An Investigation of Atomic Structures Derived from X-ray Crystallography and Cryo-Electron Microscopy Using Distal Blocks of Side-Chains


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
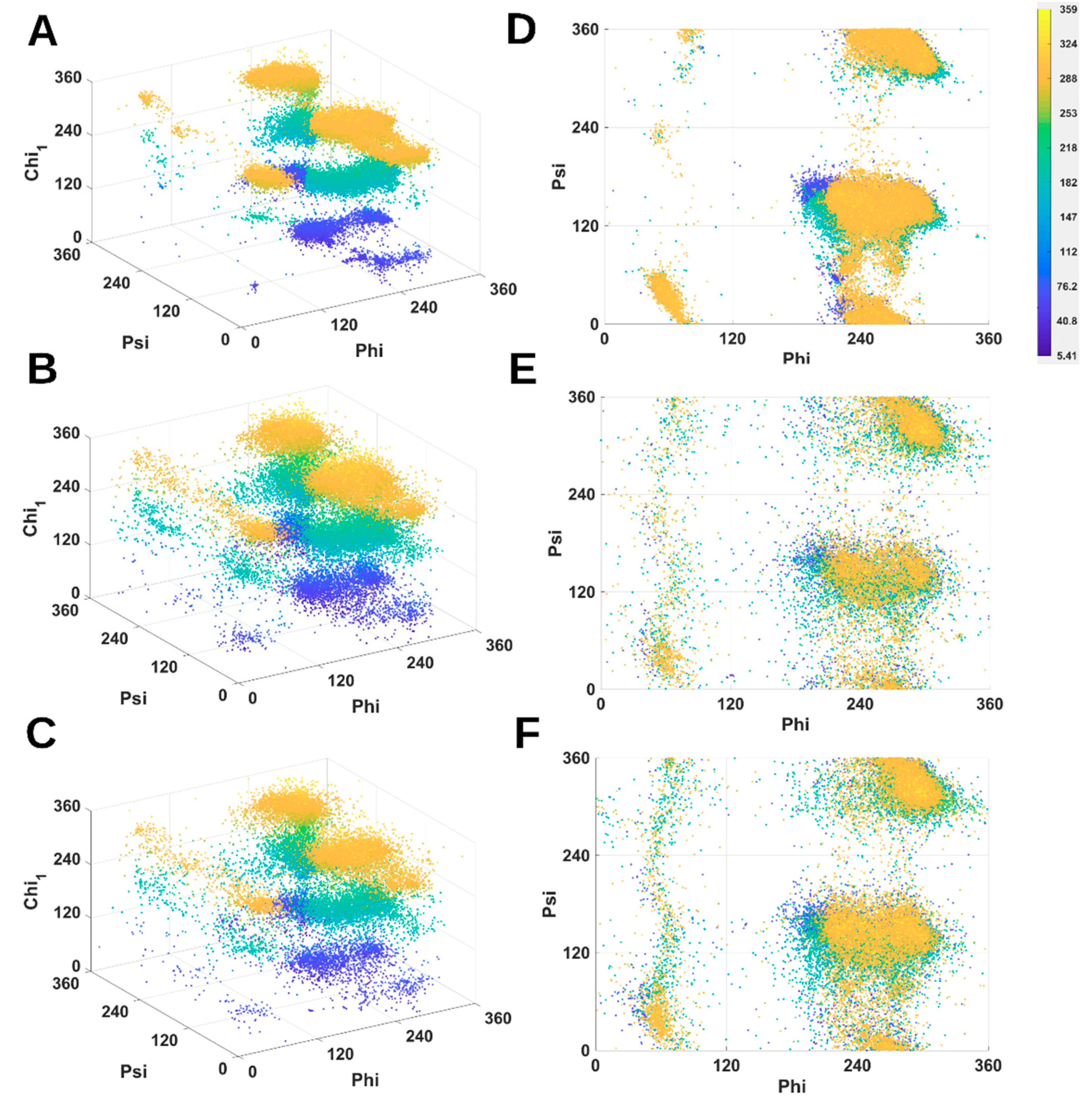
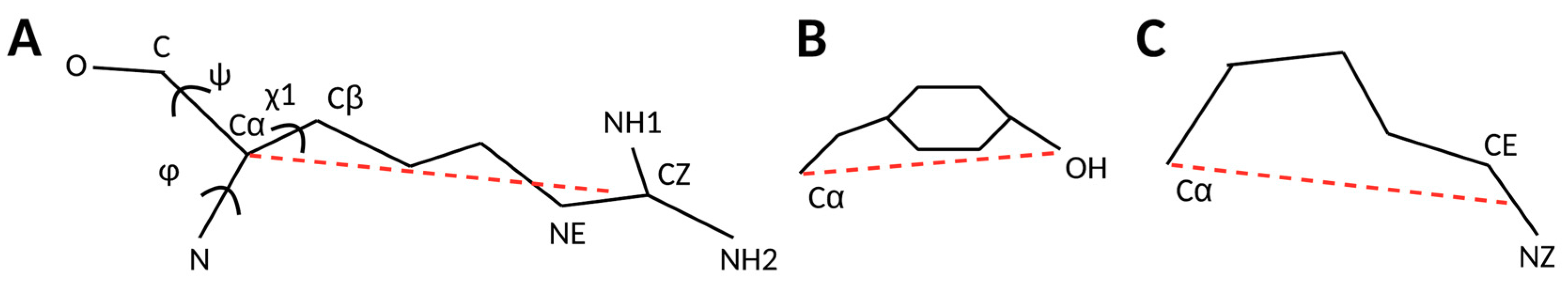
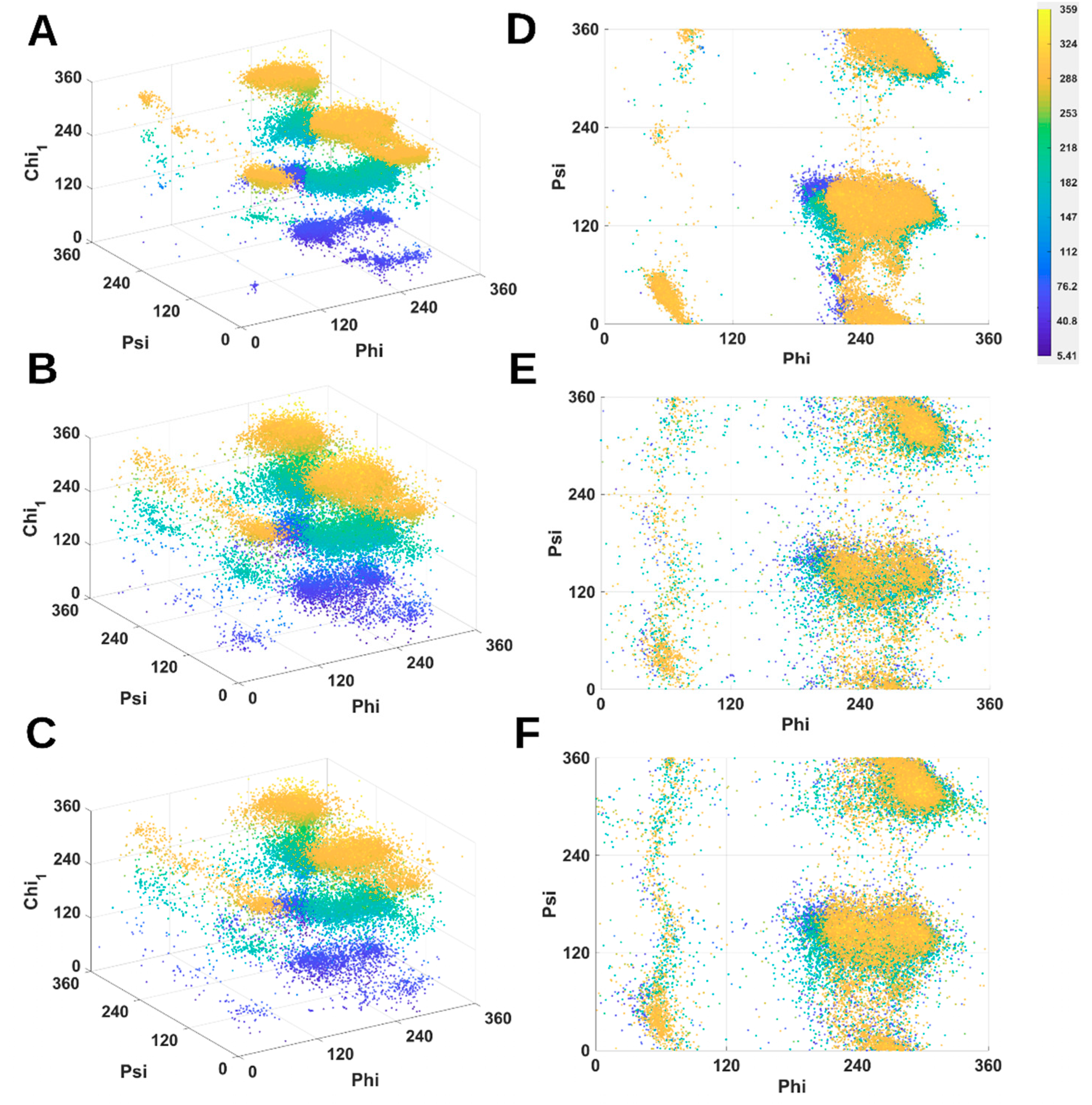
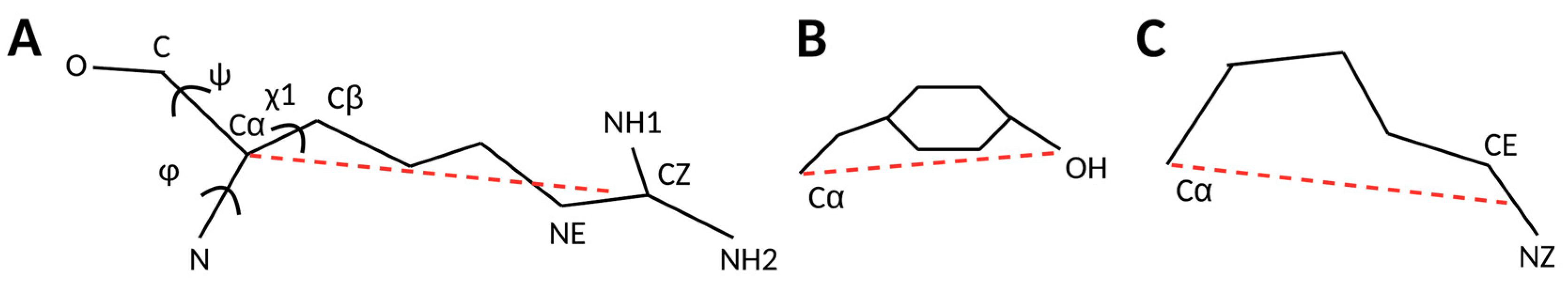
2.1. 3D Scatter Plots for Backbone and Side-Chain Torsion Angles
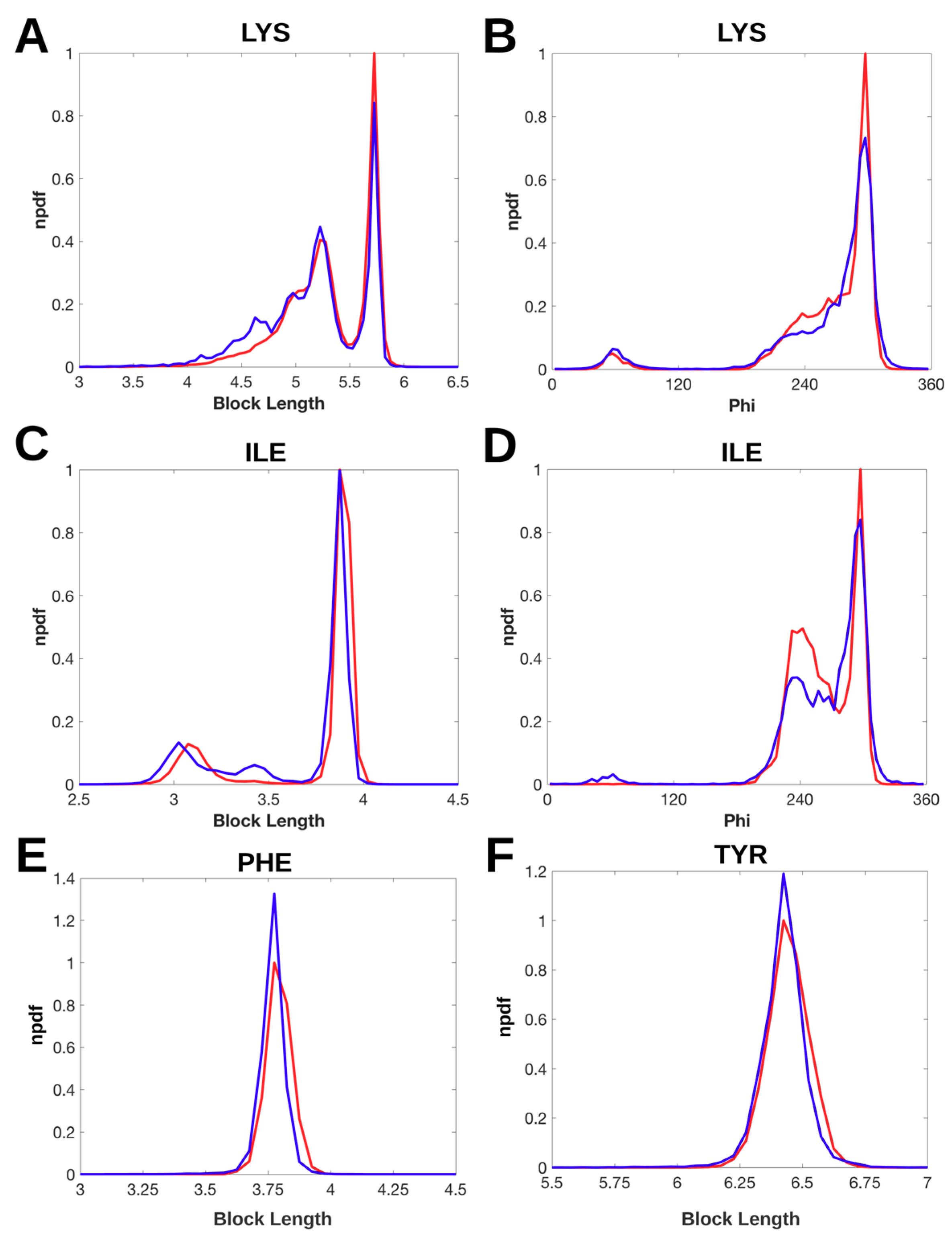
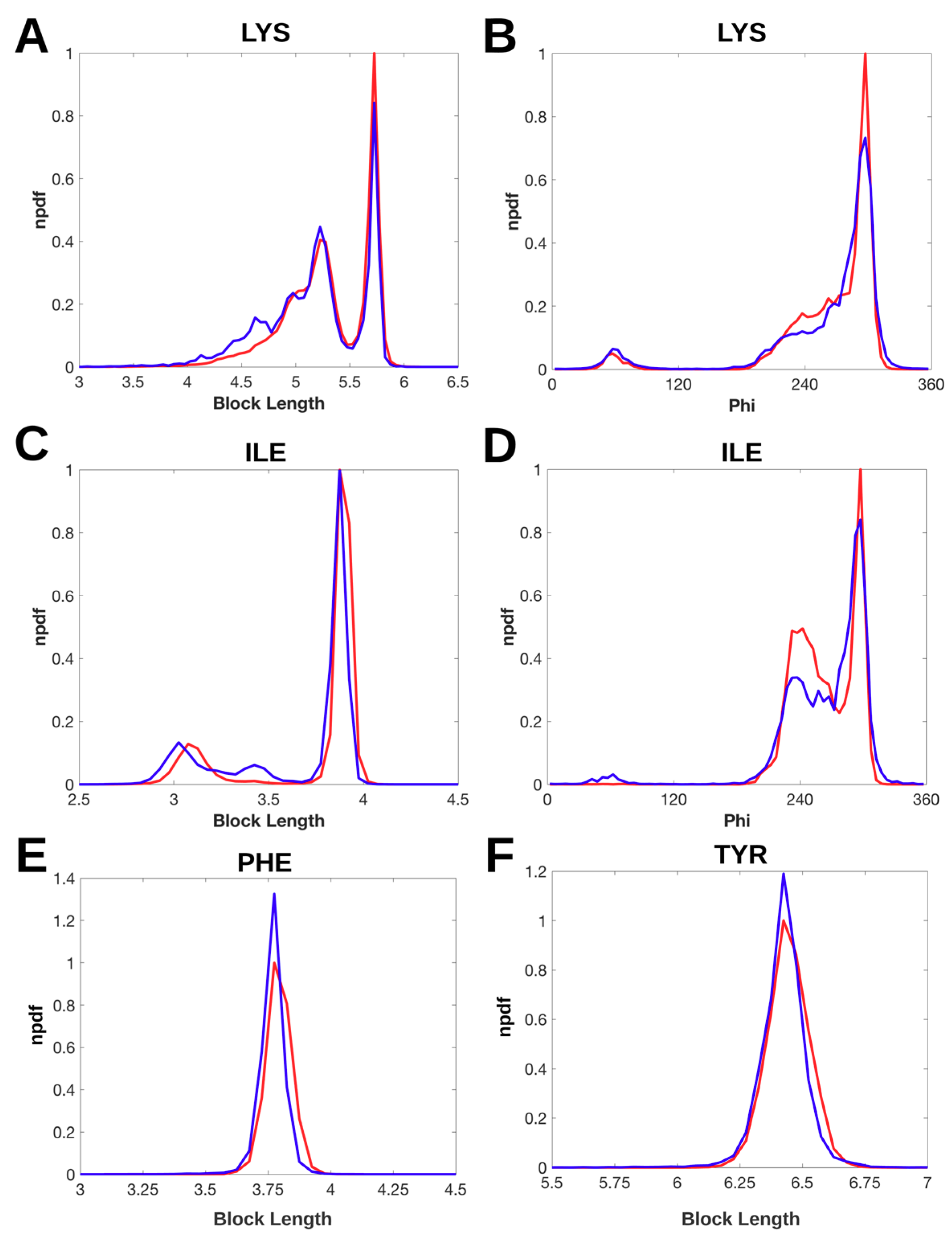
2.2. Normalized Probability Density Function for Block Lengths
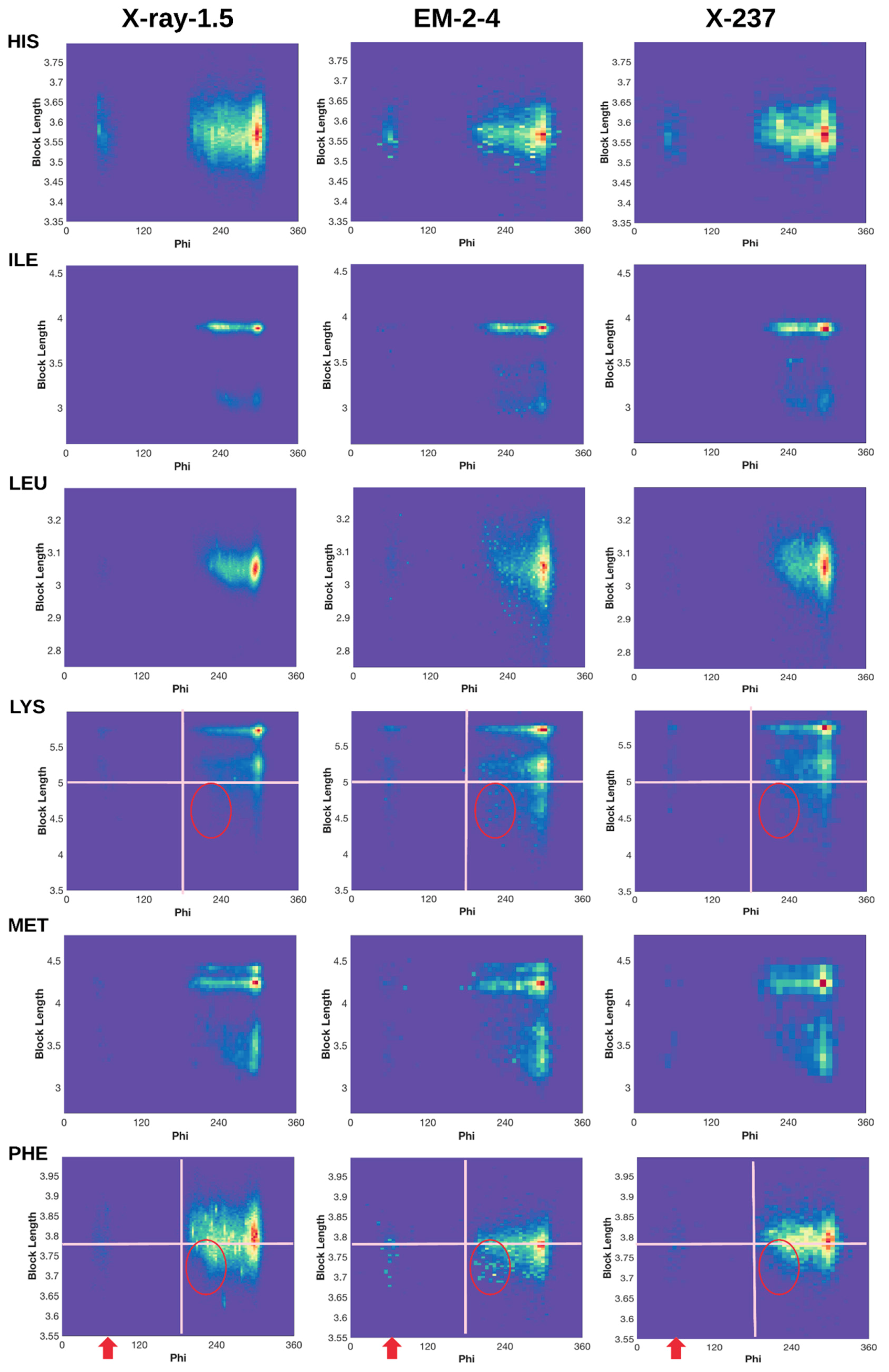
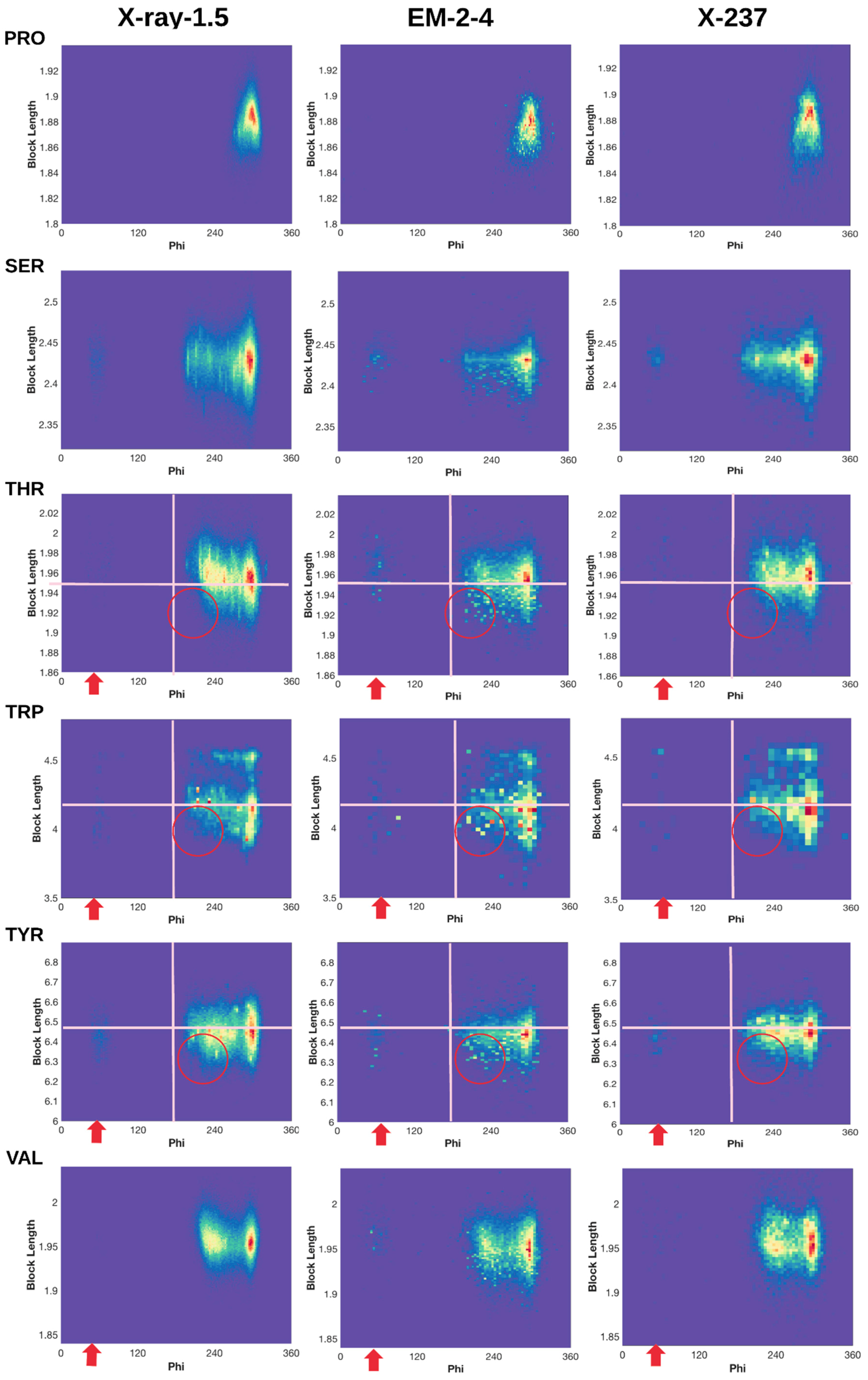
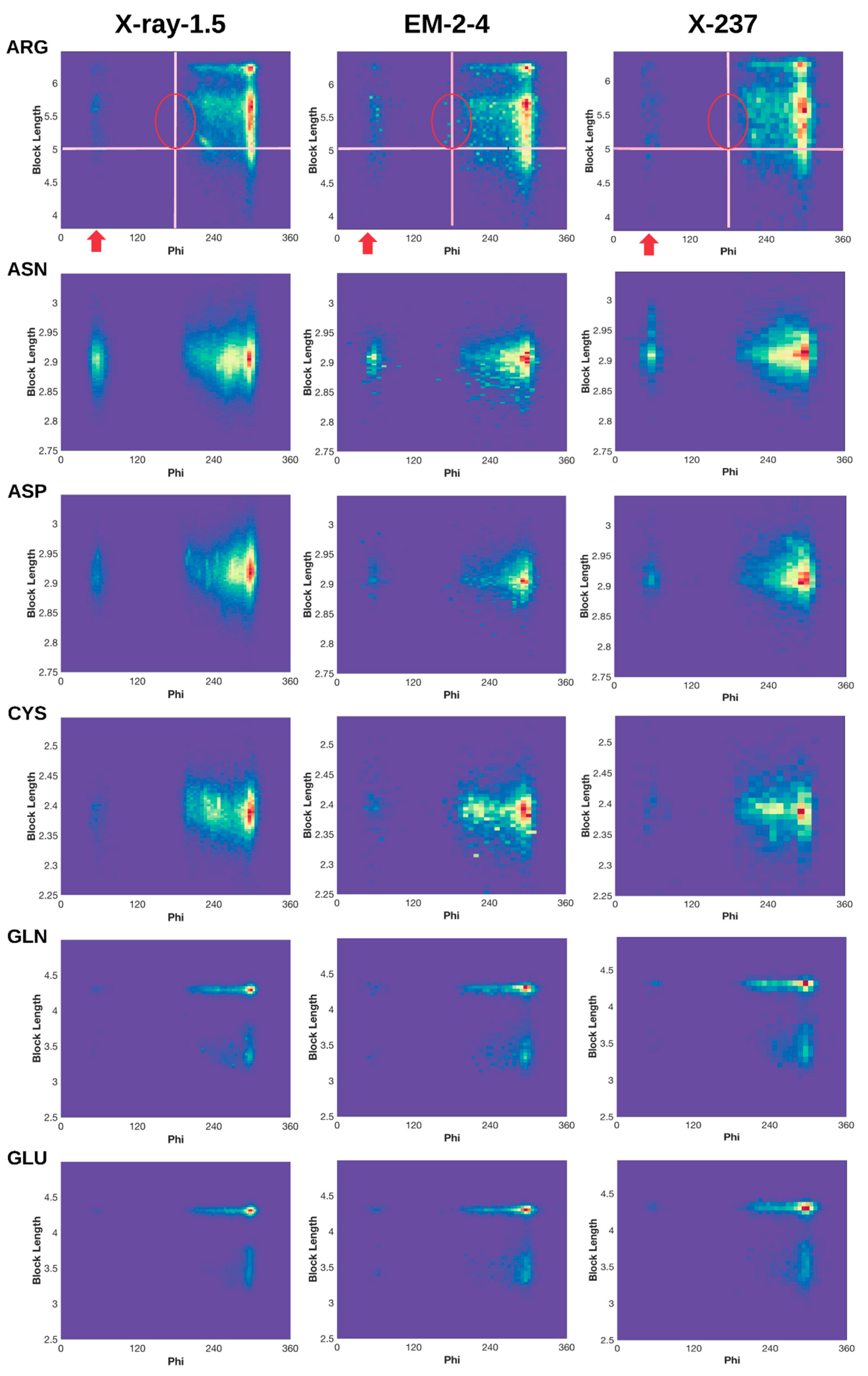
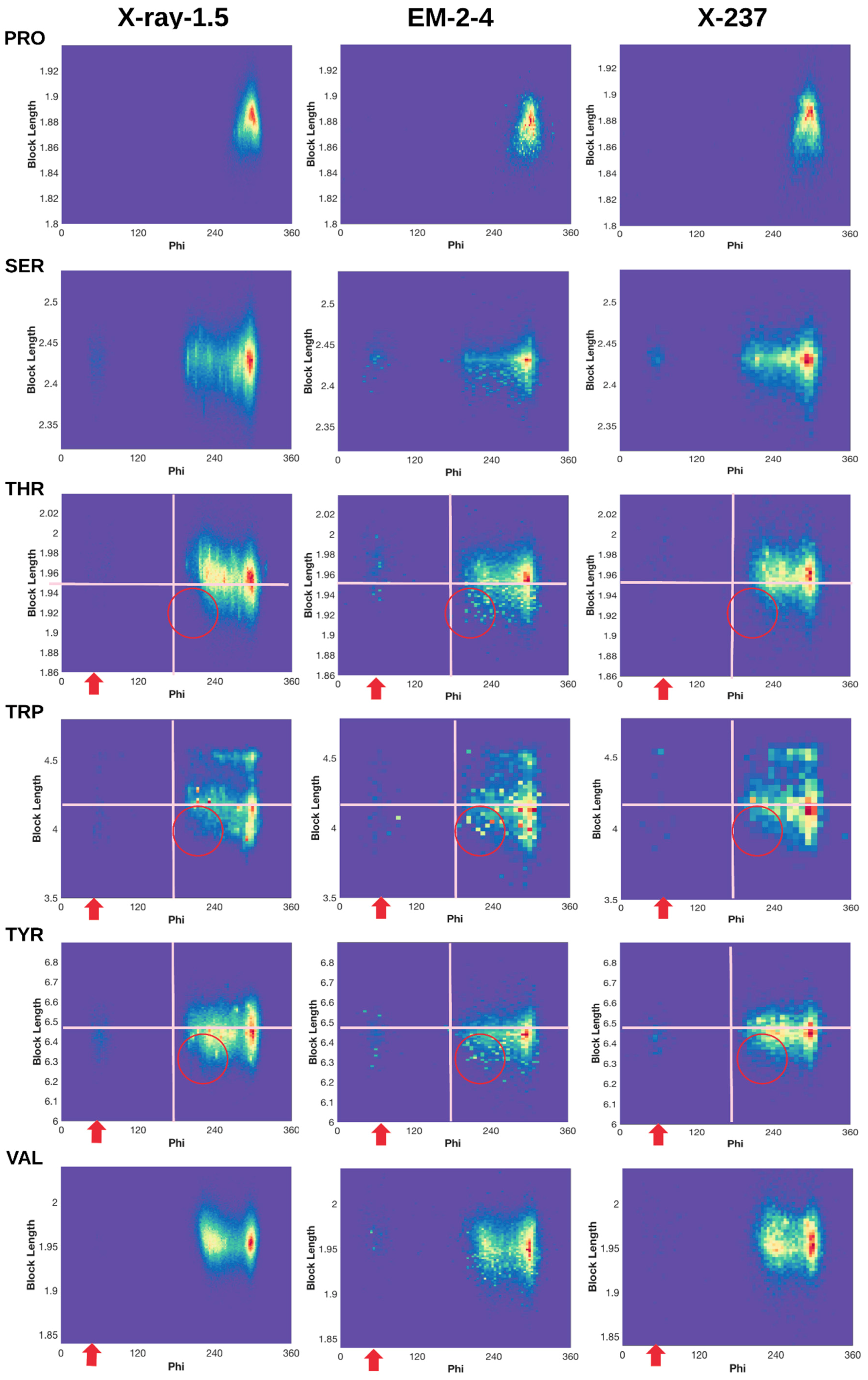
2.3. 2D Histograms of Combined Features Using the Block Length and Backbone Torsion φ
2.3.1. Conformations of Side-Chains When φ is Near 60°
2.3.2. Shorter Side-Chains When φ is Near 210°
4. Materials and Methods
5. Conclusions
Supplementary Materials
Acknowledgment
Author Contributions
Conflicts of Interest
References
- PDB RCSB. RCSB Statistics Release. 2018. Available online: https://www.rcsb.org/stats/growth/em. (accessed on 26 January 2018).
- Europe PDBi. EMDB Statistics Release. 2018. Available online: http://www.ebi.ac.uk/pdbe/emdb/statistics_releases.html/ (accessed on 26 January 2018).
- Quinlan, R.A.; Zhang, Y.; Lansbury, A.; Williamson, I.; Pohl, E.; Sun, F. Changes in the quaternary structure and function of MjHSP16.5 attributable to deletion of the IXI motif and introduction of the substitution, R107G, in the α-crystallin domain. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harauz, G.; van Heel, M. Exact Filters for General Geometry Three Dimensional Reconstruction. Optik 1986, 73, 146–156. [Google Scholar]
- Rawson, S.; Bisson, C.; Hurdiss, D.L.; Fazal, A.; McPhillie, M.J.; Sedelnikova, S.E.; Baker, P.J.; Rice, D.W.; Muench, S.P. Elucidating the structural basis for differing enzyme inhibitor potency by cryo-EM. Proc. Natl. Acad. Sci. USA 2018, 115, 1795–1800. [Google Scholar] [CrossRef] [PubMed]
- Autzen, H.E.; Myasnikov, A.G.; Campbell, M.G.; Asarnow, D.; Julius, D.; Cheng, Y. Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 2018, 359, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Shalev-Benami, M.; Zhang, Y.; Rozenberg, H.; Nobe, Y.; Taoka, M.; Matzov, D.; Zimmerman, E.; Bashan, A.; Isobe, T.; Jaffe, C.L.; et al. Atomic resolution snapshot of Leishmania ribosome inhibition by the aminoglycoside paromomycin. Nat. Commun. 2017, 8, 1589. [Google Scholar] [CrossRef] [PubMed]
- Kater, L.; Thoms, M.; Barrio-Garcia, C.; Cheng, J.; Ismail, S.; Ahmed, Y.L.; Bange, G.; Kressler, D.; Berninghausen, O.; Sinning, I.; et al. Visualizing the Assembly Pathway of Nucleolar Pre-60S Ribosomes. Cell 2017, 171, 1599-1610.e14. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; She, J.; Zeng, W.; Chen, Q.; Bai, X.C.; Jiang, Y. Structures of the calcium-activated, non-selective cation channel TRPM4. Nature 2017, 552, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Bränd’en, C.-I.; Alwyn Jones, T. Between objectivity and subjectivity. Nature 1990, 343, 687–689. [Google Scholar] [CrossRef]
- Kleywegt, G.J.; Jones, T.A. Homo Crystallographicus—Quo Vadis? Structure 2002, 10, 465–472. [Google Scholar] [CrossRef]
- Jones, T.A.; Zou, J.Y.; Cowan, S.W.; Kjeldgaard, M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 1991, 47 Pt 2, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Kleywegt, G.J.; Jones, T.A. Phi/Psi-chology: Ramachandran revisited. Structure 1996, 4, 1395–1400. [Google Scholar] [CrossRef]
- Kleywegt, G.J.; Jones, T.A. Where freedom is given, liberties are taken. Structure 1995, 3, 535–540. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Hooft, R.W.; Vriend, G.; Sander, C.; Abola, E.E. Errors in protein structures. Nature 1996, 381, 272. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., 3rd; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Bruno, I.J.; Cole, J.C.; Kessler, M.; Luo, J.; Motherwell, W.D.; Purkis, L.H.; Smith, B.R.; Taylor, R.; Cooper, R.I.; Harris, S.E.; et al. Retrieval of crystallographically-derived molecular geometry information. J. Chem. Inf. Comput. Sci. 2004, 44, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MOLPROBITY: Structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004, 32, W615–W619. [Google Scholar] [CrossRef] [PubMed]
- Kleywegt, G.J.; Harris, M.R.; Zou, J.Y.; Taylor, T.C.; Wahlby, A.; Jones, T.A. The Uppsala Electron-Density Server. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2240–2249. [Google Scholar] [CrossRef] [PubMed]
- Zwart, P.H.; Grosse-Kunstleve, R.W.; Adams, P.D. Xtriage and Fest: Automatic assessment of X-ray data and substructure structure factor estimation. CCP4 Newslett. 2005, 43. Available online: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.378.2281&rep=rep1&type=pdf (accessed on 26 January 2018).
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 1, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Hintze, B.J.; Lewis, S.M.; Richardson, J.S.; Richardson, D.C. Molprobity’s ultimate rotamer-library distributions for model validation. Proteins 2016, 84, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Weichenberger, C.X.; Pozharski, E.; Rupp, B. Twilight reloaded: The peptide experience. Acta Crystallogr. D Struct. Biol. 2017, 73 Pt 3, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Read, R.J.; Adams, P.D.; Arendall, W.B., 3rd; Brunger, A.T.; Emsley, P.; Joosten, R.P.; Kleywegt, G.J.; Krissinel, E.B.; Lütteke, T.; Otwinowski, Z.; et al. A new generation of crystallographic validation tools for the protein data bank. Structure 2011, 19, 1395–1412. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.; Sali, A.; Baker, M.L.; Carragher, B.; Devkota, B.; Downing, K.H.; Egelman, E.H.; Feng, Z.; Frank, J.; Grigorieff, N.; et al. Outcome of the first electron microscopy validation task force meeting. Structure 2012, 20, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montelione, G.T.; Nilges, M.; Bax, A.; Güntert, P.; Herrmann, T.; Richardson, J.S.; Schwieters, C.D.; Vranken, W.F.; Vuister, G.W.; Wishart, D.S.; et al. Recommendations of the wwPDB NMR Validation Task Force. Structure 2013, 21, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Gore, S.; Velankar, S.; Kleywegt, G.J. Implementing an X-ray validation pipeline for the Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2012, 68 Pt 4, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.; Grigorieff, N. Noise bias in the refinement of structures derived from single particles. Ultramicroscopy 2004, 102, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Scheres, S.H. A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 2012, 415, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Leschziner, A.E.; Nogales, E. The orthogonal tilt reconstruction method: An approach to generating single-class volumes with no missing cone for ab initio reconstruction of asymmetric particles. J. Struct. Biol. 2006, 153, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Radermacher, M. Three-dimensional reconstruction of single particles from random and nonrandom tilt series. J. Electron Microsc. Tech. 1988, 9, 359–394. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Peng, L.; Baldwin, P.R.; Mann, D.S.; Jiang, W.; Rees, I.; Ludtke, S.J. EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 2007, 157, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Penczek, P.A.; Frank, J.; Spahn, C.M. A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. J. Struct. Biol. 2006, 154, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Brünger, A.T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.G.; Bernal, R.; Pletnev, S.V. Combining electron microscopic with x-ray crystallographic structures. J. Struct. Biol. 2001, 136, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; He, J. A distance- and orientation-dependent energy function of amino acid key blocks. Biopolymers 2014, 101, 681–692. [Google Scholar] [CrossRef] [PubMed]
- MathWorks. MATLAB Central. Available online: https://www.mathworks.com/matlabcentral/fileexchange/45325-efficient-2d-histogram--no-toolboxes-needed (accessed on 26 January 2018).
- He, J.; Schmid, M.F.; Zhou, Z.H.; Rixon, F.; Chiu, W. Finding and using local symmetry in identifying lower domain movements in hexon subunits of the herpes simplex virus type 1 B capsid. J. Mol. Biol. 2001, 309, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Cao, Y. Quantifying side-chain conformational variations in protein structure. Sci. Rep. 2016, 6, 37024. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Li, M.; Dauter, Z. High-Resolution Cryo-EM Maps and Models: A Crystallographer’s Perspective. Structure 2017, 25, 1589–1597.e1. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; He, J.; Sazzed, S.; Walker, R. An Investigation of Atomic Structures Derived from X-ray Crystallography and Cryo-Electron Microscopy Using Distal Blocks of Side-Chains. Molecules 2018, 23, 610. https://doi.org/10.3390/molecules23030610
Chen L, He J, Sazzed S, Walker R. An Investigation of Atomic Structures Derived from X-ray Crystallography and Cryo-Electron Microscopy Using Distal Blocks of Side-Chains. Molecules. 2018; 23(3):610. https://doi.org/10.3390/molecules23030610
Chicago/Turabian StyleChen, Lin, Jing He, Salim Sazzed, and Rayshawn Walker. 2018. "An Investigation of Atomic Structures Derived from X-ray Crystallography and Cryo-Electron Microscopy Using Distal Blocks of Side-Chains" Molecules 23, no. 3: 610. https://doi.org/10.3390/molecules23030610