Capturing the Alternative Cleavage and Polyadenylation Sites of 14 NAC Genes in Populus Using a Combination of 3′-RACE and High-Throughput Sequencing


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
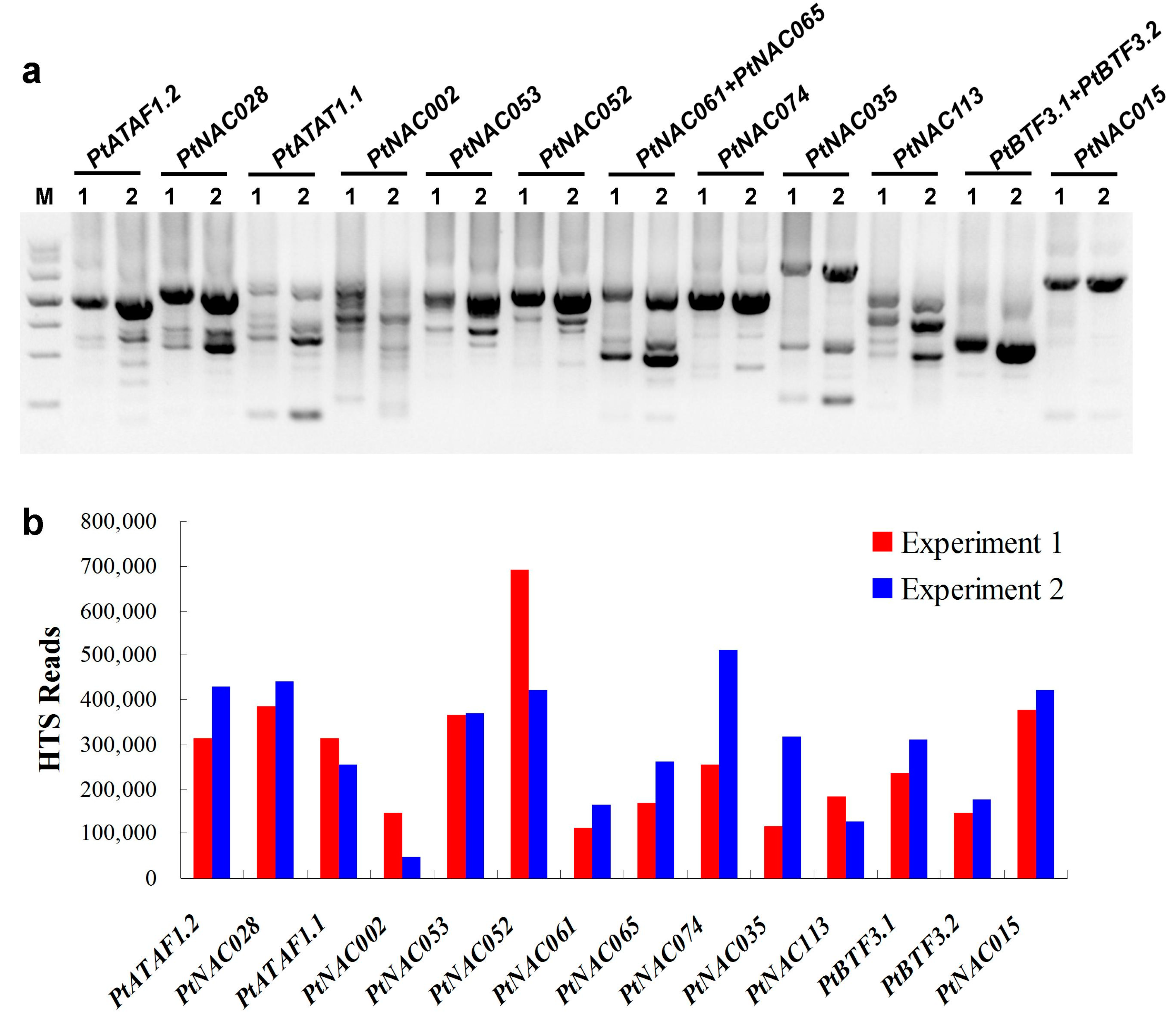
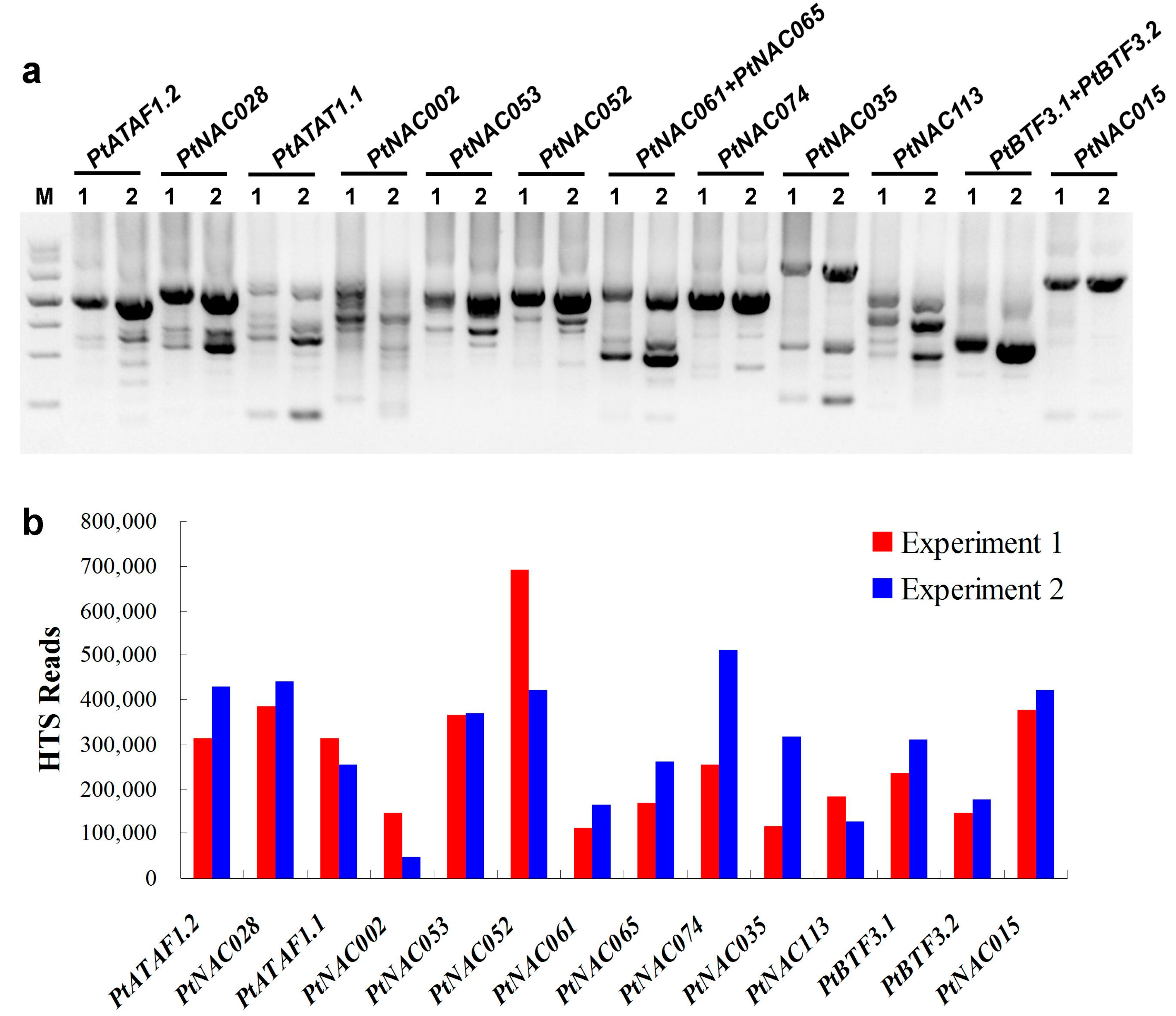
2.1. Target Transcripts Were Effectively Enriched by 3′-RACE
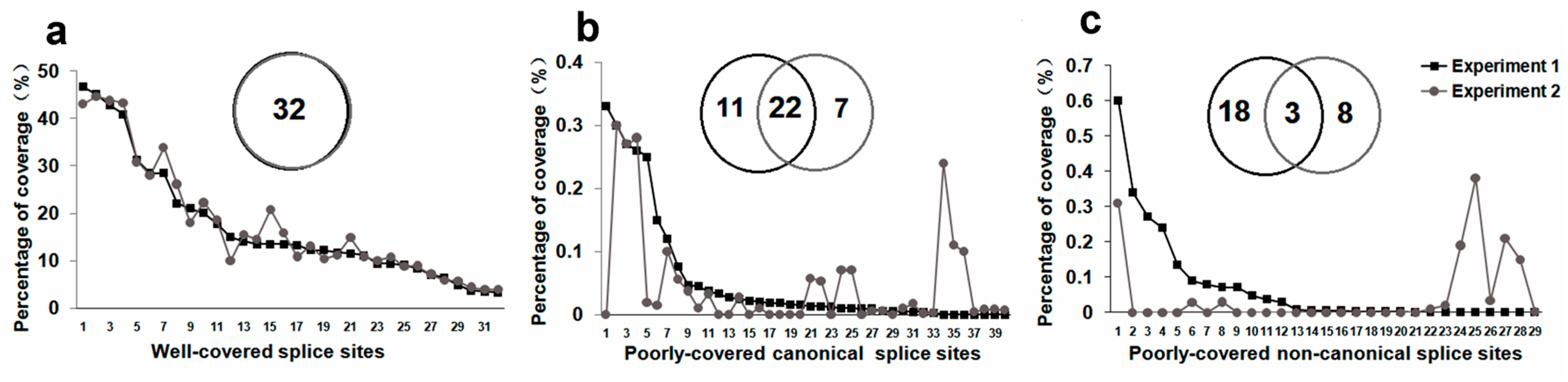
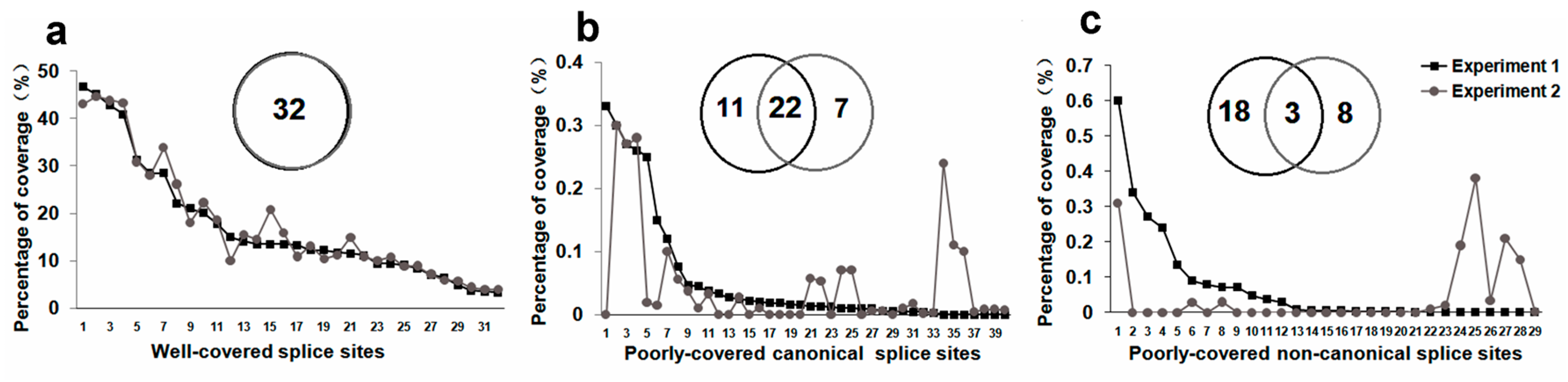
2.2. Monte Carlo Effect of Poorly Covered Splice Sites
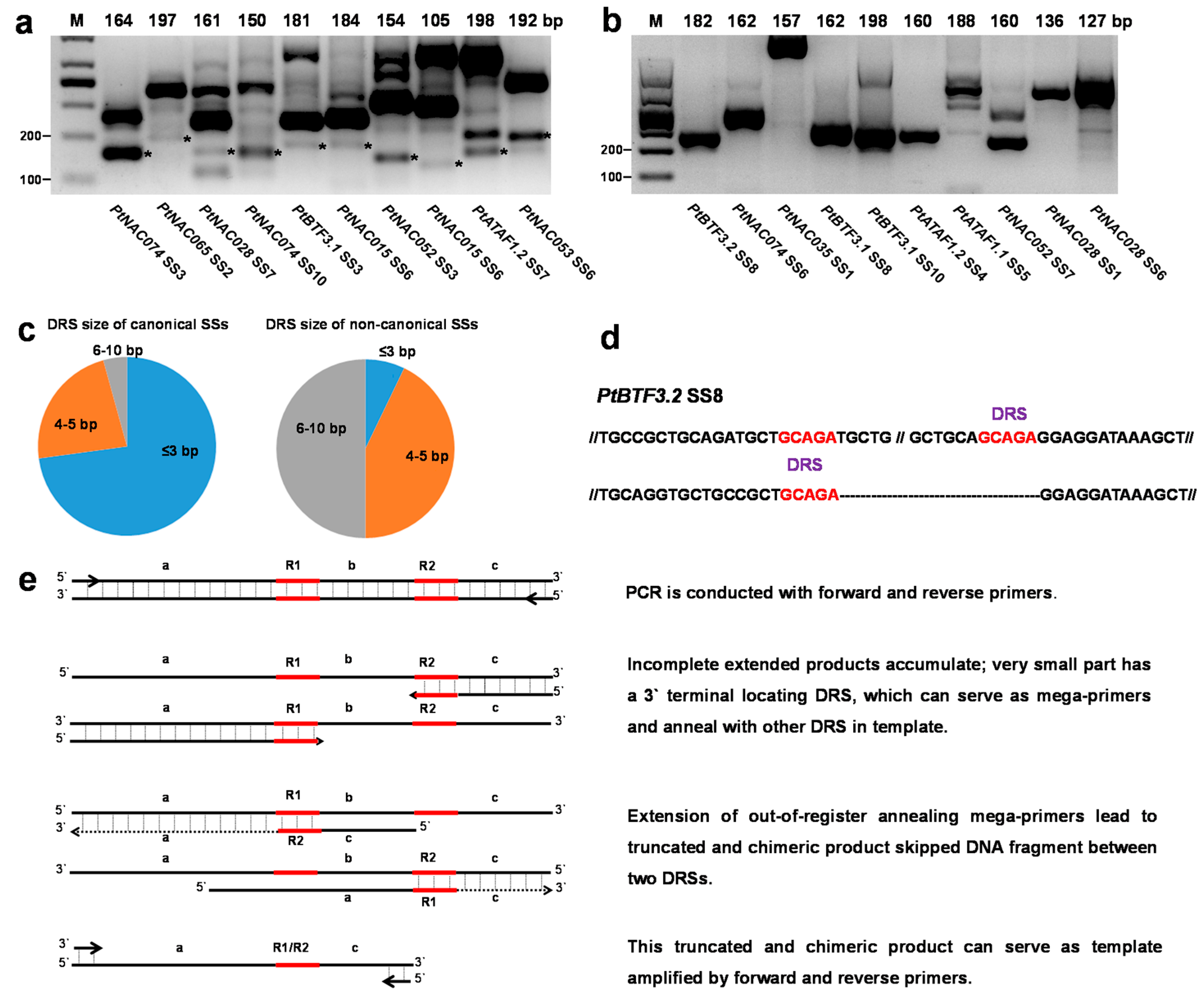
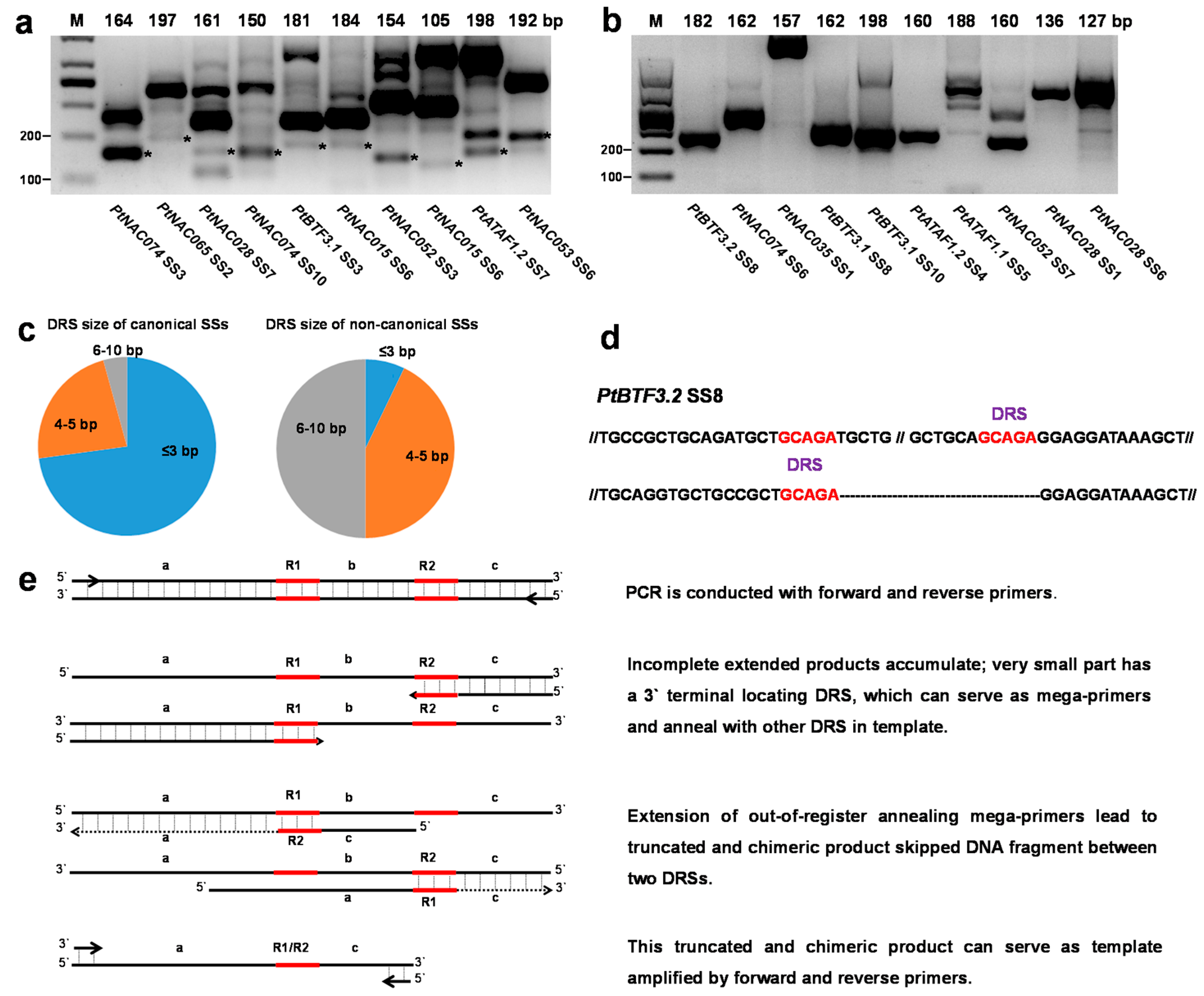
2.3. Non-Canonical SSs Probably Resulted from PCR Artifacts
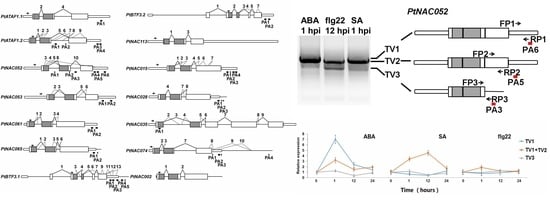
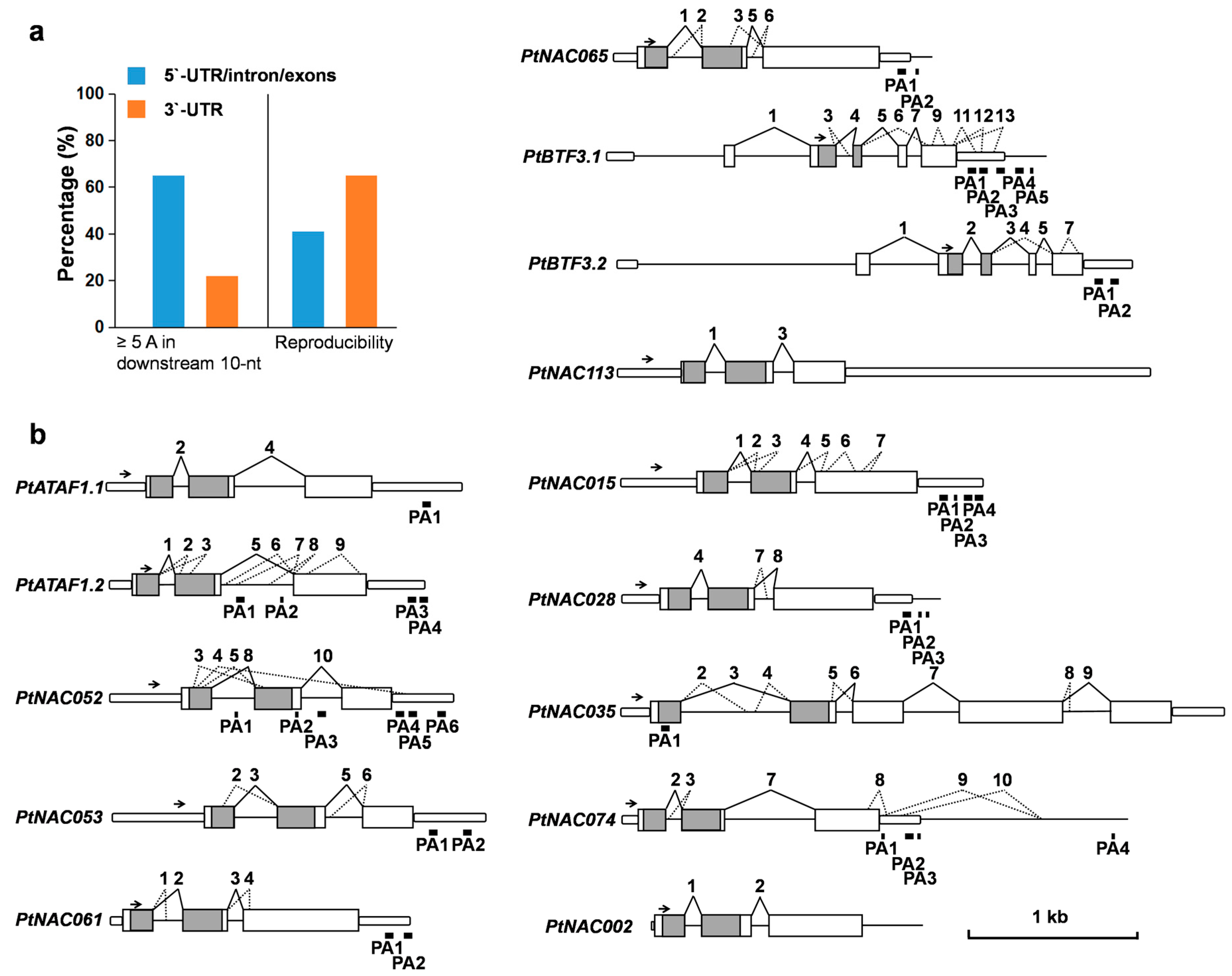
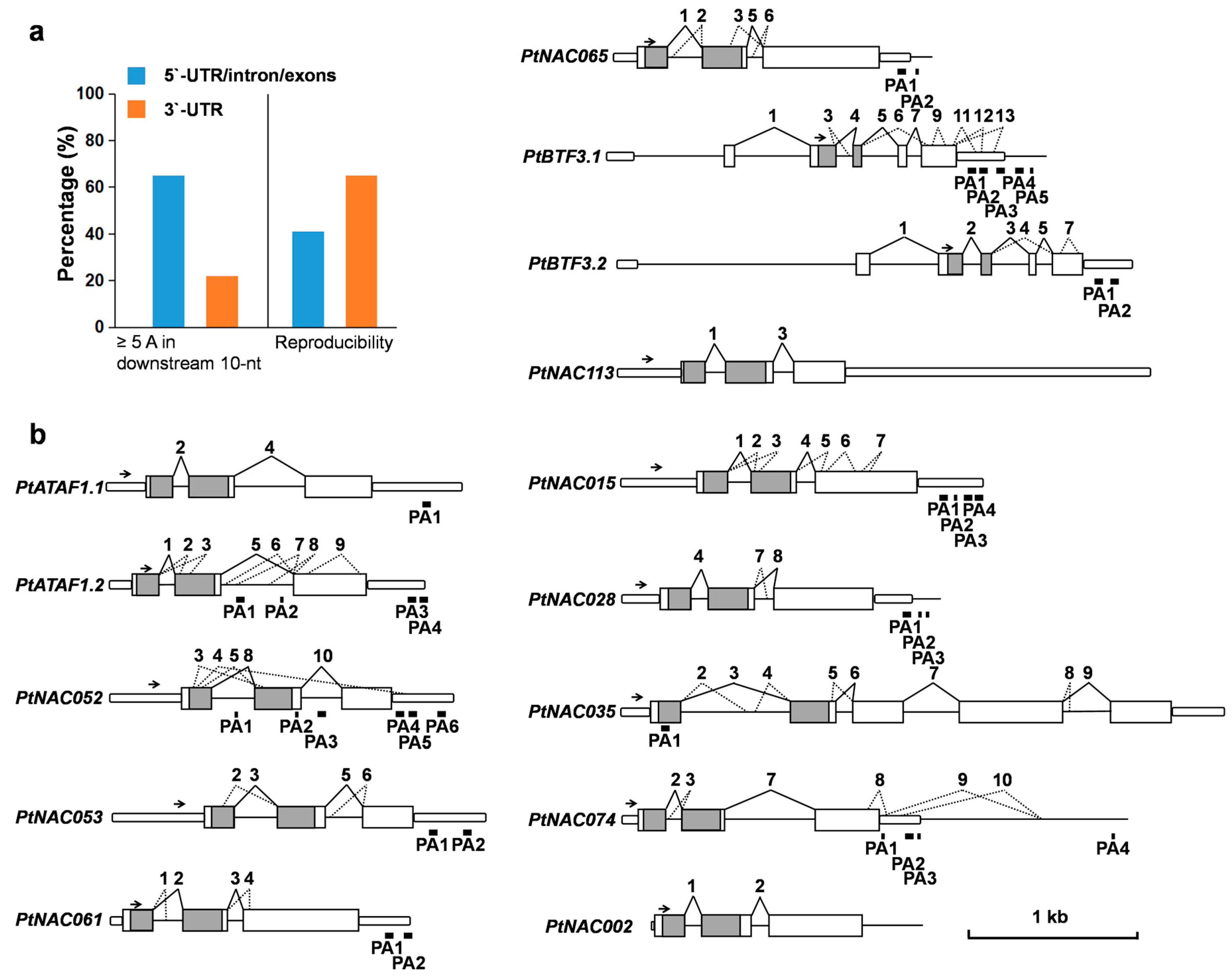
2.4. Complex AS Events in 14 NAC Transcription Factor Genes
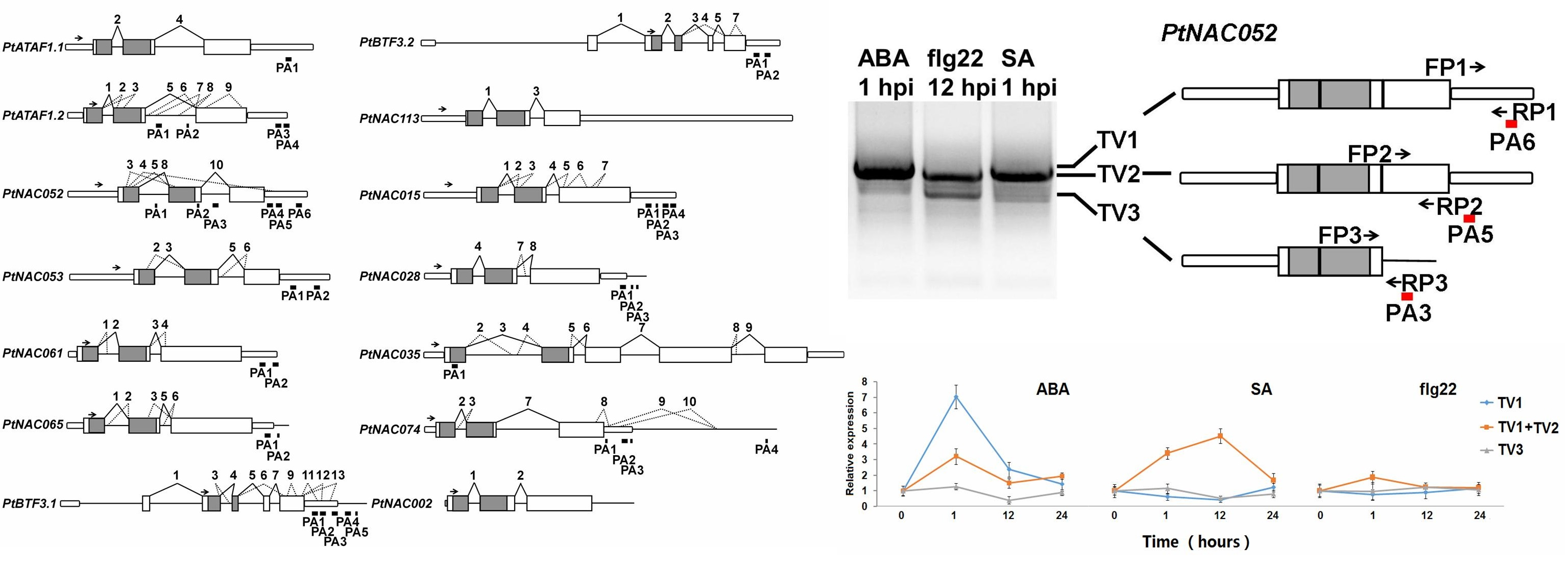
2.5. Identification of PASs by Removal of Possible Internal Priming Sites
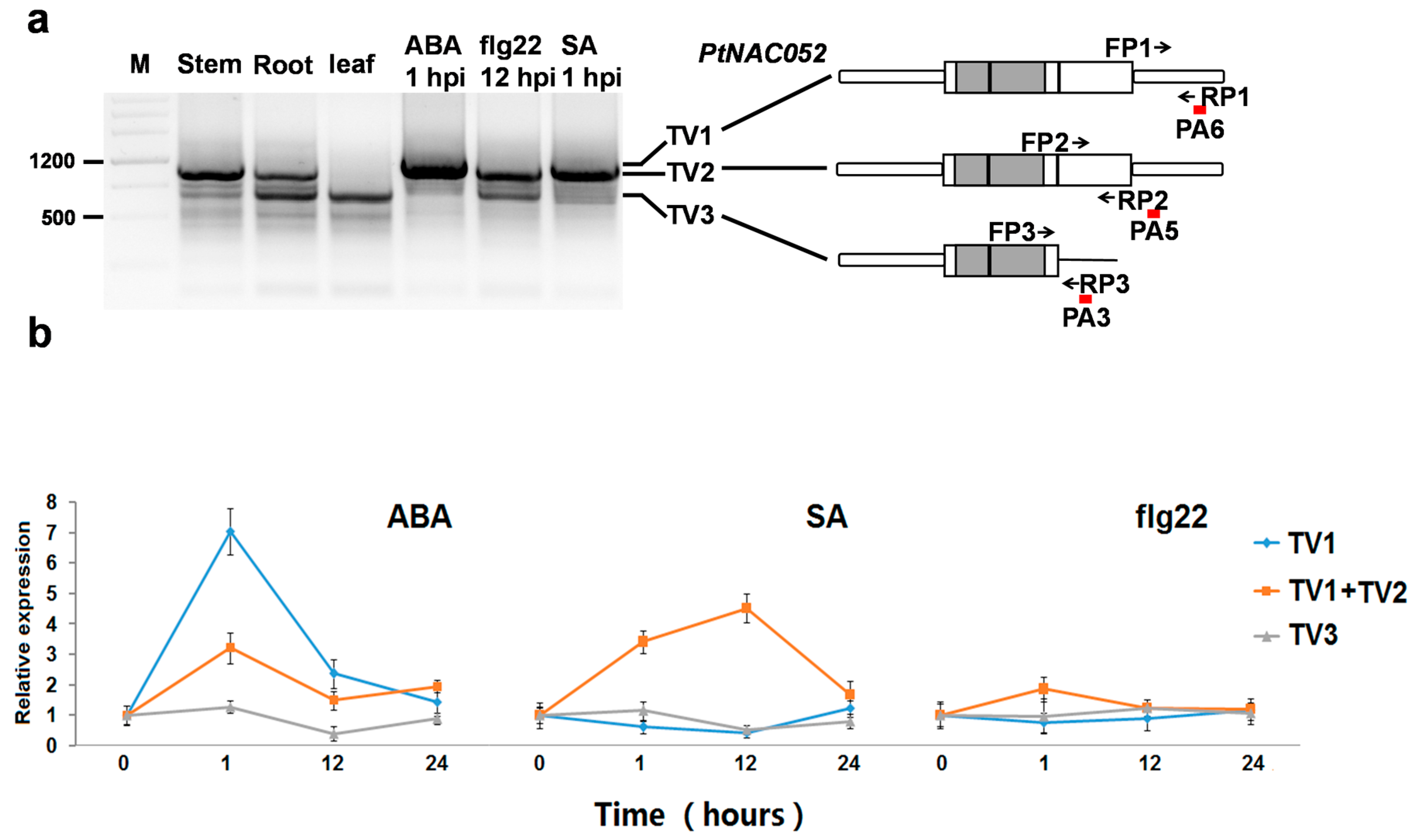
2.6. APA Transcripts Were Differentially Regulated across Tissues and in Response to Plant Hormones and Elicitors
3. Discussion
4. Materials and Methods
4.1. Plant Material and Plant Hormone and Elicitor Treatments
4.2. 3′-RACE Experiment
4.3. HTS and Data Analysis
4.4. Real-Time PCR
4.5. Availability of Supporting Data
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Proudfoot, N.J.; Furger, A.; Dye, M.J. Integrating mRNA processing with transcription. Cell 2002, 108, 501–512. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef] [PubMed]
- Staiger, D.; Brown, J.W. Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell 2013, 25, 3640–3656. [Google Scholar] [CrossRef] [PubMed]
- Filichkin, S.A.; Priest, H.D.; Givan, S.A.; Shen, R.; Bryant, D.W.; Fox, S.E.; Wong, W.K.; Mockler, T.C. Genome-wide mapping of alternative splicing in arabidopsis thaliana. Genome Res. 2010, 20, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Howard, B.E.; Hu, Q.; Babaoglu, A.C.; Chandra, M.; Borghi, M.; Tan, X.; He, L.; Winter-Sederoff, H.; Gassmann, W.; Veronese, P.; et al. High-throughput RNA sequencing of pseudomonas-infected arabidopsis reveals hidden transcriptome complexity and novel splice variants. PLoS ONE 2013, 8, e74183. [Google Scholar] [CrossRef] [PubMed]
- Marquez, Y.; Brown, J.W.; Simpson, C.; Barta, A.; Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in arabidopsis. Genome Res. 2012, 22, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, S.R.; Zhou, W.; Leonard, A.; Wang, B.B.; Beatty, M.; Zastrow-Hayes, G.; Zhao, X.; Baumgarten, A.; Li, B. Genome-wide analysis of alternative splicing in zea mays: Landscape and genetic regulation. Plant Cell 2014, 26, 3472–3487. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, S.R.; Danilevskaya, O.N.; Meng, X.; Beatty, M.; Zastrow-Hayes, G.; Harris, C.; Van Allen, B.; Habben, J.; Li, B. Genome-wide analysis of alternative splicing during development and drought stress in maize. Plant Physiol. 2016, 170, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, M.; Downie, B.; Liang, C.; Ji, G.; Li, Q.Q.; Hunt, A.G. Genome-wide landscape of polyadenylation in arabidopsis provides evidence for extensive alternative polyadenylation. Proc. Natl. Acad. Sci. USA 2011, 108, 12533–12538. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.K.; Lee, S.; Zhou, G.; Cao, X.; Wang, C.; Clark, T.; Chen, J.; Rowley, J.D.; Wang, S.M. Oligo(dt) primer generates a high frequency of truncated cdnas through internal poly(a) priming during reverse transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 6152–6156. [Google Scholar] [CrossRef] [PubMed]
- Sherstnev, A.; Duc, C.; Cole, C.; Zacharaki, V.; Hornyik, C.; Ozsolak, F.; Milos, P.M.; Barton, G.J.; Simpson, G.G. Direct sequencing of arabidopsis thaliana rna reveals patterns of cleavage and polyadenylation. Nat. Struct. Mol. Biol. 2012, 19, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.G. Rna regulatory elements and polyadenylation in plants. Front. Plant Sci. 2011, 2, 109. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Graber, J.H. Signals for pre-mrna cleavage and polyadenylation. Wiley Interdiscip. Rev. RNA 2012, 3, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Wang, H.; Xu, B.; Zhu, S.; Hu, L.; Huang, M. Discovery of a novel small secreted protein family with conserved n-terminal igy motif in dikarya fungi. BMC Genom. 2014, 15, 1151. [Google Scholar] [CrossRef] [PubMed]
- Howald, C.; Tanzer, A.; Chrast, J.; Kokocinski, F.; Derrien, T.; Walters, N.; Gonzalez, J.M.; Frankish, A.; Aken, B.L.; Hourlier, T.; et al. Combining rt-pcr-seq and rna-seq to catalog all genic elements encoded in the human genome. Genome Res. 2012, 22, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Karrer, E.E.; Lincoln, J.E.; Hogenhout, S.; Bennett, A.B.; Bostock, R.M.; Martineau, B.; Lucas, W.J.; Gilchrist, D.G.; Alexander, D. In situ isolation of mrna from individual plant cells: Creation of cell-specific cdna libraries. Proc. Natl. Acad. Sci. USA 1995, 92, 3814–3818. [Google Scholar] [CrossRef] [PubMed]
- Marquez, Y.; Hopfler, M.; Ayatollahi, Z.; Barta, A.; Kalyna, M. Unmasking alternative splicing inside protein-coding exons defines exitrons and their role in proteome plasticity. Genome Res. 2015, 25, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Subtelny, A.O.; Eichhorn, S.W.; Chen, G.R.; Sive, H.; Bartel, D.P. Poly(a)-tail profiling reveals an embryonic switch in translational control. Nature 2014, 508, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.X.; Li, Y.; Niu, Q.W.; Chua, N.H. Dehydration stress extends mrna 3’ untranslated regions with noncoding rna functions in arabidopsis. Genome Res. 2017, 27, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Hunt, A.G. A 3’ race protocol to confirm polyadenylation sites. Methods Mol. Biol. 2015, 1255, 135–144. [Google Scholar] [PubMed]
- Scotto-Lavino, E.; Du, G.; Frohman, M.A. 3’ end cdna amplification using classic race. Nat. Protoc. 2006, 1, 2742–2745. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, B.C. Revolutions in rapid amplification of cdna ends: New strategies for polymerase chain reaction cloning of full-length cdna ends. Anal. Biochem. 1995, 227, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Vitulo, N.; Forcato, C.; Carpinelli, E.C.; Telatin, A.; Campagna, D.; D’Angelo, M.; Zimbello, R.; Corso, M.; Vannozzi, A.; Bonghi, C.; et al. A deep survey of alternative splicing in grape reveals changes in the splicing machinery related to tissue, stress condition and genotype. BMC Plant Biol. 2014, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Cyrek, M.; Fedak, H.; Ciesielski, A.; Guo, Y.; Sliwa, A.; Brzezniak, L.; Krzyczmonik, K.; Pietras, Z.; Kaczanowski, S.; Liu, F.; et al. Seed dormancy in arabidopsis is controlled by alternative polyadenylation of dog1. Plant Physiol. 2016, 170, 947–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lorenzo, L.; Sorenson, R.; Bailey-Serres, J.; Hunt, A.G. Noncanonical alternative polyadenylation contributes to gene regulation in response to hypoxia. Plant Cell 2017, 29, 1262–1277. [Google Scholar] [CrossRef] [PubMed]
- Macknight, R.; Bancroft, I.; Page, T.; Lister, C.; Schmidt, R.; Love, K.; Westphal, L.; Murphy, G.; Sherson, S.; Cobbett, C.; et al. Fca, a gene controlling flowering time in arabidopsis, encodes a protein containing rna-binding domains. Cell 1997, 89, 737–745. [Google Scholar] [CrossRef]
- Michaels, S.D.; Amasino, R.M. Flowering locus c encodes a novel mads domain protein that acts as a repressor of flowering. Plant Cell 1999, 11, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, F.M.; Patton, D.A.; Meinke, D.W.; Amasino, R.M. Fpa, a gene involved in floral induction in arabidopsis, encodes a protein containing rna-recognition motifs. Plant Cell 2001, 13, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Yang, D.; Su, W.; Ma, L.; Shen, Y.; Ji, G.; Ye, X.; Wu, X.; Li, Q.Q. Genome-wide dynamics of alternative polyadenylation in rice. Genome Res. 2016, 26, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gaffney, B.; Hunt, A.G.; Li, Q.Q. Genome-wide determination of poly(a) sites in medicago truncatula: Evolutionary conservation of alternative poly(a) site choice. BMC Genom. 2014, 15, 615. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.G.; Osburn, L.; Kopsell, D.; Tuskan, G.A.; Cheng, Z.M. Micropropagation of Populus trichocarpa ‘nisqually-1’: The genotype deriving the Populus reference genome. Plant Cell Tissue Organ Cult. 2009, 99, 251–257. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Chakravarthy, S.; Velasquez, A.C.; McLane, H.L.; Zeng, L.; Nakayashiki, H.; Park, D.H.; Collmer, A.; Martin, G.B. Methods to study pamp-triggered immunity using tomato and nicotiana benthamiana. Mol. Plant-Microbe Interact. 2010, 23, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. Tophat: Discovering splice junctions with rna-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Zhang, B.; Zhuge, Q.; Zeng, Y.R.; Wang, M.X.; Huang, M.R. Expression profiles of two novel lipoxygenase genes in Populus deltoides. Plant Sci. 2006, 170, 1027–1035. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time pcr data by the comparative ct method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the total RNA of P. trichocarpa ‘Nisqually-1′ and 3′-RACE products of 14 NAC genes are available from the authors. |

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Wang, M.; Cheng, Q. Capturing the Alternative Cleavage and Polyadenylation Sites of 14 NAC Genes in Populus Using a Combination of 3′-RACE and High-Throughput Sequencing. Molecules 2018, 23, 608. https://doi.org/10.3390/molecules23030608
Wang H, Wang M, Cheng Q. Capturing the Alternative Cleavage and Polyadenylation Sites of 14 NAC Genes in Populus Using a Combination of 3′-RACE and High-Throughput Sequencing. Molecules. 2018; 23(3):608. https://doi.org/10.3390/molecules23030608
Chicago/Turabian StyleWang, Haoran, Mingxiu Wang, and Qiang Cheng. 2018. "Capturing the Alternative Cleavage and Polyadenylation Sites of 14 NAC Genes in Populus Using a Combination of 3′-RACE and High-Throughput Sequencing" Molecules 23, no. 3: 608. https://doi.org/10.3390/molecules23030608