Taming the Notch Transcriptional Regulator for Cancer Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Diversity of Notch Signaling Mechanisms
2. Diversity of Notch Signaling Activities in Cancer
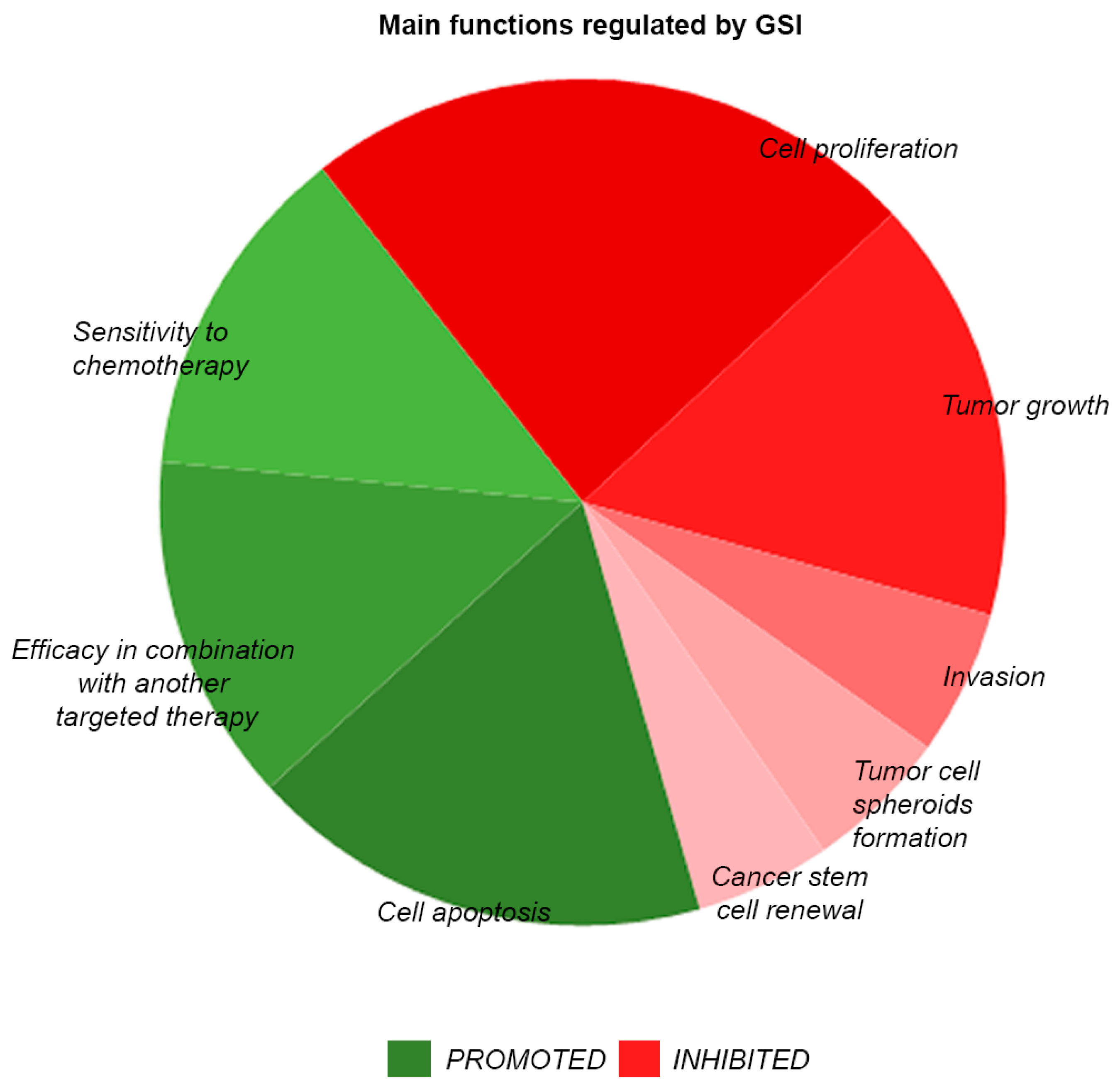
3. Advances in Targeting of Notch Signaling by Small Molecule Inhibitors
4. Combinatorial Treatments with Notch Inhibitors
5. Inhibition of Notch Signaling by Blocking Antibodies and Decoys
6. Recent Trends in Notch Targeting by Oligonucleotide-Based Methods
7. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Morgan, T.H. The Physical Basis of Heredity; JB Lippincott: Philadelphia, PA, USA, 1919. [Google Scholar]
- Dexter, J.S. The analysis of a case of continuous variation in drosophila by a study of its linkage relations. Am. Nat. 1914, 48, 712–758. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S.; Muskavitch, M.A.; Yedvobnick, B. Molecular cloning of notch, a locus affecting neurogenesis in drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1983, 80, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.; Kelley, M.R.; Young, M.W. Sequence of the notch locus of drosophila melanogaster: Relationship of the encoded protein to mammalian clotting and growth factors. Mol. Cell. Biol. 1986, 6, 3094–3108. [Google Scholar] [CrossRef] [PubMed]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The varied roles of notch in cancer. Annu. Rev. Pathol. 2017, 12, 245–275. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Golde, T.; Osborne, B. Notch signaling in cancer. Curr. Mol. Med. 2006, 6, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Shutter, J.R.; Maguire, M.; Sundberg, J.P.; Gallahan, D.; Closson, V.; Kitajewski, J.; Callahan, R.; et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000, 14, 1343–1352. [Google Scholar] [PubMed]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting notch, hedgehog, and wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Koury, J.; Zhong, L.; Hao, J. Targeting signaling pathways in cancer stem cells for cancer treatment. Stem. Cells Int. 2017, 2017, 2925869. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Del Alamo, D.; Rouault, H.; Schweisguth, F. Mechanism and significance of cis-inhibition in notch signalling. Curr. Biol. 2011, 21, R40–R47. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, K.; Perez, L.; Clausen, H.; Cohen, S. Glycosyltransferase activity of fringe modulates notch-delta interactions. Nature 2000, 406, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Cornejo, H.; Saucedo-Correa, G.; Oviedo-Boyso, J.; Valdez-Alarcon, J.J.; Baizabal-Aguirre, V.M.; Cajero-Juarez, M.; Bravo-Patino, A. The csl proteins, versatile transcription factors and context dependent corepressors of the notch signaling pathway. Cell Div. 2016, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.; Uosaki, H.; Shenje, L.T.; Kwon, C. Non-canonical notch signaling: Emerging role and mechanism. Trends Cell Biol. 2012, 22, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, F.; Osborne, B.A. Non-canonical notch signaling in cancer and immunity. Front. Oncol. 2014, 4, 345. [Google Scholar] [CrossRef] [PubMed]
- Blokzijl, A.; Dahlqvist, C.; Reissmann, E.; Falk, A.; Moliner, A.; Lendahl, U.; Ibanez, C.F. Cross-talk between the notch and tgf-beta signaling pathways mediated by interaction of the notch intracellular domain with smad3. J. Cell Biol. 2003, 163, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.R.; Hsieh, R.H.; Hsu, K.W.; Wu, M.Z.; Tseng, M.J.; Mai, R.T.; Wu Lee, Y.H.; Yeh, T.S. The cbf1-independent notch1 signal pathway activates human c-myc expression partially via transcription factor yy1. Carcinogenesis 2007, 28, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Linke, S.; Dias, J.M.; Gradin, K.; Wallis, T.P.; Hamilton, B.R.; Gustafsson, M.; Ruas, J.L.; Wilkins, S.; Bilton, R.L.; et al. Interaction with factor inhibiting hif-1 defines an additional mode of cross-coupling between the notch and hypoxia signaling pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 3368–3373. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Felli, M.P.; Palermo, R.; Di Mario, G.; Calce, A.; Di Giovine, M.; Frati, L.; Gulino, A.; Screpanti, I. Notch3 and pre-tcr interaction unveils distinct nf-kappab pathways in t-cell development and leukemia. EMBO J. 2006, 25, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Mutvei, A.P.; Chivukula, I.V.; Andersson, E.R.; Ramskold, D.; Sandberg, R.; Lee, K.L.; Kronqvist, P.; Mamaeva, V.; Ostling, P.; et al. Non-canonical notch signaling activates il-6/jak/stat signaling in breast tumor cells and is controlled by p53 and ikkalpha/ikkbeta. Oncogene 2013, 32, 4892–4902. [Google Scholar] [CrossRef] [PubMed]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.L.; Reynolds, T.C.; Smith, S.D.; Sklar, J. Tan-1, the human homolog of the drosophila notch gene, is broken by chromosomal translocations in t lymphoblastic neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.T.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of notch1 in human t cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Weaver, K.L.; Capobianco, A.J. Notch signalling in solid tumours: A little bit of everything but not all the time. Nat. Rev. Cancer 2011, 11, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Meunier, A.; Flores, A.N.; McDermott, N.; Rivera-Figueroa, K.; Perry, A.; Lynch, T.; Redalen, K.R.; Marignol, L. Hypoxia regulates notch-3 mrna and receptor activation in prostate cancer cells. Heliyon 2016, 2, e00104. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. Notch1 directly regulates c-myc and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Wang, Q.; Wang, Y.; Huang, K.; Yang, C.; Li, Y.; Yi, K.; Kang, C. Egfr/c-myc axis regulates tgfbeta/hippo/notch pathway via epigenetic silencing mir-524 in gliomas. Cancer Lett. 2017, 406, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.; Banerjee, L.; Cheung, C.W.; Mansour, M.R.; Jenkinson, S.; Gale, R.E.; Khwaja, A. Pi3k/mtor inhibition upregulates notch-myc signalling leading to an impaired cytotoxic response. Leukemia 2013, 27, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, M.V. The love-hate relationship between ras and notch. Genes Dev. 2005, 19, 1825–1839. [Google Scholar] [CrossRef] [PubMed]
- Kefas, B.; Comeau, L.; Floyd, D.H.; Seleverstov, O.; Godlewski, J.; Schmittgen, T.; Jiang, J.; diPierro, C.G.; Li, Y.; Chiocca, E.A.; et al. The neuronal microrna mir-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J. Neurosci. 2009, 29, 15161–15168. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Cheng, Y.S. Mir-34a inhibits pancreatic cancer progression through snail1-mediated epithelial-mesenchymal transition and the notch signaling pathway. Sci. Rep. 2017, 7, 38232. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Hao, X.; Zhang, M.; Tang, W.; Yang, M.; Li, L.; Xiang, D.; Desano, J.T.; Bommer, G.T.; Fan, D.; et al. Microrna mir-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS ONE 2009, 4, e6816. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Zhang, Y.; Wang, L. Microrna-206 targets notch3, activates apoptosis, and inhibits tumor cell migration and focus formation. J. Biol. Chem. 2009, 284, 31921–31927. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.; Shimizu, M.; Izrailit, J.; Ng, N.F.; Buchman, Y.; Pan, J.G.; Dering, J.; Reedijk, M. Cyclin d1 is a direct target of jag1-mediated notch signaling in breast cancer. Breast Cancer Res. Treat. 2010, 123, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Lanner, F.; Main, H.; Andersson, E.R.; Bergmann, O.; Sahlgren, C.; Heldring, N.; Hermanson, O.; Hansson, E.M.; Lendahl, U. Notch induces cyclin-d1-dependent proliferation during a specific temporal window of neural differentiation in es cells. Dev. Biol. 2010, 348, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Subramanyam, D.; Dey, D.; Kumar, R.V.; Rangarajan, A. Cooperation of notch and ras/mapk signaling pathways in human breast carcinogenesis. Mol. Cancer 2009, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Meurette, O.; Stylianou, S.; Rock, R.; Collu, G.M.; Gilmore, A.P.; Brennan, K. Notch activation induces akt signaling via an autocrine loop to prevent apoptosis in breast epithelial cells. Cancer Res. 2009, 69, 5015–5022. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, F.; Lu, J.; Li, T.; Yang, H.; Wu, C.; Liu, Y. Notch-1 signaling promotes the malignant features of human breast cancer through nf-kappab activation. PLoS ONE 2014, 9, e95912. [Google Scholar]
- Gutierrez, A.; Look, A.T. Notch and pi3k-akt pathways intertwined. Cancer Cell 2007, 12, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.J.; Zhang, L.; Wang, J.; Su, Q.; Feng, Q.; Zhang, X.H.; Mani, S.A.; Paulter, R.; Creighton, C.J.; Ittmann, M.M.; et al. Notch promotes tumor metastasis in a prostate-specific pten-null mouse model. J. Clin. Invest. 2016, 126, 2626–2641. [Google Scholar] [CrossRef] [PubMed]
- Phin, S.; Moore, M.W.; Cotter, P.D. Genomic rearrangements of pten in prostate cancer. Front. Oncol. 2013, 3, 240. [Google Scholar] [CrossRef] [PubMed]
- Lotan, T.L.; Carvalho, F.L.; Peskoe, S.B.; Hicks, J.L.; Good, J.; Fedor, H.; Humphreys, E.; Han, M.; Platz, E.A.; Squire, J.A.; et al. Pten loss is associated with upgrading of prostate cancer from biopsy to radical prostatectomy. Mod. Pathol. 2015, 28, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of pten induces resistance to notch1 inhibition in t-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Wang, H.; Xia, X.; Rao, Y.; Ma, X.; Ma, D.; Wu, P.; Chen, G. Loss of e-cadherin promotes prostate cancer metastasis via upregulation of metastasis-associated gene 1 expression. Oncol. Lett. 2012, 4, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Zhao, X.; Zhang, X.; Luo, M.; Zuo, X.; Huang, S.; Wang, Y.; Gu, S. Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a slug-dependent manner. Mol. Cancer 2015, 14, 28. [Google Scholar] [CrossRef] [PubMed]
- Fender, A.W.; Nutter, J.M.; Fitzgerald, T.L.; Bertrand, F.E.; Sigounas, G. Notch-1 promotes stemness and epithelial to mesenchymal transition in colorectal cancer. J. Cell Biochem. 2015, 116, 2517–2527. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Sarkar, F.H. The role of notch signaling pathway in epithelial-mesenchymal transition (emt) during development and tumor aggressiveness. Curr. Drug Targets 2010, 11, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.; Gurrapu, S.; Cagnoni, G.; Capparuccia, L.; Tamagnone, L. Plexind1 is a novel transcriptional target and effector of notch signaling in cancer cells. PLoS ONE 2016, 11, e0164660. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Finisguerra, V.; Capparuccia, L.; Camperi, A.; Swiercz, J.M.; Rizzolio, S.; Rolny, C.; Christensen, C.; Bertotti, A.; Sarotto, I.; et al. Sema3e-plexin d1 signaling drives human cancer cell invasiveness and metastatic spreading in mice. J. Clin. Invest. 2010, 120, 2684–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, C.H.; Murray, K.D.; Jou, M.F.; Hsu, S.M.; Cheng, H.J.; Huang, P.H. Sema3e/plexin-d1 mediated epithelial-to-mesenchymal transition in ovarian endometrioid cancer. PLoS ONE 2011, 6, e19396. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Oh, W.J.; Gaiano, N.; Yoshida, Y.; Gu, C. Semaphorin 3e-plexin-d1 signaling regulates vegf function in developmental angiogenesis via a feedback mechanism. Genes Dev. 2011, 25, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.L.; Kissil, J.L. Notch signaling in pancreatic cancer: Oncogene or tumor suppressor? Trends Mol. Med. 2013, 19, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Yap, L.F.; Lee, D.; Khairuddin, A.; Pairan, M.F.; Puspita, B.; Siar, C.H.; Paterson, I.C. The opposing roles of notch signalling in head and neck cancer: A mini review. Oral. Dis. 2015, 21, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761–17766. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, A.; Talora, C.; Okuyama, R.; Nicolas, M.; Mammucari, C.; Oh, H.; Aster, J.C.; Krishna, S.; Metzger, D.; Chambon, P.; et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001, 20, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Rampias, T.; Vgenopoulou, P.; Avgeris, M.; Polyzos, A.; Stravodimos, K.; Valavanis, C.; Scorilas, A.; Klinakis, A. A new tumor suppressor role for the notch pathway in bladder cancer. Nat. Med. 2014, 20, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Su, H.; Li, X.; Guo, G.; Cheng, L.; Qin, R.; Qing, G.; Liu, H. The notch ligand jagged2 promotes pancreatic cancer metastasis independent of notch signaling activation. Mol. Cancer Ther. 2015, 14, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Ortica, S.; Tarantino, N.; Aulner, N.; Israel, A.; Gupta-Rossi, N. The 4 notch receptors play distinct and antagonistic roles in the proliferation and hepatocytic differentiation of liver progenitors. FASEB J. 2014, 28, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.; Zhang, Z.; Zhou, Y.; Wang, W.; Li, Y.; Zhang, H.; Dong, G.; Zhao, Q.; Ji, G. Notch1 and notch2 have opposite prognostic effects on patients with colorectal cancer. Ann. Oncol. 2011, 22, 2440–2447. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.; Zheng, J.; Wang, W.; Zhao, Q.; Li, Y.; Li, J.; Xie, H.; Zhang, H.; Dong, G.; Xu, C.; et al. Notch2 expression is decreased in colorectal cancer and related to tumor differentiation status. Ann. Surg. Oncol. 2009, 16, 3259–3266. [Google Scholar] [CrossRef] [PubMed]
- Choy, L.; Hagenbeek, T.J.; Solon, M.; French, D.; Finkle, D.; Shelton, A.; Venook, R.; Brauer, M.J.; Siebel, C.W. Constitutive notch3 signaling promotes the growth of basal breast cancers. Cancer Res. 2017, 77, 1439–1452. [Google Scholar] [CrossRef] [PubMed]
- Danza, G.; Di Serio, C.; Ambrosio, M.R.; Sturli, N.; Lonetto, G.; Rosati, F.; Rocca, B.J.; Ventimiglia, G.; del Vecchio, M.T.; Prudovsky, I.; et al. Notch3 is activated by chronic hypoxia and contributes to the progression of human prostate cancer. Int. J. Cancer 2013, 133, 2577–2586. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, N.; Song, W.; You, N.; Li, Q.; Sun, W.; Zhang, Y.; Wang, D.; Dou, K. The significance of notch1 compared with notch3 in high metastasis and poor overall survival in hepatocellular carcinoma. PLoS ONE 2013, 8, e57382. [Google Scholar] [CrossRef] [PubMed]
- Kayamori, K.; Katsube, K.; Sakamoto, K.; Ohyama, Y.; Hirai, H.; Yukimori, A.; Ohata, Y.; Akashi, T.; Saitoh, M.; Harada, K.; et al. Notch3 is induced in cancer-associated fibroblasts and promotes angiogenesis in oral squamous cell carcinoma. PLoS ONE 2016, 11, e0154112. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi-Elias, P.; Hu, T.; Jenkins, D.; Firestone, B.; Gans, S.; Kurth, E.; Capodieci, P.; Deplazes-Lauber, J.; Petropoulos, K.; Thiel, P.; et al. Characterization of activating mutations of notch3 in t-cell acute lymphoblastic leukemia and anti-leukemic activity of notch3 inhibitory antibodies. Oncogene 2016, 35, 6077–6086. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Severson, E.; Pear, W.S.; Liu, X.S.; Aster, J.C.; Blacklow, S.C. The common oncogenomic program of notch1 and notch3 signaling in t-cell acute lymphoblastic leukemia. PLoS ONE 2017, 12, e0185762. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Kong, Y.; Xu, M.; Zhang, H. Notch3 functions as a tumor suppressor by controlling cellular senescence. Cancer Res. 2013, 73, 3451–3459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, X.; Luo, J.; Xiao, W.; Ye, X.; Chen, M.; Li, Y.; Zhang, G.J. Notch3 inhibits epithelial-mesenchymal transition by activating kibra-mediated hippo/yap signaling in breast cancer epithelial cells. Oncogenesis 2016, 5, e269. [Google Scholar] [CrossRef] [PubMed]
- Nagamatsu, I.; Onishi, H.; Matsushita, S.; Kubo, M.; Kai, M.; Imaizumi, A.; Nakano, K.; Hattori, M.; Oda, Y.; Tanaka, M.; et al. Notch4 is a potential therapeutic target for triple-negative breast cancer. Anticancer Res. 2014, 34, 69–80. [Google Scholar] [PubMed]
- Bui, Q.T.; Im, J.H.; Jeong, S.B.; Kim, Y.M.; Lim, S.C.; Kim, B.; Kang, K.W. Essential role of notch4/stat3 signaling in epithelial-mesenchymal transition of tamoxifen-resistant human breast cancer. Cancer Lett. 2017, 390, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.J.; Chen, Y.Y.; Zhang, X.; Liu, F.Q.; Yue, T.T.; Ye, B.; Yao, J. Notch4 inhibition reduces migration and invasion and enhances sensitivity to docetaxel by inhibiting akt/fascin in pancreatic cancer cells. Oncol. Lett. 2016, 12, 3499–3505. [Google Scholar] [CrossRef] [PubMed]
- Bonyadi Rad, E.; Hammerlindl, H.; Wels, C.; Popper, U.; Ravindran Menon, D.; Breiteneder, H.; Kitzwoegerer, M.; Hafner, C.; Herlyn, M.; Bergler, H.; et al. Notch4 signaling induces a mesenchymal-epithelial-like transition in melanoma cells to suppress malignant behaviors. Cancer Res. 2016, 76, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Kulic, I.; Robertson, G.; Chang, L.; Baker, J.H.; Lockwood, W.W.; Mok, W.; Fuller, M.; Fournier, M.; Wong, N.; Chou, V.; et al. Loss of the notch effector rbpj promotes tumorigenesis. J. Exp. Med. 2015, 212, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Ganesan, K.; Tan, L.K.; Laban, M.; Wu, J.; Zhao, X.D.; Li, H.; Leung, C.H.; Zhu, Y.; Wei, C.L.; et al. A precisely regulated gene expression cassette potently modulates metastasis and survival in multiple solid cancers. PLoS Genet. 2008, 4, e1000129. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Wu, Q.; Kim, L.; Miller, T.E.; Liau, B.B.; Mack, S.C.; Yang, K.; Factor, D.C.; Fang, X.; Huang, Z.; et al. Rbpj maintains brain tumor-initiating cells through cdk9-mediated transcriptional elongation. J. Clin. Invest. 2016, 126, 2757–2772. [Google Scholar] [CrossRef] [PubMed]
- De Kloe, G.E.; De Strooper, B. Small molecules that inhibit notch signaling. Methods Mol. Biol. 2014, 1187, 311–322. [Google Scholar] [PubMed]
- Liebler, S.S.; Feldner, A.; Adam, M.G.; Korff, T.; Augustin, H.G.; Fischer, A. No evidence for a functional role of bi-directional notch signaling during angiogenesis. PLoS ONE 2012, 7, e53074. [Google Scholar] [CrossRef] [PubMed]
- Vidal, G.A.; Naresh, A.; Marrero, L.; Jones, F.E. Presenilin-dependent gamma-secretase processing regulates multiple erbb4/her4 activities. J. Biol. Chem. 2005, 280, 19777–19783. [Google Scholar] [CrossRef] [PubMed]
- Murakami, D.; Okamoto, I.; Nagano, O.; Kawano, Y.; Tomita, T.; Iwatsubo, T.; De Strooper, B.; Yumoto, E.; Saya, H. Presenilin-dependent gamma-secretase activity mediates the intramembranous cleavage of cd44. Oncogene 2003, 22, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.E.; Albright, C.F. Recent progress in the medicinal chemistry of gamma-secretase inhibitors. Curr. Top. Med. Chem. 2008, 8, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Dovey, H.F.; John, V.; Anderson, J.P.; Chen, L.Z.; de Saint Andrieu, P.; Fang, L.Y.; Freedman, S.B.; Folmer, B.; Goldbach, E.; Holsztynska, E.J.; et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J. Neurochem. 2001, 76, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Fu, J.; Zheng, S.; Yao, J.; Wang, S.; Liu, D.D.; Yuan, Y.; Sulman, E.P.; Lang, F.F.; Colman, H.; et al. A high notch pathway activation predicts response to gamma secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem Cells 2014, 32, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Gavai, A.V.; Quesnelle, C.; Norris, D.; Han, W.C.; Gill, P.; Shan, W.; Balog, A.; Chen, K.; Tebben, A.; Rampulla, R.; et al. Discovery of clinical candidate bms-906024: A potent pan-notch inhibitor for the treatment of leukemia and solid tumors. ACS Med. Chem. Lett. 2015, 6, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Bolos, V.; Mira, E.; Martinez-Poveda, B.; Luxan, G.; Canamero, M.; Martinez, A.C.; Manes, S.; de la Pompa, J.L. Notch activation stimulates migration of breast cancer cells and promotes tumor growth. Breast Cancer Res. 2013, 15, R54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, F.; Zhu, S.; Ruan, J.; Muftuoglu, Y.; Zhang, L.; Yuan, Q. Combination therapy of ry10-4 with the gamma-secretase inhibitor dapt shows promise in treating her2-amplified breast cancer. Oncotarget 2016, 7, 4142–4154. [Google Scholar] [PubMed]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. Notch pathway blockade depletes cd133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Yeatman, T.; Weber, J.; Coppola, D.; Schell, M.J.; Han, G.; Almhanna, K.; Kim, R.; Valone, T.; Jump, H.; et al. A phase ii study of ro4929097 in metastatic colorectal cancer. Eur. J. Cancer 2012, 48, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Samon, J.B.; Castillo-Martin, M.; Hadler, M.; Ambesi-Impiobato, A.; Paietta, E.; Racevskis, J.; Wiernik, P.H.; Rowe, J.M.; Jakubczak, J.; Randolph, S.; et al. Preclinical analysis of the gamma-secretase inhibitor pf-03084014 in combination with glucocorticoids in t-cell acute lymphoblastic leukemia. Mol. Cancer Ther. 2012, 11, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- De Jesus-Acosta, A.; Laheru, D.; Maitra, A.; Arcaroli, J.; Rudek, M.A.; Dasari, A.; Blatchford, P.J.; Quackenbush, K.; Messersmith, W. A phase ii study of the gamma secretase inhibitor ro4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Invest. New Drugs 2014, 32, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.; Demuth, T.; Guthrie, T.; Wen, P.Y.; Mason, W.P.; Chinnaiyan, P.; Butowski, N.; Groves, M.D.; Kesari, S.; Freedman, S.J.; et al. Phase i pharmacologic and pharmacodynamic study of the gamma secretase (notch) inhibitor mk-0752 in adult patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 2307–2313. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, J.; Grim, J.; Strack, P.; Rao, S.; Tibbitts, D.; Winter, C.; Hardwick, J.; Welcker, M.; Meijerink, J.P.; Pieters, R.; et al. Fbw7 mutations in leukemic cells mediate notch pathway activation and resistance to gamma-secretase inhibitors. J. Exp. Med. 2007, 204, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Yashiro-Ohtani, Y.; Wang, H.; Zang, C.; Arnett, K.L.; Bailis, W.; Ho, Y.; Knoechel, B.; Lanauze, C.; Louis, L.; Forsyth, K.S.; et al. Long-range enhancer activity determines myc sensitivity to notch inhibitors in t cell leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E4946–E4953. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Sperling, R.A.; Seimers, E.; Sethuraman, G.; Mohs, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Sun, X.; et al. Peripheral and central effects of gamma-secretase inhibition by semagacestat in alzheimer’s disease. Alzheimers Res. Ther. 2015, 7, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of alzheimer’s disease. N. Eng. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Ryeom, S.W. The cautionary tale of side effects of chronic notch1 inhibition. J. Clin. Invest. 2011, 121, 508–509. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Turkoz, A.; Jackson, E.N.; Corbo, J.C.; Engelbach, J.A.; Garbow, J.R.; Piwnica-Worms, D.R.; Kopan, R. Notch1 loss of heterozygosity causes vascular tumors and lethal hemorrhage in mice. J. Clin. Invest. 2011, 121, 800–808. [Google Scholar] [CrossRef] [PubMed]
- VanDussen, K.L.; Carulli, A.J.; Keeley, T.M.; Patel, S.R.; Puthoff, B.J.; Magness, S.T.; Tran, I.T.; Maillard, I.; Siebel, C.; Kolterud, A.; et al. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development 2012, 139, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Astudillo, L.; Da Silva, T.G.; Wang, Z.; Han, X.; Jin, K.; VanWye, J.; Zhu, X.; Weaver, K.; Oashi, T.; Lopes, P.E.; et al. The small molecule imr-1 inhibits the notch transcriptional activation complex to suppress tumorigenesis. Cancer Res. 2016, 76, 3593–3603. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, M.R.; Biskup, E.; Manfe, V.; Savorani, C.; Liszewski, W.; Wiren, J.; Specht, L.; Gniadecki, R. Chemotherapeutic treatment is associated with notch1 induction in cutaneous t-cell lymphoma. Leuk. Lymphoma 2017, 58, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Ma, Y.; Zhang, Z.; Zhao, J.; Kobayashi, H.; Zhang, L.; Fu, L. Expression of stat3 and notch1 is associated with cisplatin resistance in head and neck squamous cell carcinoma. Oncol. Rep. 2010, 23, 671–676. [Google Scholar] [PubMed]
- Meng, R.D.; Shelton, C.C.; Li, Y.M.; Qin, L.X.; Notterman, D.; Paty, P.B.; Schwartz, G.K. Gamma-secretase inhibitors abrogate oxaliplatin-induced activation of the notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. 2009, 69, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Qian, C. Inhibition of notch3 enhances sensitivity to gemcitabine in pancreatic cancer through an inactivation of pi3k/akt-dependent pathway. Med. Oncol. 2010, 27, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Stephen, S.L.; Hanby, A.M.; Horgan, K.; Perry, S.L.; Richardson, J.; Roundhill, E.A.; Valleley, E.M.; Verghese, E.T.; Williams, B.J.; et al. Chemotherapy induces notch1-dependent mrp1 up-regulation, inhibition of which sensitizes breast cancer cells to chemotherapy. BMC Cancer 2015, 15, 634. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.L.; Chen, C.; Yang, Y.; Wang, C.; Yang, T.; Yang, X.; Liu, S.C. Gamma secretase inhibitor enhances sensitivity to doxorubicin in mda-mb-231 cells. Int. J. Clin. Exp. Pathol. 2015, 8, 4378–4387. [Google Scholar] [PubMed]
- Dong, Y.; Li, A.; Wang, J.; Weber, J.D.; Michel, L.S. Synthetic lethality through combined notch-epidermal growth factor receptor pathway inhibition in basal-like breast cancer. Cancer Res. 2010, 70, 5465–5474. [Google Scholar] [CrossRef] [PubMed]
- Pandya, K.; Meeke, K.; Clementz, A.G.; Rogowski, A.; Roberts, J.; Miele, L.; Albain, K.S.; Osipo, C. Targeting both notch and erbb-2 signalling pathways is required for prevention of erbb-2-positive breast tumour recurrence. Br. J. Cancer 2011, 105, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Dai, J.; Keller, J.M.; Mizokami, A.; Xia, S.; Keller, E.T. Notch pathway inhibition using pf-03084014, a gamma-secretase inhibitor (gsi), enhances the antitumor effect of docetaxel in prostate cancer. Clin. Cancer Res. 2015, 21, 4619–4629. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Nakada, M.; Teng, L.; Furuta, T.; Sabit, H.; Hayashi, Y.; Demuth, T.; Hirao, A.; Sato, H.; Zhao, G.; et al. Combination therapy using notch and akt inhibitors is effective for suppressing invasion but not proliferation in glioma cells. Neurosci. Lett. 2013, 534, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Ambrogio, C.; Gomez-Lopez, G.; Falcone, M.; Vidal, A.; Nadal, E.; Crosetto, N.; Blasco, R.B.; Fernandez-Marcos, P.J.; Sanchez-Cespedes, M.; Ren, X.; et al. Combined inhibition of ddr1 and notch signaling is a therapeutic strategy for kras-driven lung adenocarcinoma. Nat. Med. 2016, 22, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, F.; Clifton, N.; Sullivan, D.M.; Dalton, W.S.; Gabrilovich, D.I.; Nefedova, Y. Combined inhibition of notch signaling and bcl-2/bcl-xl results in synergistic antimyeloma effect. Mol. Cancer Ther. 2010, 9, 3200–3209. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; Del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. C-myc is an important direct target of notch1 in t-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.; Falk, A.; Dyson, M.R.; Melidoni, A.N.; Parthiban, K.; Young, J.L.; Roake, W.; McCafferty, J. Generation of anti-notch antibodies and their application in blocking notch signalling in neural stem cells. Methods 2012, 58, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Yen, W.C.; Fischer, M.M.; Axelrod, F.; Bond, C.; Cain, J.; Cancilla, B.; Henner, W.R.; Meisner, R.; Sato, A.; Shah, J.; et al. Targeting notch signaling with a notch2/notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clin. Cancer Res. 2015, 21, 2084–2095. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Smith, L.S.; Bendell, J.C.; Rangwala, F.A.; Schmidt, W.; Stephenson, J.; Kapoun, A.; Xu, L.; Hill, D.; Zhou, L.; et al. Phase ib of anticancer stem cell antibody omp-59r5 (anti-notch2/3) in combination with nab-paclitaxel and gemcitabine (nab-p+gem) in patients (pts) with untreated metastatic pancreatic cancer (mpc). J. Clin. Oncol. 2014, 32, 220. [Google Scholar] [CrossRef]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; de Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.; Cattaruzza, F.; Yen, W.-C.; Yeung, P.; Fischer, M.; Cancilla, B.; O’Young, G.; Tam, R.; Liu, Y.-W.; Gurney, A.; et al. Abstract 4652: Effects of anti-dll4 treatment on non-small cell lung cancer (nsclc) human xenograft tumors. Cancer Res. 2016, 76, 4652. [Google Scholar] [CrossRef]
- Ridgway, J.; Zhang, G.; Wu, Y.; Stawicki, S.; Liang, W.C.; Chanthery, Y.; Kowalski, J.; Watts, R.J.; Callahan, C.; Kasman, I.; et al. Inhibition of dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 2006, 444, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Hu, W.; Hu, L.; Previs, R.A.; Dalton, H.J.; Yang, X.Y.; Sun, Y.; McGuire, M.; Rupaimoole, R.; Nagaraja, A.S.; et al. Dll4 inhibition plus aflibercept markedly reduces ovarian tumor growth. Mol. Cancer Ther. 2016, 15, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, Y.; Hernandez, S.L.; Das, I.; Ahn, A.; Huang, J.; Vorontchikhina, M.; Sharma, A.; Kanamaru, E.; Borisenko, V.; Desilva, D.M.; et al. A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res. 2008, 68, 4727–4735. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, T.; Goto, H.; Mitsuhashi, A.; Tabata, S.; Ogawa, H.; Uehara, H.; Saijo, A.; Kakiuchi, S.; Maekawa, Y.; Yasutomo, K.; et al. Dll4-fc, an inhibitor of dll4-notch signaling, suppresses liver metastasis of small cell lung cancer cells through the downregulation of the nf-kappab activity. Mol. Cancer Ther. 2012, 11, 2578–2587. [Google Scholar] [CrossRef] [PubMed]
- Baladron, V.; Ruiz-Hidalgo, M.J.; Nueda, M.L.; Diaz-Guerra, M.J.; Garcia-Ramirez, J.J.; Bonvini, E.; Gubina, E.; Laborda, J. Dlk acts as a negative regulator of notch1 activation through interactions with specific egf-like repeats. Exp. Cell Res. 2005, 303, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.; Berger, C.; Moll, I.; Adam, M.G.; Schwarz, F.; Mohr, K.; Augustin, H.G.; Fischer, A. Soluble notch ligand and receptor peptides act antagonistically during angiogenesis. Cardiova. Res. 2015, 107, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Kangsamaksin, T.; Murtomaki, A.; Kofler, N.M.; Cuervo, H.; Chaudhri, R.A.; Tattersall, I.W.; Rosenstiel, P.E.; Shawber, C.J.; Kitajewski, J. Notch decoys that selectively block dll/notch or jag/notch disrupt angiogenesis by unique mechanisms to inhibit tumor growth. Cancer Discov. 2015, 5, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Moellering, R.E.; Cornejo, M.; Davis, T.N.; Del Bianco, C.; Aster, J.C.; Blacklow, S.C.; Kung, A.L.; Gilliland, D.G.; Verdine, G.L.; Bradner, J.E. Direct inhibition of the notch transcription factor complex. Nature 2009, 462, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Lundin, K.E.; Gissberg, O.; Smith, C.I. Oligonucleotide therapies: The past and the present. Hum. Gene Ther. 2015, 26, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.; Ishii, H.; Aono, H.; Takai, M.; Honda, T.; Aratani, S.; Fukamizu, A.; Nakamura, H.; Yoshino, S.; Kobata, T.; et al. Role of notch-1 intracellular domain in activation of rheumatoid synoviocytes. Arthritis Rheum. 2001, 44, 1545–1554. [Google Scholar] [CrossRef]
- Shan, L.; Aster, J.C.; Sklar, J.; Sunday, M.E. Notch-1 regulates pulmonary neuroendocrine cell differentiation in cell lines and in transgenic mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L500–L509. [Google Scholar] [CrossRef] [PubMed]
- Zimrin, A.B.; Pepper, M.S.; McMahon, G.A.; Nguyen, F.; Montesano, R.; Maciag, T. An antisense oligonucleotide to the notch ligand jagged enhances fibroblast growth factor-induced angiogenesis in vitro. J. Biol. Chem. 1996, 271, 32499–32502. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chan, S.L.; Miele, L.; Yao, P.J.; Mackes, J.; Ingram, D.K.; Mattson, M.P.; Furukawa, K. Involvement of notch signaling in hippocampal synaptic plasticity. Proc. Natl. Acad. Sci. USA 2004, 101, 9458–9462. [Google Scholar] [CrossRef] [PubMed]
- Souilhol, C.; Cormier, S.; Monet, M.; Vandormael-Pournin, S.; Joutel, A.; Babinet, C.; Cohen-Tannoudji, M. Nas transgenic mouse line allows visualization of notch pathway activity in vivo. Genesis 2006, 44, 277–286. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamagnone, L.; Zacchigna, S.; Rehman, M. Taming the Notch Transcriptional Regulator for Cancer Therapy. Molecules 2018, 23, 431. https://doi.org/10.3390/molecules23020431
Tamagnone L, Zacchigna S, Rehman M. Taming the Notch Transcriptional Regulator for Cancer Therapy. Molecules. 2018; 23(2):431. https://doi.org/10.3390/molecules23020431
Chicago/Turabian StyleTamagnone, Luca, Serena Zacchigna, and Michael Rehman. 2018. "Taming the Notch Transcriptional Regulator for Cancer Therapy" Molecules 23, no. 2: 431. https://doi.org/10.3390/molecules23020431