Investigation of Hydro-Lipophilic Properties of N-Alkoxyphenylhydroxynaphthalenecarboxamides †

 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
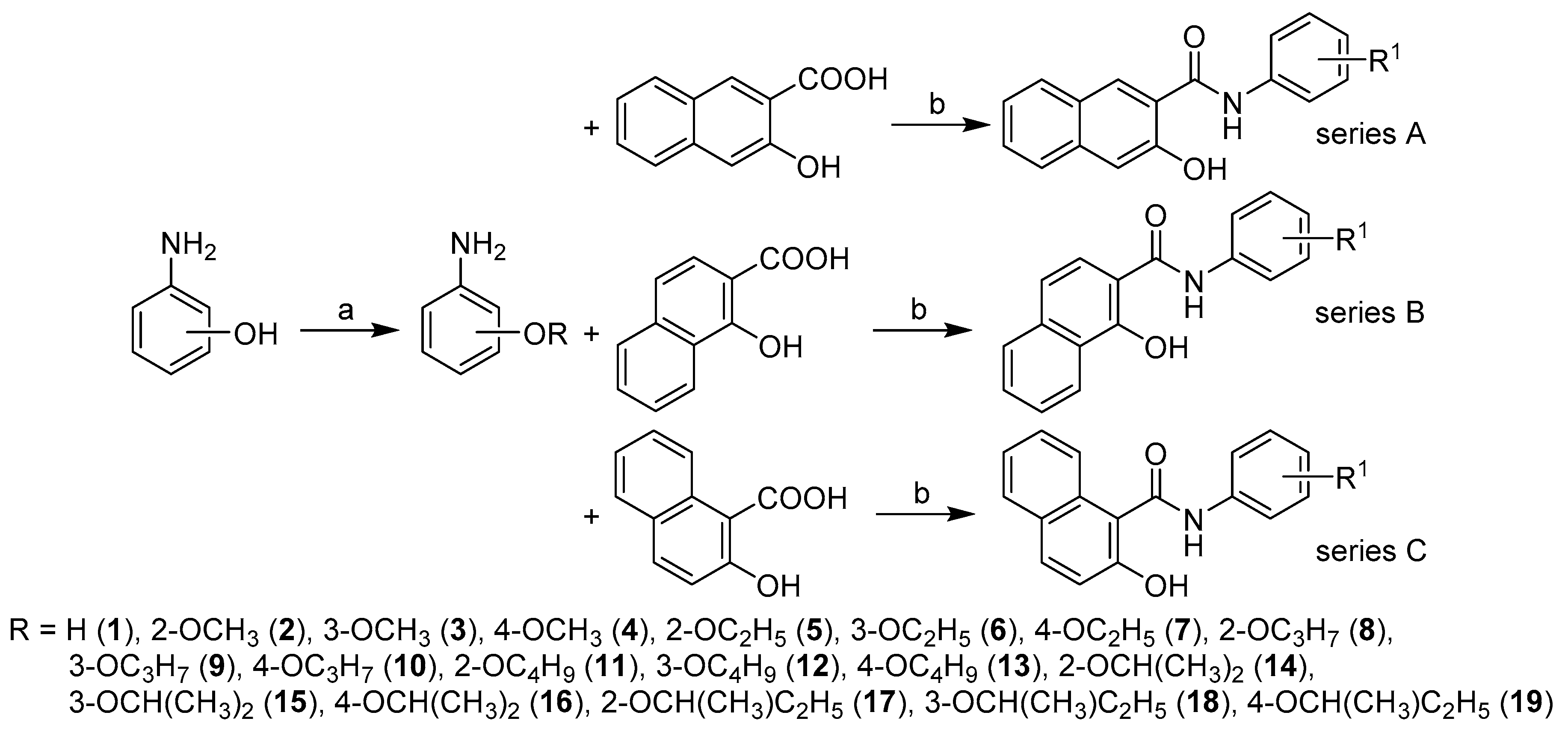
2.1. Chemistry
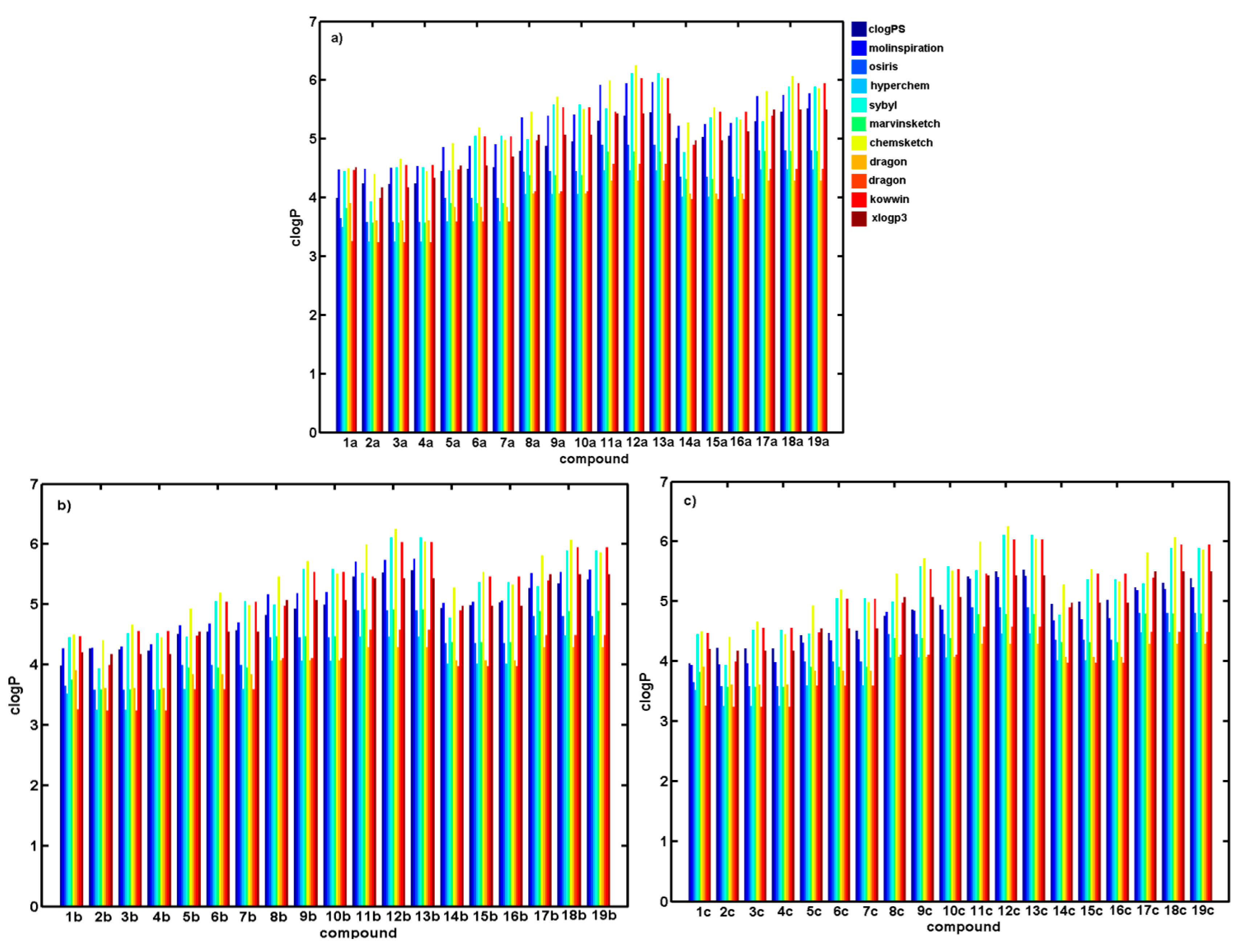
2.2. Consensus Lipophilicity Estimation
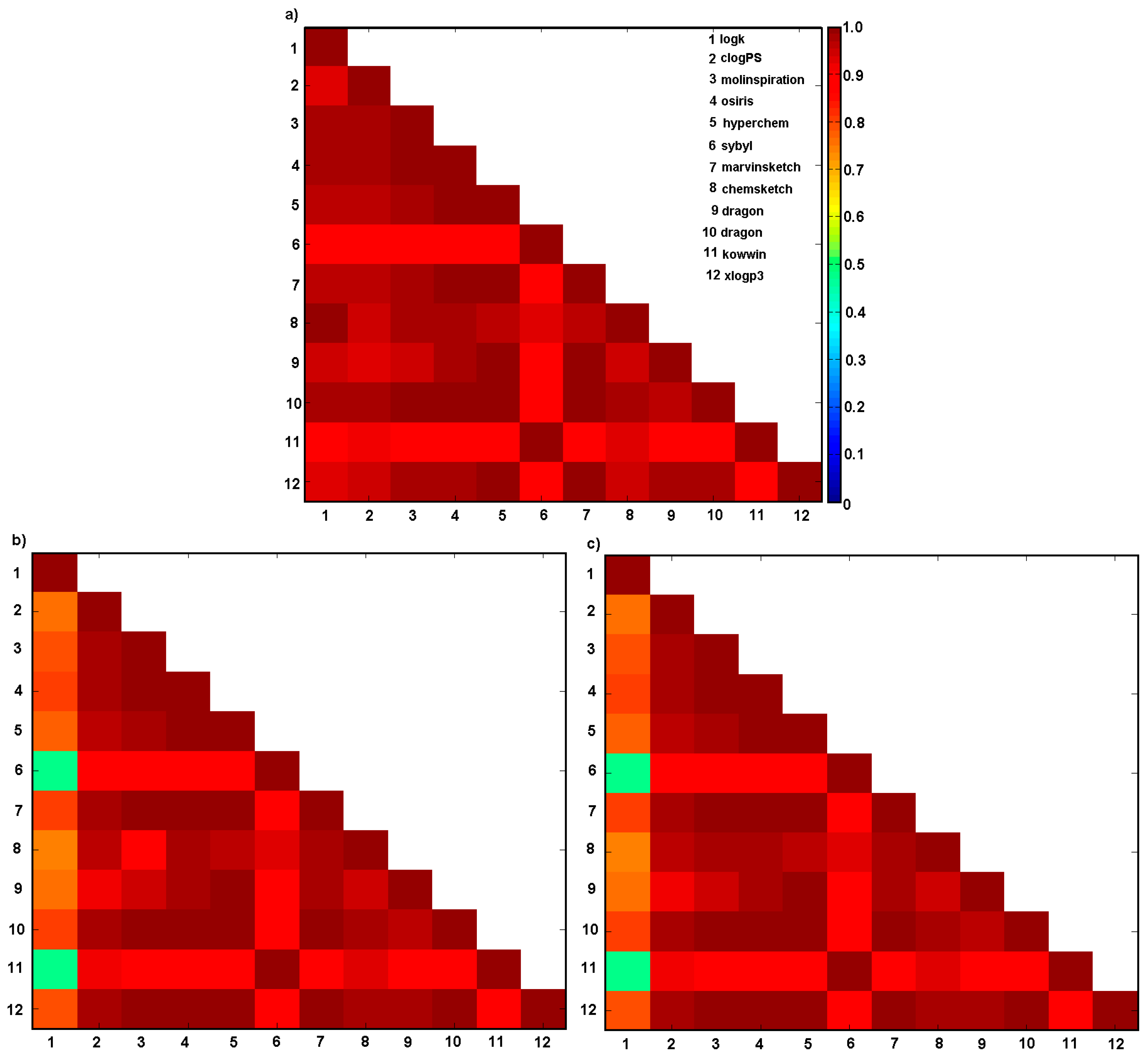
2.3. Descriptor-Based Similarity Assessment
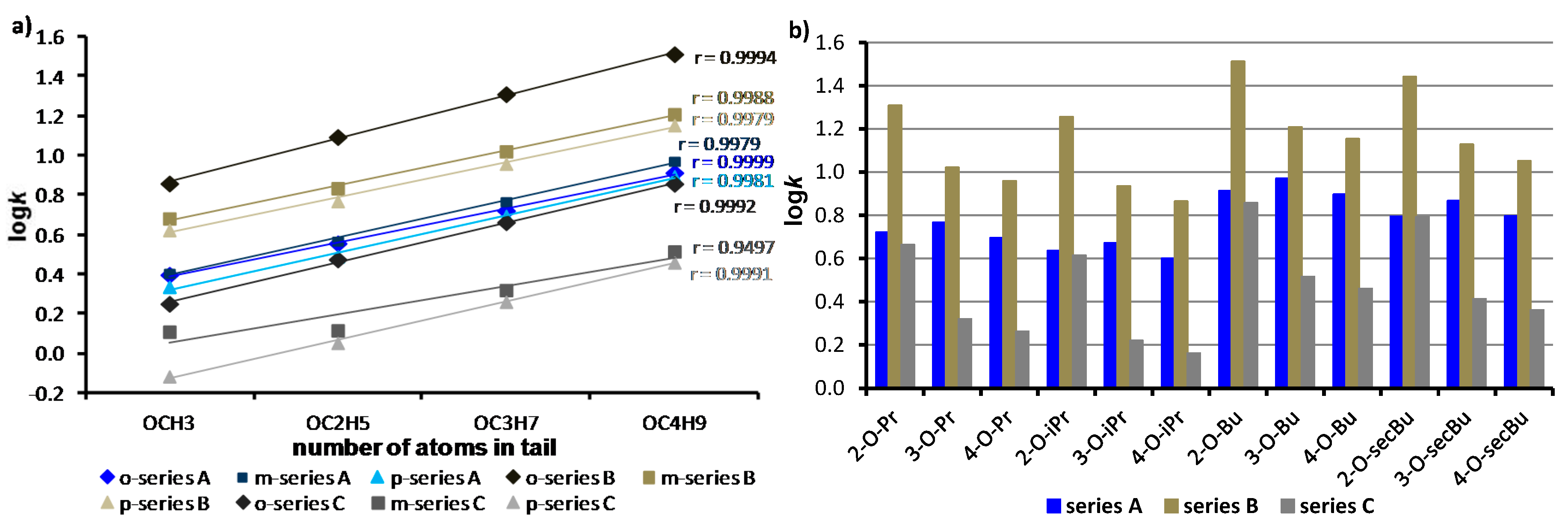
2.4. Experimental Lipophilicity and Biological Effects
3. Experimental Section
3.1. Synthesis
3.2. Lipophilicity Determination by HPLC (Capacity Factor k/Calculated Logk)
3.3. Theoretical Lipophilicity Estimation
3.4. Iterative PLS-Based Variable Elimination
- Step 1.
- Standard PLS analysis with LOO-CV to assess the performance of the PLS model
- Step 2.
- Elimination of a matrix column with the lowest abs(mean(b)/std(b)) value
- Step 3.
- Standard PLS analysis of the new matrix without the column rejected in step 2
- Step 4.
- Recurrent repetition of steps 1–3 to maximize the LOO parameter
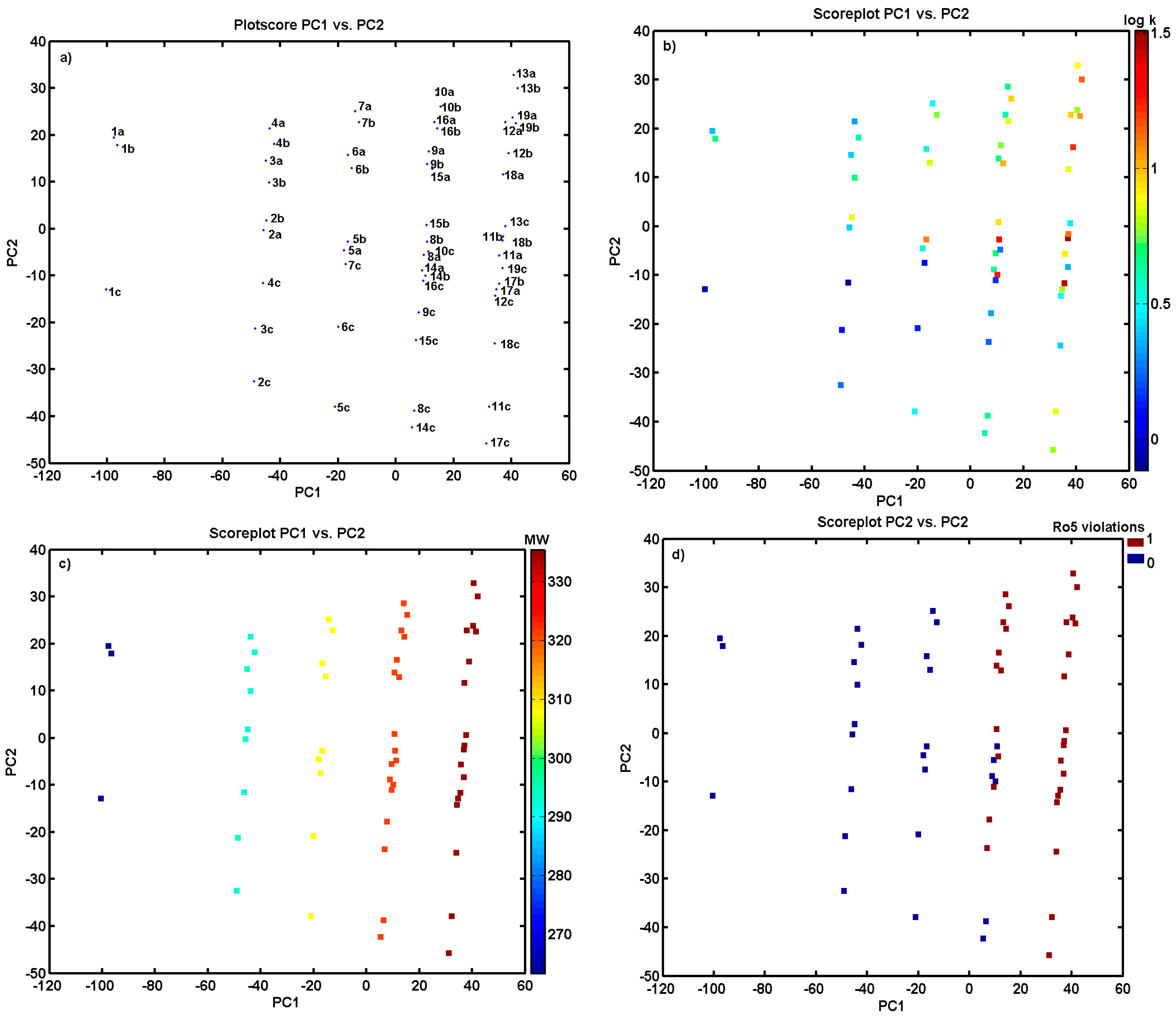
3.5. Principal Component Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J.; Kurczyk, A.; Bak, A.; Musiol, R. Privileged structures-dream or reality: Preferential organization of azanaphthalene scaffold. Curr. Med. Chem. 2012, 19, 1921–1945. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.; Csollei, J.; Jampilek, J.; Stanzel, L.; Zadrazilova, I.; Hosek, J.; Pospisilova, S.; Cizek, A.; Coffey, A.; O’Mahony, J. The structure–antimicrobial activity relationships of promising class of the compounds containing N-arylpiperazine scaffold. Molecules 2016, 21, 1274. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Malik, I.; Csollei, J.; Jampilek, J.; Stolarikova, J.; Solovic, I.; Mikus, P.; Keltosova, S.; Kollar, P.; O’Mahony, J.; et al. Synthesis and in vitro antimycobacterial activity of the novel N-arylpiperazines containing ethane-1,2-diyl connecting chain. Molecules 2017, 22, 2100. [Google Scholar] [CrossRef] [PubMed]
- Kubinyi, H. QSAR: Hansch Analysis and Related Approaches; Wiley-VCH: Weinheim, Germany, 1993. [Google Scholar]
- Kerns, E.H.; Di, L. Drug-Like Properties: Concepts, Structure Design and Methods: From ADME to Toxicity Optimization; Academic Press: San Diego, CA, USA, 2008. [Google Scholar]
- Pliska, V. Lipophilicity in Drug Action and Toxicology, 1st ed.; Pliska, V., Testa, B., van der Waterbeemd, H., Eds.; Methods and Principles in Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 1996; Volume 4. [Google Scholar]
- Avdeef, A. Absorption and Drug Development: Solubility, Permeability, and Charge State, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Hansch, C.; Mahoney, P.P.; Fujita, T.; Muir, R.M. Correlation of biological activity of phenoxyacetic acids with Hammett substituent constants and partition coefficients. Nature 1962, 194, 178–180. [Google Scholar] [CrossRef]
- Hansch, C.; Fujita, T. ρ-σ-π Analysis. A method for the correlation of biological activity and chemical structure. J. Am. Chem. Soc. 1964, 86, 1616–1626. [Google Scholar] [CrossRef]
- Leo, A.; Hansch, C.; Elkins, D. partition coefficients and their uses. Chem. Rev. 1971, 71, 525–616. [Google Scholar] [CrossRef]
- Valko, K. Application of high-performance liquid chromatography based measurements of lipophilicity to model biological distribution. J. Chromatogr. A 2004, 1037, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Carlson, R.M.; Carlson, R.E.; Kopperman, H.L. Determination of partition coefficients by liquid chromatography. J. Chromatogr. A 1975, 107, 219–223. [Google Scholar] [CrossRef]
- Mirrlees, M.S.; Moulton, S.J.; Murphy, C.T.; Taylor, P. Direct measurement of octanol-water partition coefficients by high-pressure liquid chromatography. J. Med. Chem. 1976, 19, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Unger, S.H.; Cook, J.R.; Holenberg, J.S. Simple procedure for determining octanol-aqueous partition, distribution, and ionization coefficients by reversed phase high pressure liquid chromatography. J. Pharm. Sci. 1978, 67, 1364–1367. [Google Scholar] [CrossRef] [PubMed]
- Braumann, T. Determination of hydrophobic parameters by reversed-phase liquid chromatography: Theory, experimental techniques, and application in studies on quantitative structure-activity relationships. J. Chromatogr. A 1986, 373, 191–225. [Google Scholar] [CrossRef]
- Giaginis, C.; Tsantili-Kakoulidou, A. Current state of the art in HPLC methodology for lipophilicity assessment of basic drugs. A review. J. Liq. Chromatogr. Relat. 2008, 31, 79–96. [Google Scholar] [CrossRef]
- Valko, K.; Du, C.M.; Bevan, C.; Reynolds, D.P.; Abraham, M.H. Rapid method for the estimation of octanol/water partition coefficient (log P(oct)) from gradient RP-HPLC retention and a hydrogen bond acidity term (zetaalpha(2)(H)). Curr. Med. Chem. 2001, 8, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Cimpan, G.; Irimie, F.; Gocan, S.; Claessens, H.A. Role of stationary phase and eluent composition on the determination of log P values of N-hydroxyethylamide of aryloxyalkylen and pyridine carboxylic acids by reversed-phase high-performance liquid chromatography. J. Chromatogr. B 1998, 714, 247–261. [Google Scholar] [CrossRef]
- Gocan, S.; Cimpan, G.; Comer, J. Lipophilicity measurements by liquid chromatography. Adv. Chromatogr. 2006, 44, 79–176. [Google Scholar] [PubMed]
- Kucerova-Chlupacova, M.; Opletalova, V.; Jampilek, J.; Dolezel, J.; Dohnal, J.; Pour, M.; Kunes, J.; Vorisek, V. New hydrophobicity constants of substituents in pyrazine rings derived from RP-HPLC study. Collect. Czechoslov. Chem. Commun. 2008, 73, 1–18. [Google Scholar] [CrossRef]
- Musilek, K.; Jampilek, J.; Dohnal, J.; Jun, D.; Gunn-Moore, F.; Dolezal, M.; Kuca, K. RP-HPLC determination of the lipophilicity of bispyridinium reactivators of acetylcholinesterase bearing a but-2-ene connecting linker. Anal. Bioanal. Chem. 2008, 391, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Musiol, R.; Jampilek, J.; Podeszwa, B.; Finster, J.; Tabak, D.; Dohnal, J.; Polanski, J. RP-HPLC determination of drug lipophilicity in series of quinoline derivatives. Cent. Eur. J. Chem. 2009, 7, 586–597. [Google Scholar]
- Tengler, J.; Kapustikova, I.; Stropnicky, O.; Mokry, P.; Oravec, M.; Csollei, J.; Jampilek, J. Synthesis of New (arylcarbonyloxy)aminopropanol derivatives and the determination of their physico-chemical properties. Cent. Eur. J. Chem. 2013, 11, 1757–1767. [Google Scholar] [CrossRef]
- Hartmann, T.; Schmitt, J. Lipophilicity-beyond octanol/water: A short comparison of modern technologies. Drug Discov. Today Technol. 2004, 1, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Nasal, A.; Siluk, D.; Kaliszan, R. Chromatographic retention parameters in medicinal chemistry and molecular pharmacology. Curr. Med. Chem. 2003, 10, 381–426. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Kozik, V.; Smolinski, S.; Jampilek, J. In silico estimation of basic activity-relevant parameters for a set of drug absorption promoters. SAR QSAR Environ. Res. 2017, 28, 427–449. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Zadrazilova, I.; Pesko, M.; Keltosova, S.; Tengler, J.; Gonec, T.; Bobal, P.; Kauerova, T.; Oravec, M.; Kollar, P.; et al. Antibacterial and herbicidal activity of ring-substituted 3-hydroxynaphthalene-2-carbox-anilides. Molecules 2013, 18, 7977–7997. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Zadrazilova, I.; Nevin, E.; Kauerova, T.; Pesko, M.; Kos, J.; Oravec, M.; Kollar, P.; Coffey, A.; O’Mahony, J.; et al. Synthesis and biological evaluation of N-alkoxyphenyl-3-hydroxynaphthalene-2-carbox-anilides. Molecules 2015, 20, 9767–9787. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Kos, J.; Zadrazilova, I.; Pesko, M.; Keltosova, S.; Tengler, J.; Bobal, P.; Kollar, P.; Cizek, A.; Kralova, K.; et al. Antimycobacterial and herbicidal activity of ring-substituted 1-hydroxynaphthalene-2-carboxanilides. Bioorg. Med. Chem. 2013, 21, 6531–6541. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Kos, J.; Zadrazilova, I.; Pesko, M.; Govender, R.; Keltosova, S.; Chambel, B.; Pereira, D.; Kollar, P.; Imramovsky, A.; et al. Antibacterial and herbicidal activity of ring-substituted 2-hydroxynaphthalene-1-carboxanilides. Molecules 2013, 18, 9397–9419. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Pospisilova, S.; Kauerova, T.; Kos, J.; Dohanosova, J.; Oravec, M.; Kollar, P.; Coffey, A.; Liptaj, T.; Cizek, A.; et al. N-Alkoxyphenylhydroxynaphthalenecarboxamides and their antimycobacterial activity. Molecules 2016, 21, 1068. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Kralova, K.; Pesko, M.; Jampilek, J. Antimycobacterial N-alkoxyphenylhydroxynaphthalene-carboxamides affecting photosystem II. Bioorg. Med. Chem. Lett. 2017, 27, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- De Marco, A.; De Candia, M.; Carotti, A.; Cellamare, S.; De Candia, E.; Altomare, C. Lipophilicity-related inhibition of blood platelet aggregation by nipecotic acid anilides. Eur. J. Pharm. Sci. 2004, 22, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Opletalova, V.; Kalinowski, D.S.; Vejsova, M.; Kunes, J.; Pour, M.; Jampilek, J.; Buchta, V.; Richardson, D.R. Identification and characterization of thiosemicarbazones with antifungal and antitumor effects: Cellular iron chelation mediating cytotoxic activity. Chem. Res. Toxicol. 2008, 21, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Opletalova, V.; Dolezel, J.; Kralova, K.; Pesko, M.; Kunes, J.; Jampilek, J. Synthesis and characterization of (Z)-5-arylmethylidenerhodanines with photosynthesis-inhibiting properties. Molecules 2011, 16, 5207–5227. [Google Scholar] [CrossRef] [PubMed]
- Norrington, F.E.; Hyde, R.M.; Williams, S.G.; Wootton, R. Physicochemical-activity relations in practice I. A rational and self-consistent data bank. J. Med. Chem. 1975, 18, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Dearden, J.C. Partitioning and lipophilicity in quantitative structure-activity relationships. Environ. Health Perspect. 1985, 61, 203–228. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Unger, S.H.; Kim, K.H.; Nikaitani, D.; Lien, E.J. “Aromatic” substituent constants for structure-activity correlations. J. Med. Chem. 1973, 16, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory--design and description. J. Comput. Aided Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Polanski, J. Modeling robust QSAR 3: SOM-4D-QSAR with iterative variable elimination IVE-PLS: Application to steroid, azo dye, and benzoic acid series. J. Chem. Inf. Model. 2007, 47, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Centner, V.; Massart, D.L.; de Noord, O.E.; de Jong, S.; Vandeginste, B.M.V.; Sterna, C. Elimination of uninformative variables for multivariate calibration. Anal. Chem. 1996, 68, 3851–3858. [Google Scholar] [CrossRef] [PubMed]
- Smolinski, A.; Drobek, L.; Dombek, V.; Bak, A. Modeling of experimental data on trace elements and organic compounds content in industrial waste dumps. Chemosphere 2016, 162, 189–198. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of compounds are available from authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| No. | logk | logP a | miLogP b | ClogP c | ClogP d | ClogP e | ClogP f | ClogP g | MlogP h | AlogP i | ClogP j | ClogP k |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 0.3927 | 3.99 | 4.48 | 3.65 | 3.51 | 4.45 | 3.82 | 4.50 | 3.91 | 3.26 | 4.47 | 4.52 |
| 2a | 0.3982 | 4.24 | 4.49 | 3.58 | 3.25 | 3.93 | 3.57 | 4.40 | 3.61 | 3.24 | 3.99 | 4.17 |
| 3a | 0.4055 | 4.23 | 4.51 | 3.58 | 3.25 | 4.52 | 3.57 | 4.66 | 3.61 | 3.24 | 4.55 | 4.17 |
| 4a | 0.3374 | 4.24 | 4.53 | 3.58 | 3.25 | 4.52 | 3.57 | 4.45 | 3.61 | 3.24 | 4.55 | 4.33 |
| 5a | 0.5570 | 4.45 | 4.86 | 3.99 | 3.59 | 4.46 | 3.91 | 4.93 | 3.84 | 3.59 | 4.48 | 4.54 |
| 6a | 0.5682 | 4.49 | 4.88 | 3.99 | 3.59 | 5.05 | 3.91 | 5.19 | 3.84 | 3.59 | 5.04 | 4.54 |
| 7a | 0.4916 | 4.52 | 4.91 | 3.99 | 3.59 | 5.05 | 3.91 | 4.98 | 3.84 | 3.59 | 5.04 | 4.70 |
| 8a | 0.7221 | 4.79 | 5.36 | 4.44 | 4.06 | 4.99 | 4.38 | 5.46 | 4.07 | 4.11 | 4.97 | 5.07 |
| 9a | 0.7672 | 4.88 | 5.39 | 4.45 | 4.06 | 5.58 | 4.38 | 5.72 | 4.07 | 4.11 | 5.54 | 5.07 |
| 10a | 0.6963 | 4.95 | 5.41 | 4.45 | 4.06 | 5.58 | 4.38 | 5.51 | 4.07 | 4.11 | 5.54 | 5.07 |
| 11a | 0.9136 | 5.31 | 5.92 | 4.90 | 4.46 | 5.52 | 4.78 | 5.99 | 4.29 | 4.57 | 5.46 | 5.43 |
| 12a | 0.9711 | 5.39 | 5.95 | 4.90 | 4.46 | 6.12 | 4.78 | 6.25 | 4.29 | 4.57 | 6.03 | 5.43 |
| 13a | 0.8961 | 5.45 | 5.97 | 4.90 | 4.46 | 6.12 | 4.78 | 6.04 | 4.29 | 4.57 | 6.03 | 5.43 |
| 14a | 0.6360 | 5.01 | 5.22 | 4.35 | 4.01 | 4.77 | 4.32 | 5.28 | 4.07 | 3.97 | 4.90 | 4.97 |
| 15a | 0.6723 | 5.03 | 5.25 | 4.35 | 4.01 | 5.36 | 4.32 | 5.54 | 4.07 | 3.97 | 5.46 | 4.97 |
| 16 | 0.6017 | 5.05 | 5.27 | 4.35 | 4.01 | 5.36 | 4.32 | 5.33 | 4.07 | 3.97 | 5.46 | 5.13 |
| 17a | 0.7956 | 5.30 | 5.73 | 4.80 | 4.48 | 5.30 | 4.79 | 5.81 | 4.29 | 4.49 | 5.39 | 5.50 |
| 18a | 0.8670 | 5.46 | 5.75 | 4.80 | 4.48 | 5.89 | 4.79 | 6.07 | 4.29 | 4.49 | 5.95 | 5.50 |
| 19a | 0.7977 | 5.52 | 5.77 | 4.80 | 4.48 | 5.89 | 4.79 | 5.86 | 4.29 | 4.49 | 5.95 | 5.50 |

| No. | logk | logP a | miLogP b | ClogP c | ClogP d | ClogP e | ClogP f | ClogP g | MlogP h | AlogP i | ClogP j | ClogP k |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1b | 0.6755 | 3.98 | 4.27 | 3.65 | 3.51 | 4.45 | 3.75 | 4.50 | 3.91 | 3.26 | 4.47 | 4.20 |
| 2b | 0.8593 | 4.27 | 4.28 | 3.58 | 3.25 | 3.93 | 3.59 | 4.40 | 3.61 | 3.24 | 3.99 | 4.17 |
| 3b | 0.6828 | 4.25 | 4.30 | 3.58 | 3.25 | 4.52 | 3.59 | 4.66 | 3.61 | 3.24 | 4.55 | 4.17 |
| 4b | 0.6239 | 4.23 | 4.33 | 3.58 | 3.25 | 4.52 | 3.59 | 4.45 | 3.61 | 3.24 | 4.55 | 4.17 |
| 5b | 1.0940 | 4.51 | 4.65 | 3.99 | 3.59 | 4.46 | 3.95 | 4.93 | 3.84 | 3.59 | 4.48 | 4.54 |
| 6b | 0.8353 | 4.54 | 4.68 | 3.99 | 3.59 | 5.05 | 3.95 | 5.19 | 3.84 | 3.59 | 5.04 | 4.54 |
| 7b | 0.7700 | 4.56 | 4.70 | 3.99 | 3.59 | 5.05 | 3.95 | 4.98 | 3.84 | 3.59 | 5.04 | 4.54 |
| 8b | 1.3103 | 4.82 | 5.16 | 4.45 | 4.06 | 4.99 | 4.47 | 5.46 | 4.07 | 4.11 | 4.97 | 5.07 |
| 9b | 1.0215 | 4.93 | 5.18 | 4.45 | 4.06 | 5.58 | 4.47 | 5.72 | 4.07 | 4.11 | 5.54 | 5.07 |
| 10b | 0.9588 | 4.99 | 5.20 | 4.45 | 4.06 | 5.58 | 4.47 | 5.51 | 4.07 | 4.11 | 5.54 | 5.07 |
| 11b | 1.5122 | 5.46 | 5.71 | 4.90 | 4.46 | 5.52 | 4.92 | 5.99 | 4.29 | 4.57 | 5.46 | 5.43 |
| 12b | 1.2088 | 5.53 | 5.74 | 4.90 | 4.46 | 6.11 | 4.92 | 6.25 | 4.29 | 4.57 | 6.03 | 5.43 |
| 13b | 1.1537 | 5.56 | 5.76 | 4.90 | 4.46 | 6.11 | 4.92 | 6.04 | 4.29 | 4.57 | 6.03 | 5.43 |
| 14b | 1.2556 | 4.94 | 5.02 | 4.35 | 4.01 | 4.77 | 4.37 | 5.28 | 4.07 | 3.97 | 4.90 | 4.97 |
| 15b | 0.9355 | 4.98 | 5.04 | 4.35 | 4.01 | 5.36 | 4.37 | 5.54 | 4.07 | 3.97 | 5.46 | 4.97 |
| 16b | 0.8648 | 5.03 | 5.06 | 4.35 | 4.01 | 5.36 | 4.37 | 5.33 | 4.07 | 3.97 | 5.46 | 4.97 |
| 17b | 1.4424 | 5.27 | 5.52 | 4.80 | 4.48 | 5.30 | 4.89 | 5.81 | 4.29 | 4.49 | 5.39 | 5.50 |
| 18b | 1.1291 | 5.35 | 5.54 | 4.80 | 4.48 | 5.89 | 4.89 | 6.07 | 4.29 | 4.49 | 5.95 | 5.50 |
| 19b | 1.0518 | 5.41 | 5.57 | 4.80 | 4.48 | 5.89 | 4.89 | 5.86 | 4.29 | 4.49 | 5.95 | 5.50 |

| No. | logk | logP a | miLogP b | ClogP c | ClogP d | ClogP e | ClogP f | ClogP g | MlogP h | AlogP i | ClogP j | ClogP k |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1c | −0.0581 | 3.96 | 3.93 | 3.65 | 3.51 | 4.45 | 3.82 | 4.50 | 3.91 | 3.26 | 4.47 | 4.20 |
| 2c | 0.2518 | 4.22 | 3.94 | 3.58 | 3.25 | 3.93 | 3.57 | 4.40 | 3.61 | 3.24 | 3.99 | 4.17 |
| 3c | 0.1106 | 4.21 | 3.96 | 3.58 | 3.25 | 4.52 | 3.57 | 4.66 | 3.61 | 3.24 | 4.55 | 4.17 |
| 4c | −0.1149 | 4.21 | 3.98 | 3.58 | 3.25 | 4.52 | 3.57 | 4.45 | 3.61 | 3.24 | 4.55 | 4.17 |
| 5c | 0.4759 | 4.43 | 4.31 | 3.99 | 3.59 | 4.46 | 3.91 | 4.93 | 3.84 | 3.59 | 4.48 | 4.54 |
| 6c | 0.1175 | 4.47 | 4.34 | 3.99 | 3.59 | 5.05 | 3.91 | 5.19 | 3.84 | 3.59 | 5.04 | 4.54 |
| 7c | 0.0542 | 4.51 | 4.36 | 3.99 | 3.59 | 5.05 | 3.91 | 4.98 | 3.84 | 3.59 | 5.04 | 4.54 |
| 8c | 0.6639 | 4.75 | 4.82 | 4.45 | 4.06 | 4.99 | 4.38 | 5.46 | 4.07 | 4.11 | 4.97 | 5.07 |
| 9c | 0.3209 | 4.86 | 4.84 | 4.45 | 4.06 | 5.58 | 4.38 | 5.72 | 4.07 | 4.11 | 5.54 | 5.07 |
| 10c | 0.2622 | 4.94 | 4.86 | 4.45 | 4.06 | 5.58 | 4.38 | 5.51 | 4.07 | 4.11 | 5.54 | 5.07 |
| 11c | 0.8578 | 5.41 | 5.37 | 4.90 | 4.46 | 5.52 | 4.78 | 5.99 | 4.29 | 4.57 | 5.46 | 5.43 |
| 12c | 0.5161 | 5.50 | 5.40 | 4.90 | 4.46 | 6.11 | 4.78 | 6.25 | 4.29 | 4.57 | 6.03 | 5.43 |
| 13c | 0.4604 | 5.53 | 5.42 | 4.90 | 4.46 | 6.11 | 4.78 | 6.04 | 4.29 | 4.57 | 6.03 | 5.43 |
| 14c | 0.6145 | 4.95 | 4.68 | 4.35 | 4.01 | 4.77 | 4.32 | 5.28 | 4.07 | 3.97 | 4.90 | 4.97 |
| 15c | 0.2214 | 4.99 | 4.70 | 4.35 | 4.01 | 5.36 | 4.32 | 5.54 | 4.07 | 3.97 | 5.46 | 4.97 |
| 16c | 0.1619 | 5.02 | 4.72 | 4.35 | 4.01 | 5.36 | 4.32 | 5.33 | 4.07 | 3.97 | 5.46 | 4.97 |
| 17c | 0.7927 | 5.23 | 5.18 | 4.80 | 4.48 | 5.30 | 4.79 | 5.81 | 4.29 | 4.49 | 5.39 | 5.50 |
| 18c | 0.4129 | 5.31 | 5.20 | 4.80 | 4.48 | 5.89 | 4.79 | 6.07 | 4.29 | 4.49 | 5.95 | 5.50 |
| 19c | 0.3621 | 5.38 | 5.23 | 4.80 | 4.48 | 5.89 | 4.79 | 5.86 | 4.29 | 4.49 | 5.95 | 5.50 |
| Comp. | R | πA | πB | πC | π ± SD | π+ [39] | π− [39] | π ‡ |
|---|---|---|---|---|---|---|---|---|
| 1 | H | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 2-OCH3 | 0.09 | 0.09 | 0.05 | 0.08 ± 0.02 | −0.33 | −0.13 | −0.34 |
| 3 | 3-OCH3 | 0.04 | 0.04 | 0.02 | 0.03 ± 0.01 | 0.12 | 0.12 | −0.24 |
| 4 | 4-OCH3 | −0.15 | −0.15 | −0.19 | −0.16 ± 0.02 | −0.03 | −0.12 | −0.19 |
| 5 | 2-OC2H5 | 0.40 | 0.40 | 0.39 | 0.40 ± 0.01 | 0.17 | 0.37 | 0.19 |
| 6 | 3-OC2H5 | 0.36 | 0.36 | 0.34 | 0.35 ± 0.01 | 0.62 | 0.62 | 0.29 |
| 7 | 4-OC2H5 | 0.15 | 0.15 | 0.27 | 0.19 ± 0.07 | 0.47 | 0.35 | 0.34 |
| 8 | 2-OC3H7 | 0.74 | 0.74 | 0.73 | 0.74 ± 0.01 | 0.67 | 0.87 | 0.72 |
| 9 | 3-OC3H7 | 0.69 | 0.69 | 0.65 | 0.68 ± 0.02 | 1.12 | 1.12 | 0.82 |
| 10 | 4-OC3H7 | 0.75 | 0.75 | 0.72 | 0.74 ± 0.02 | 0.97 | 0.85 | 0.87 |
| 11 | 2-OC4H9 | 1.08 | 1.08 | 1.04 | 1.07 ± 0.02 | 1.17 | 1.37 | 1.26 |
| 12 | 3-OC4H9 | 1.02 | 1.02 | 1.00 | 1.01 ± 0.01 | 1.62 | 1.62 | 1.36 |
| 13 | 4-OC4H9 | 1.08 | 1.08 | 1.05 | 1.07 ± 0.02 | 1.47 | 1.35 | 1.41 |
| 14 | 2-OCH(CH3)2 | 0.66 | 0.66 | 0.66 | 0.66 ± 0.01 | 0.55 | 0.75 | 0.54 |
| 15 | 3-OCH(CH3)2 | 0.61 | 0.61 | 0.57 | 0.60 ± 0.02 | 1.00 | 1.00 | 0.64 |
| 16 | 4-OCH(CH3)2 | 0.59 | 0.59 | 0.55 | 0.58 ± 0.02 | 0.85 | 0.73 | 0.69 |
| 17 | 2-OCH(CH3)CH2CH3 | 1.00 | 1.00 | 0.98 | 0.99 ± 0.01 | N.F. | N.F. | 1.07 |
| 18 | 3-OCH(CH3)CH2CH3 | 0.95 | 0.95 | 0.91 | 0.94 ± 0.02 | N.F. | N.F. | 1.17 |
| 19 | 4-OCH(CH3)CH2CH3 | 0.09 | 0.94 | 0.91 | 0.93 ± 0.02 | N.F. | N.F. | 1.22 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapustikova, I.; Bak, A.; Gonec, T.; Kos, J.; Kozik, V.; Jampilek, J.
Investigation of Hydro-Lipophilic Properties of N-Alkoxyphenylhydroxynaphthalenecarboxamides
Kapustikova I, Bak A, Gonec T, Kos J, Kozik V, Jampilek J.
Investigation of Hydro-Lipophilic Properties of N-Alkoxyphenylhydroxynaphthalenecarboxamides
Kapustikova, Iva, Andrzej Bak, Tomas Gonec, Jiri Kos, Violetta Kozik, and Josef Jampilek.
2018. "Investigation of Hydro-Lipophilic Properties of N-Alkoxyphenylhydroxynaphthalenecarboxamides
Kapustikova, I., Bak, A., Gonec, T., Kos, J., Kozik, V., & Jampilek, J.
(2018). Investigation of Hydro-Lipophilic Properties of N-Alkoxyphenylhydroxynaphthalenecarboxamides