, 781–782. [Google Scholar] [CrossRef]
- Welsch, S.; Gröger, C.; Sierka, M.; Scheer, M. An Organometallic Nanosized Capsule Consisting of cyclo-P5 Units and Copper(I) Ions. Angew. Chem. Int. Ed. 2011, 50, 1435–1438. [Google Scholar] [CrossRef]
- Elsayed Moussa, M.; Attenberger, B.; Peresypkina, E.V.; Scheer, M. Neutral two-dimensional organometallic-organic hybrid polymers based on pentaphosphaferrocene, bipyridyl linkers and CuCl. Dalton Trans. 2018, 47, 1014–1017. [Google Scholar] [CrossRef]
- Elsayed Moussa, M.; Welsch, S.; Lochner, M.; Peresypkina, E.V.; Virovets, A.V.; Scheer, M. Organometallic-Organic Hybrid Polymers Assembled from Pentaphosphaferrocene, Bipyridyl Linkers, and CuI ions. Eur. J. Inorg. Chem. 2018, 23, 2689–2694. [Google Scholar] [CrossRef]
- Scherer, O.J.; Brück, T.; Wolmershäuser, G. Pentaphosphaferrocene als Komplexliganden. Chem. Ber. 1989, 122, 2049–2054. [Google Scholar] [CrossRef]
- Rink, B.; Scherer, O.J.; Heckmann, G.; Wolmershäuser, G. Neutrale 30-Valenzelektronen-Tripeldeckerkomplexe mit cyclo-E5-Mitteldeck (E = P, As). Chem. Ber. 1992, 125, 1011–1016. [Google Scholar] [CrossRef]
- Kudinov, A.R.; Rybinskaya, M.I. New triple-decker complexes prepared by the stacking reactions of cationic metallofragments with sandwich compounds. Russ. Chem. Bull. 1999, 48, 1636–1642. [Google Scholar] [CrossRef]
- Kudinov, A.R.; Loginov, D.A.; Starikova, Z.A.; Petrovskii, P.V.; Corsini, M.; Zanello, P. Iron- and Ruthenium-Containing Triple-Decker Complexes with a Central Pentaphospholyl Ligand—X-ray Structures of [(η-C5H5)Fe(µ-η:η-P5)Ru(η-C5Me5)]PF6 and [(η-C5H5)Fe(µ-η:η-P5)Ru(η-C5Me5)]PF6. Eur. J. Inorg. Chem. 2002, 2002, 3018–3027. [Google Scholar] [CrossRef]
- Kudinov, A.R.; Petrovskii, P.V.; Rybinskaya, M.I. Synthesis and the fluxional behavior of the 30-electron cationic iron-molybdenum triple-decker complex with a central pentaphospholyl ligand, [(η-C7H7)Mo(µ-η:η-P5)Fe(η-C5Me5)]BF4. Russ. Chem. Bull. 1999, 48, 1374–1376. [Google Scholar] [CrossRef]
- Mädl, E.; Peresypkina, E.V.; Timoshkin, A.Y.; Scheer, M. Triple-decker sandwish complexes with a bent cyclo-P5 middle-deck. Chem. Commun. 2016, 52, 12298–12301. [Google Scholar] [CrossRef] [PubMed]
- Heinl, S.; Balázs, G.; Bodensteiner, M.; Scheer, M. Synthesis and characterization of manganese triple-decker complexes. Dalton Trans. 2016, 45, 1962–1966. [Google Scholar] [CrossRef] [PubMed]
- Scherer, O.J.; Schwalb, J.; Wolmershäuser, G.; Kaim, W.; Gross, R. cyclo-P5 as Complex Ligand―the Phosphorus Analogue of the Cyclopentadienyl Ligand. Angew. Chem. Int. Ed. 1986, 25, 363–364. [Google Scholar] [CrossRef]
- Goh, L.Y.; Wong, R.C.S.; Chu, C.K.; Hambley, T.W. Reaction of [{Cr(cp)(CO)3}2](cp = η5-C5H5) with elemental phosphorus. Isolation of [Cr2(cp)2(P5)] as a thermolysis product and its X-ray crystal structure. J. Chem. Soc. Dalton Trans. 1990, 977–982. [Google Scholar] [CrossRef]
- Fleischmann, M.; Dielmann, F.; Gregoriades, L.J.; Peresypkina, E.V.; Virovets, A.V.; Huber, S.; Timoshkin, A.Y.; Balázs, G.; Scheer, M. Redox and Coordination Behavior of the Hexaphosphabenzene Ligand in [(Cp*Mo)2(µ, η6:η6-P6)] Towards the “Naked” Cations Cu+, Ag+, and Tl+. Angew. Chem. Int. Ed. 2015, 54, 13110–13115. [Google Scholar] [CrossRef]
- Klingler, R.J.; Butler, W.M.; Curtis, M.D. Molecular Structure of Dicyclopentadienyltetracarbonyldimolybdenum (Mo≡Mo). Semibridging Carbonyls as Four-Electron Donors in Complexes with Metal-Metal Multiple Bonds. J. Am. Chem. Soc. 1978, 100, 5034–5038. [Google Scholar] [CrossRef]
- Ghisolfi, A.; Fliedel, C.; de Frémont, P.; Braunstein, P. Mono- and polynuclear Ag(I) complexes of N-functionalized bis(diphenylphosphino)amine DPPA-type ligands: Synthesis, solid-state structures and reactivity. Dalton Trans. 2017, 46, 5571–5586. [Google Scholar] [CrossRef] [PubMed]
- Croizat, P.; Sculfort, S.; Welter, R.; braunstein, P. Hexa- and Octanuclear Heterometallic Clusters with Copper-, Silver-, or Gold-Molybdenum Bonds and d10-d10 Interactions. Organometallics 2016, 35, 3949–3958. [Google Scholar] [CrossRef]
- Krossing, I. The Facile Preparation of Weakly Coordinating Anions: Structure and Characterisation of Silverpolyfluoroalkoxyaluminates AgAl(ORF)4, Calculation of the Alkoxide Ion Affinity. Chem. Eur. J. 2001, 7, 490–502. [Google Scholar] [CrossRef]
- Santiso-Quiñones, G.; Higelin, A.; Schaefer, J.; Brückner, R.; Knapp, C.; Krossing, I. Cu[Al(ORF)4] Starting Materials and their Application in the Preparation of {Cu(Sn)]+; (n = 12, 8) Complexes. Chem. Eur. J. 2009, 15, 6663–6677. [Google Scholar] [CrossRef]
- CrysAlisPro Software System, Rigaku Oxford Diffraction, version 1.171.38; Software for Data Reduction of the X-Ray Data; Rigaku: Tokyo, Japan, 2015.
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta Cryst. 1995, A51, 887–897. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with ShelXL. Acta Cryst. 2015, C27, 3–8. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 2’–5 are available from the authors. |









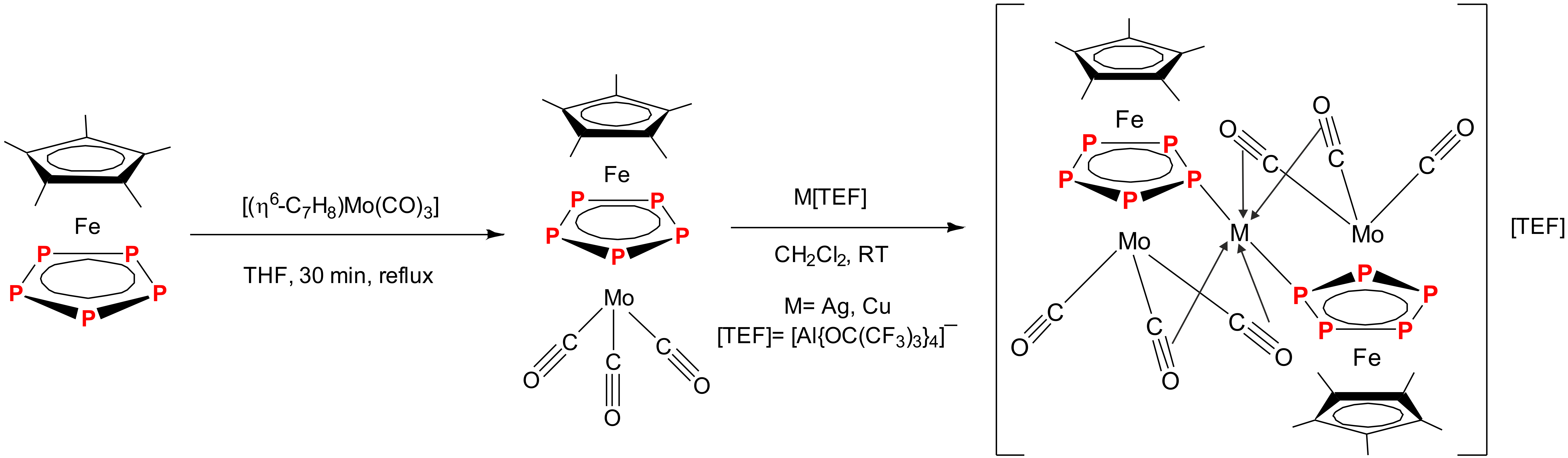{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2’ | M = Ag [X] = [BF4] 3 | M = Ag [X] = [TEF] 4 | M = Cu [X] = [TEF] 5’ | M = Cu [X] = [TEF] 5’’ | |
|---|---|---|---|---|---|
| (P,P)/Å | 2.154(1) | 2.160 | 2.158 | 2.156 | 2.154 |
| (M,P)/Å | ― | 2.742 | 2.695 | 2.687 | 2.662 |
| (Mo,M)/Å | ― | 2.837 | 2.824 | 2.680 | 2.673 |
| (P,M,P)/° | ― | 175.09(3) | 177.12(7) | 179.61(9) | 176.89(8) |
| (Mo,M,Mo)/° | ― | 155.93(3) | 173.56(3) | 174.92(4) | 173.93(4) |
| δ31P{1H}/ppm CD2Cl2, RT | 9.7 | 25.2 | 30.1 | 31.7 | |
| ῦCO/cm-1 KBr | 1963, 1955, 1894, 1881 | 1963, 1955, 1892, 1880 | 1980 (br), 1928, 1914 | 1993, 1961, 1923, 1910 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsayed Moussa, M.; Welsch, S.; Dütsch, L.; Piesch, M.; Reichl, S.; Seidl, M.; Scheer, M. The Triple-Decker Complex [Cp*Fe(µ,η5:η5-P5)Mo(CO)3] as a Building Block in Coordination Chemistry. Molecules 2019, 24, 325. https://doi.org/10.3390/molecules24020325
Elsayed Moussa M, Welsch S, Dütsch L, Piesch M, Reichl S, Seidl M, Scheer M. The Triple-Decker Complex [Cp*Fe(µ,η5:η5-P5)Mo(CO)3] as a Building Block in Coordination Chemistry. Molecules. 2019; 24(2):325. https://doi.org/10.3390/molecules24020325
Chicago/Turabian StyleElsayed Moussa, Mehdi, Stefan Welsch, Luis Dütsch, Martin Piesch, Stephan Reichl, Michael Seidl, and Manfred Scheer. 2019. "The Triple-Decker Complex [Cp*Fe(µ,η5:η5-P5)Mo(CO)3] as a Building Block in Coordination Chemistry" Molecules 24, no. 2: 325. https://doi.org/10.3390/molecules24020325