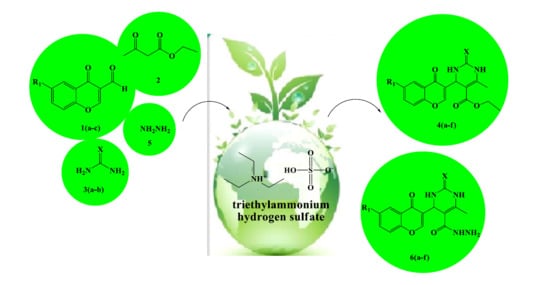
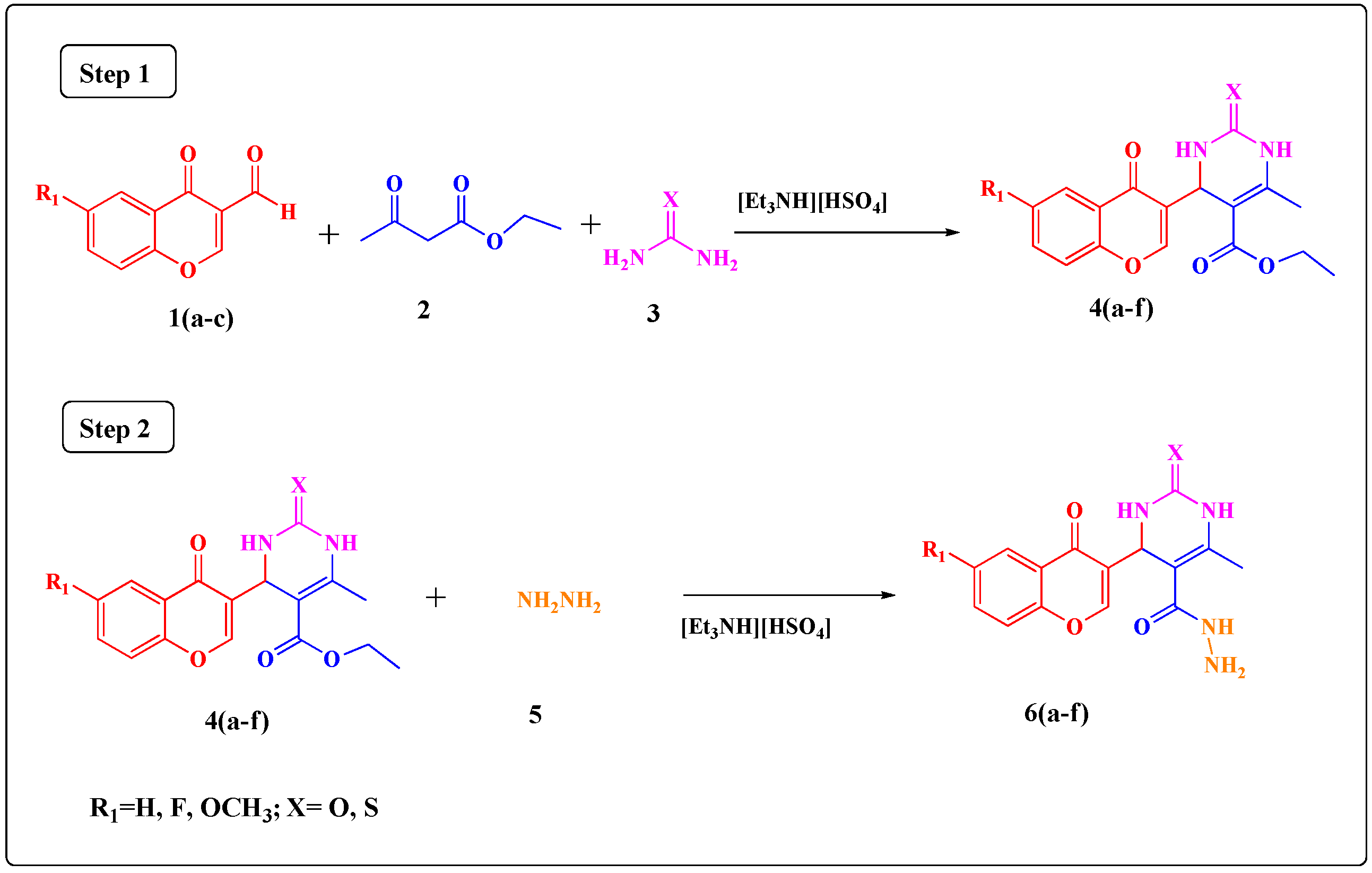
3.2. Synthesis of Ethyl 4-(6-Substituted-4-oxo-4H-chromen-3-yl)-6-methyl-2-thioxo/oxo-1,2,3,4-tetrahydro- pyrimidine-5-carboxylate Derivatives 4(a–f)
A mixture of substituted 4-oxo-4H-chromene-3-carbaldehydes 1(a–c) (1 mmol), ethyl acetoacetate (2) (1 mmol), urea/thiourea (3) (1 mmol) and [Et3NH][HSO4] (15 mol %) was refluxed under solvent-free conditions at 100 °C for the required time while the reactions were monitored by TLC. After completion of the reaction, the reaction mixture was poured into crushed ice and stirred for 5 min. The solid obtained was filtered, washed with cold water and then recrystallized from ethanol to afford the pure product. The ionic liquid present in the filtrate was recovered and was used for other reactions.
Ethyl 6-methyl-2-oxo-4-(4-oxo-4H-chromen-3-yl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate 4a. Yield 95%; M.P.: 270–272 °C; IR (KBr vmax in cm−1): 3238 (N–H stretching), 3005 (C–H stretching), 2900 (–CH3 stretching), 2815 (alkyl CH stretching), 1746 (C=O stretching), 1601 (C=C stretching), 1454 (CH bending of CH2), 1356 (C–N stretching), 1002 (–O– stretching); 1H-NMR (DMSO-d6, δH ppm): 1.14 (t, J = 7.10 Hz, 3 H, CH3), 2.28 (s, 3 H, CH3), 4.05 (q, J = 7.04 Hz, 2 H, CH2), 4.72 (d, J = 1.49 Hz, 1H, CH), 7.41–7.59 (m, 3H, aromatic), 7.75 (s, 1H, NH), 8.05 (s, 1H, aromatic), 8.10 (d, J = 1.44 Hz, 1H, aromatic), 9.12 (s, 1H, NH); 13C-NMR (DMSO-d6, δC ppm): 14.45, 18.51, 45.91, 61.37, 101.94, 118.07, 121.61, 123.87, 126.27, 126.68, 133.85, 149.33, 150.72, 155.65, 155.77, 167.37, 172.17; MS: m/z: 329.21 [M + 1]+; Anal. Calcd. for C17H16N2O5: C, 62.19; H, 4.91; N, 8.53. Found: C, 62.20; H, 4.93; N, 8.50.
Ethyl 6-methyl-4-(4-oxo-4H-chromen-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate 4b. Yield 87%; M.P.: 210–212 °C; IR (KBr vmax in cm−1): 3235 (N–H stretching), 3005 (C–H stretching), 2905 (–CH3 stretching), 2815 (CH alkyl stretching), 1600 (C=C stretching), 1454 (CH bending of CH2), 1360 (C–N stretching), 1140 (C=S stretching), 1002 (–O– stretching); 1H-NMR (DMSO-d6, δH ppm): 1.17 (t, J = 7.10 Hz, 3 H, CH3), 2.29 (s, 3 H, CH3), 4.74 (d, J = 1.49 Hz, 1H, CH), 7.47–7.74 (m, 3H, aromatic), 8.03 (s, 1H, NH), 8.06 (s, 1H, aromatic), 8.12 (d, J = 1.44 Hz, 1H, aromatic), 8.89 (s, 1H, NH); 13C-NMR (DMSO-d6, δC ppm): 14.50, 18.44, 50.62, 61.77, 106.24, 118.71, 121.32, 123.99, 126.12, 133.91, 150.11, 155.60, 160.39, 167.33, 171.94, 177.89; MS: m/z: 345.01 [M + 1]+; Anal. Calcd. for C17H16N2O4S: C, 59.29; H, 4.68; N, 8.13. Found: C, 59.31; H, 4.69; N, 8.10.
Ethyl 4-(6-fluoro-4-oxo-4H-chromen-3-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate 4c. Yield 92%; M.P.: 200–202 °C; IR (KBr vmax in cm−1): 3230 (N–H stretching), 3000 (C–H stretching), 2900 (–CH3 stretching), 2815 (CH alkyl stretching), 1745 (C=O stretching), 1600 (C=C stretching), 1454 (CH bending of CH2), 1364 (C–N stretching), 1002 (–O– stretching); 1H-NMR (DMSO-d6, δH ppm): 1.15 (t, J = 7.10 Hz, 3 H, CH3), 2.29 (s, J = 2.11 Hz, 3 H, CH3), 4.09 (q, J = 7.04 Hz, 2 H, CH2), 4.74 (d, J = 1.51 Hz, 1H, CH), 6.91–7.11 (m, 2 H, aromatic), 7.43 (s, 1 H, NH), 7.74 (d, J = 2.93 Hz, 1 H, aromatic) 8.06 (s, 1 H, aromatic), 9.13 (s, 1H, NH); 13C-NMR (DMSO-d6, δC ppm): 14.22, 18.11, 45.51, 61.70, 101.99, 111.52, 118.71, 119.23, 122.00, 126.88, 149.14, 150.79, 155.13, 156.11, 164.19, 167.19, 172.10; MS: m/z: 347.11 [M + 1]+; Anal. Calcd. for C17H15FN2O5: C, 58.96; H, 4.37; N, 8.09. Found: C, 58.99; H, 4.39; N, 8.04.
Ethyl 4-(6-methoxy-4-oxo-4H-chromen-3-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate 4d. Yield 88%; M.P.: 230–232 °C; IR (KBr vmax in cm−1): 3238 (N–H stretching), 3005 (C–H stretching), 2900 (–CH3 stretching), 2845 (–OCH3 stretching), 2815 (alkyl CH stretching), 1742 (C=O stretching), 1600 (C=C stretching), 1454 (CH bending of CH2), 1362 (C–N stretching), 1002 (–O– stretching); 1H-NMR (DMSO-d6, δH ppm): 1.17 (t, J = 7.10 Hz, 3 H, CH3), 2.29 (s, J = 2.11 Hz, 3 H, CH3), 3.80 (s, 3 H, O CH3), 4.10 (q, J = 7.04 Hz, 2 H, CH2) 4.74 (d, J = 1.51 Hz, 1H, CH), 7.37 (d, J = 8.90 Hz, 1H), 7.42 (s, 1H, NH), 7.48–7.52 (m, 2 H), 8.06 (s, 1H, aromatic), 9.13 (s, 1 H, NH); 13C-NMR (DMSO-d6, δC ppm): 14.45, 18.51, 45.91, 55.08, 61.37, 101.94, 107.44, 117.98, 119.35, 122.42, 149.33, 150.72, 152.63, 155.65, 156.18, 167.37, 171.94; MS: m/z: 359.53 [M + 1]+; Anal. Calcd. for C18H18N2O6: C, 60.33; H, 5.06; N, 7.82. Found: C, 60.35; H, 5.09; N, 7.80.
Ethyl 4-(6-methoxy-4-oxo-4H-chromen-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate 4e. Yield 86%; M.P.: 222–224 °C; IR (KBr vmax in cm−1): 3235 (N–H stretching), 3008 (C–H stretching), 2908 (–CH3 stretching), 2845 (–OCH3 stretching), 2813 (alkyl CH stretching), 1740 (C=O stretching), 1600 (C=C stretching), 1454 (CH bending of CH2), 1360 (C–N stretching), 1148 (C=S stretching), 1002 (–O– stretching); 1H-NMR (DMSO-d6, δH ppm): 1.17 (t, J = 7.10 Hz, 3 H, CH3), 2.29 (s, 3 H, CH3), 3.80 (s, 3 H, O CH3), 4.11 (q, J = 7.04 Hz, 2 H, CH2), 4.73 (d, J = 1.49 Hz, 1 H), 7.37 (d, J = 8.90 Hz, 1 H), 7.36 (s, 1 H, NH), 7.48–8.08 (m, 3 H), 8.89 (s, 1 H, NH); 13C-NMR (DMSO-d6, δC ppm): 14.45, 18.51, 49.08, 55.80, 61.37, 104.43, 107.44, 117.98, 119.35, 122.42, 126.68, 149.33, 152.63, 154.52, 156.18, 167.37, 171.94, 177.89; MS: m/z: 375.17 [M + 1]+; Anal. Calcd. for C18H18N2O5S: C, 57.74; H, 4.85; N, 7.48. Found: C, 57.77; H, 4.86; N, 7.46.
Ethyl 4-(6-fluoro-4-oxo-4H-chromen-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate 4f. Yield 92%; M.P.: 228–230 °C; IR (KBr vmax in cm−1): 3230 (N–H stretching), 3000 (C–H stretching), 2910 (–CH3 stretching), 2810 (alkyl CH stretching), 1742 (C=O stretching), 1605 (C=C stretching), 1450 (CH bending of CH2), 1360 (C–N stretching), 1140 (C=S stretching), 1005 (–O– stretching); 1H-NMR (DMSO-d6, δH ppm): 1.15 (t, J = 7.10 Hz, 3 H, CH3), 2.29 (s, 3 H, CH3), 4.11 (q, J = 7.04 Hz, 2 H, CH2), 4.73 (d, J = 1.49 Hz, 1 H, CH), 6.91–7.74 (m, 3 H, aromatic), 7.76 (s, 1 H, NH), 8.08 (s, 1 H, aromatic), 8.89 (s, 1 H, NH); 13C-NMR (DMSO-d6, δC ppm): 14.44, 18.46, 50.66, 61.79, 106.33, 111.50, 118.73, 119.31, 122.18, 126.89, 149.15, 152.36, 160.19, 162.58, 167.79, 171.96, 177.88; MS: m/z: 363.15 [M + 1]+; Anal. Calcd. for C17H15FN2O4S: C, 56.35; H, 4.17; N, 7.73. Found: C, 56.37; H, 4.19; N, 7.70.
3.3. Synthesis of 4-(6-Substituted-4-oxo-4H-chromen-3-yl)-6-methyl-2-thioxo/oxo-1,2,3,4-tetrahydro- pyrimidine-5-carbohydrazide Derivatives 6(a–f)
A mixture of ethyl4-(6-substituted-4-oxo-4H-chromen-3-yl)-6-methyl-2-thioxo/oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate derivative 4(a–f) (1 mmol), hydrazine hydrate (5) (1.2 mmol) and [Et3NH][HSO4] (5 mol %) was refluxed under solvent-free conditions at 90 °C for the required time, while reaction completion was monitored by TLC. After completion of the reaction, the reaction mixture was poured into crushed ice and stirred for 5 min. The solid obtained was filtered, washed with cold water and then recrystallized from ethanol to afford the pure product. The ionic liquid present in the filtrate was recovered and was used for other reactions.
6-Methyl-2-oxo-4-(4-oxo-4H-chromen-3-yl)-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide 6a. Yield 88%; M.P.: 256–258 °C; IR (KBr vmax in cm−1): 3520 (NH2 stretching), 3450 (N–H stretching), 3000 (C–H stretching), 2900 (–CH3 stretching), 1742 (C=O stretching), 1605 (C=C stretching), 1368 (C–N stretching), 1005 (–O– stretching); 1H-NMR (DMSO-d6, δH ppm): 2.03 (s, 3H, CH3), 3.13 (s, 2 H, NH2), 4.74 (d, J = 1.51 Hz, 1H, CH), 7.08 (s, 1 H, NH), 7.42 (s, 1H, NH), 7.47–8.13 (m, 5H, aromatic), 9.39 (s, 1 H, NH); 13C-NMR (DMSO-d6, δC ppm): 18.49, 46.11, 102.31, 118.71, 121.61, 123.44, 126.23, 126.85, 133.88, 149.45, 150.71, 155.36, 159.17, 168.62, 172.49; MS: m/z: 315.25 [M + 1]+; Anal. Calcd. for C15H14N4O4: C, 57.32; H, 4.49; N, 17.83. Found: C, 57.32; H, 4.49; N, 17.83.
6-Methyl-4-(4-oxo-4H-chromen-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide 6b. Yield 88%; M.P.: 198–200 °C; IR (KBr vmax in cm−1): 3515 (–NH2 stretching), 3450 (N–H stretching), 3000 (C–H stretching), 2908 (–CH3 stretching), 1742 (C=O stretching), 1600 (C=C stretching), 1360 (C–N stretching), 1145 (C=S stretching), 1002 (–O– stretching); 1H-NMR (DMSO-d6, δH ppm): 2.26 (s, 3 H, CH3), 3.13 (s, 2H, NH2), 4.74 (d, J = 1.49 Hz, 1 H, CH), 7.08 (s, 1 H, NH), 7.47 (s, 1 H, NH), 7.59–8.13 (m, 5 H, aromatic), 8.89 (s, 1H, NH); 13C-NMR (DMSO-d6, δC ppm): 18.97, 51.16, 106.11, 118.23, 121.14, 123.43, 126.28, 126.81, 133.69, 150.11, 155.49, 160.19, 168.66, 172.32, 177.80; MS: m/z: 331.29 [M + 1]+; Anal. Calcd. for C15H14N4O3S: C, 54.53; H, 4.27; N, 16.96. Found: C, 54.55; H, 4.29; N, 16.93.
4-(6-Fluoro-4-oxo-4H-chromen-3-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide 6c. Yield 95%; M.P.: 210–212 °C; IR (KBr vmax in cm−1): 3510 (NH2 stretching), 3455 (N–H stretching), 3000 (C–H stretching), 2910 (–CH3 stretching), 1742 (C=O stretching), 1605 (C=C stretching), 1365 (C–N stretching), 1005 (–O– stretching); 1H-NMR (DMSO-d6, δH ppm): 2.03 (s, 3 H, CH3), 3.13 (s, 2H, NH2), 4.74 (d, J = 1.51 Hz, 1 H, CH), 6.91 (d, J = 8.93 Hz, 1H), 7.08 (s, 1H, NH), 7.10–8.06 (m, 4 H, aromatic), 9.39 (s, 1H, NH); 13C-NMR (DMSO-d6, δC ppm): 18.26, 46.83, 106.12, 109.11, 118.36, 121.33, 123.39, 126.10, 147.77, 150.18, 155.60, 155.99, 162.45, 168.20, 172.45; MS: m/z: 333.55 [M + 1]+; Anal. Calcd. for C15H13FN4O4: C, 54.22; H, 3.94; N, 16.86. Found: C, 54.25; H, 3.96; N, 16.83.
4-(6-Fluoro-4-oxo-4H-chromen-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide 6d. Yield 90%; M.P.: 146–148 °C; IR (KBr vmax in cm−1): 3512 (NH2 stretching), 3458 (N–H stretching), 3000 (C–H stretching), 2912 (–CH3 stretching), 1742 (C=O stretching), 1605 (C=C stretching), 1360 (C–N stretching), 1140 (C=S stretching), 1005 (–O– stretching); 1H-NMR (DMSO-d6, δH ppm): 2.27 (s, 3H, CH3), 3.14 (s, 2H, NH2), 4.74 (d, J = 1.49 Hz, 1H, CH), 6.92 (d, J = 8.93 Hz, 1H), 7.09 (s, 1H, NH), 7.11–8.09 (m, 4H, aromatic), 8.90 (s, 1 H, NH); 13C-NMR (DMSO-d6, δC ppm): 18.10, 52.61, 106.21, 109.14, 118.24, 121.39, 123.41, 126.52, 150.19, 155.62, 160.41, 162.56, 168.21, 171.96, 177.81; MS: m/z: 349.72 [M + 1]+; Anal. Calcd. for C15H13FN4O3S: C, 54.22; H, 3.94; N, 16.86. Found: C, 54.27; H, 3.99; N, 16.80.
4-(6-Methoxy-4-oxo-4H-chromen-3-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide 6e. Yield 82%; M.P.: 208–210 °C; IR (KBr vmax in cm−1): 3520 (NH2 stretching), 3452 (N–H stretching), 3000 (C–H stretching), 2905 (–CH3 stretching), 2845 (–OCH3 stretching), 1742 (C=O stretching), 1605 (C=C stretching), 1362 (C–N stretching), 1005 (–O– stretching); 1H-NMR (DMSO-d6, δH ppm): 2.03 (s, 3H, CH3), 3.13 (s, 2H, NH2), 3.80 (s, 3 H, OCH3), 4.74 (d, J = 1.51 Hz, 1 H), 7.08 (s, 1H, NH), 7.37–8.06 (m, 5 H, aromatic), 9.39 (s, 1H, NH); 13C-NMR (DMSO-d6, δC ppm): 18.92, 46.48, 56.82, 106.66, 109.11, 118.24, 121.48, 123.49, 125.76, 147.41, 150.62, 155.12, 156.30, 168.61, 172.27; MS: m/z: 345.02 [M + 1]+; Anal. Calcd. for C16H16N4O5: C, 55.81; H, 4.68; N, 16.27. Found: C, 55.84; H, 4.69; N, 16.25.
4-(6-Methoxy-4-oxo-4H-chromen-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide 6f. Yield 80%; M.P.: 158–160 °C; IR (KBr vmax in cm−1): 3522 (NH2 stretching), 3455 (N–H stretching), 3000 (C–H stretching), 2910 (–CH3 stretching), 2845 (–OCH3 stretching), 1742 (C=O stretching), 1605 (C=C stretching), 1360 (C–N stretching), 1146 (C=S stretching), 1005 (–O– stretching); 1H-NMR (DMSO-d6, δH ppm): 2.26 (s, 3H, CH3), 3.13 (s, 2H, NH2), 3.80 (s, 3 H, OCH3), 4.74 (d, J = 1.49 Hz, 1 H), 7.08 (s, 1H, NH), 7.37–8.08 (m, 5H, aromatic), 8.89 (s, 1H, NH); 13C-NMR (DMSO-d6, δC ppm): 18.90, 51.50, 105.66, 106.41, 118.90, 121.71, 123.44, 126.47, 150.53, 150.66, 156.30, 160.31, 168.22, 171.96, 177.98; MS: m/z: 361.10 [M + 1]+; Anal. Calcd. for C16H16N4O4S: C, 53.32; H, 4.47; N, 15.55. Found: C, 53.35; H, 4.49; N, 15.51.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}