Chiral Thioxanthones as Modulators of P-glycoprotein: Synthesis and Enantioselectivity Studies

 ,
,  ,
,  ,
,  and
and 
Abstract
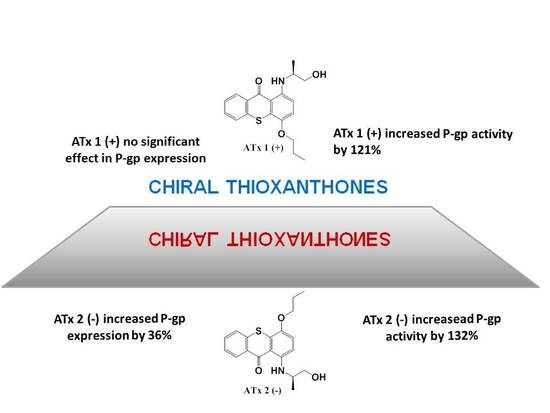
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
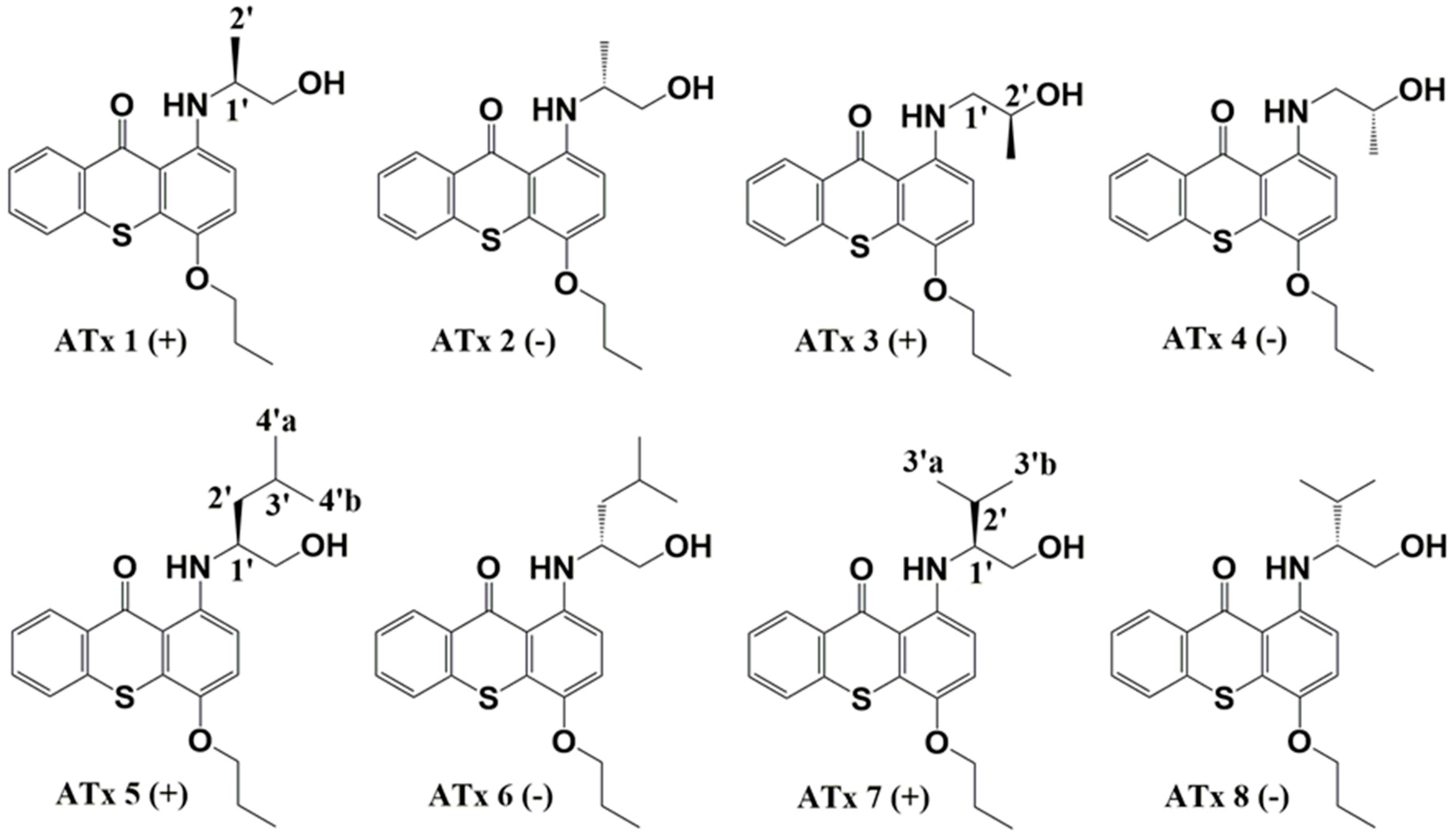
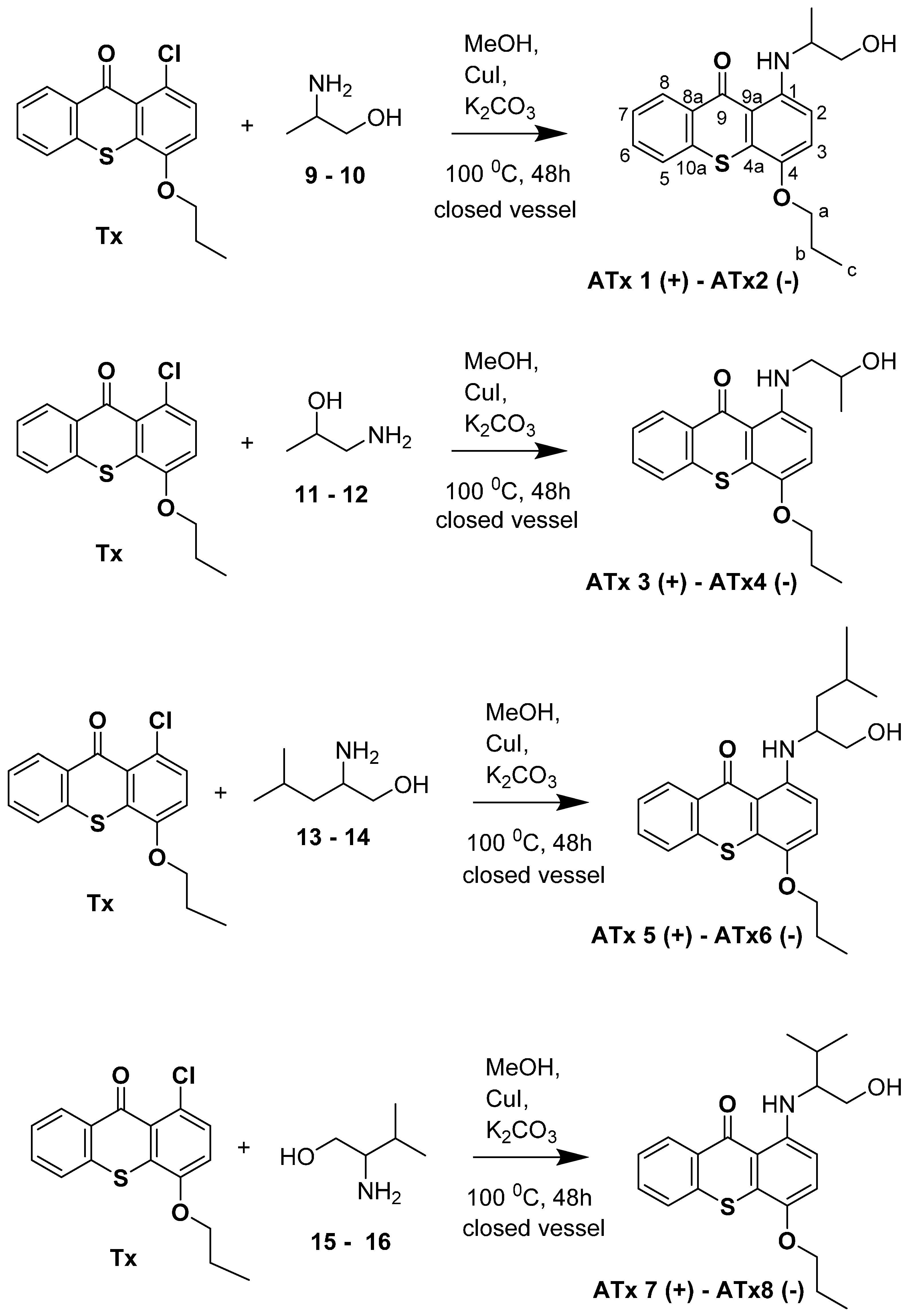
2.1. Synthesis of Thioxanthones
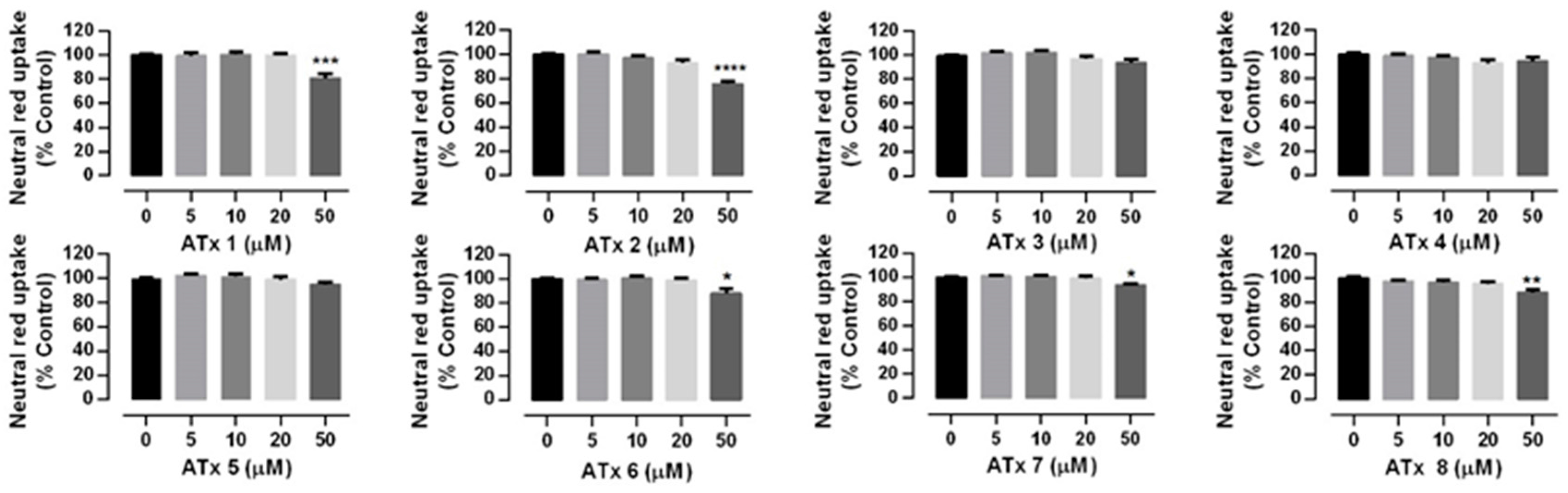
2.2. Chiral Thioxanthones Cytotoxicity Assays
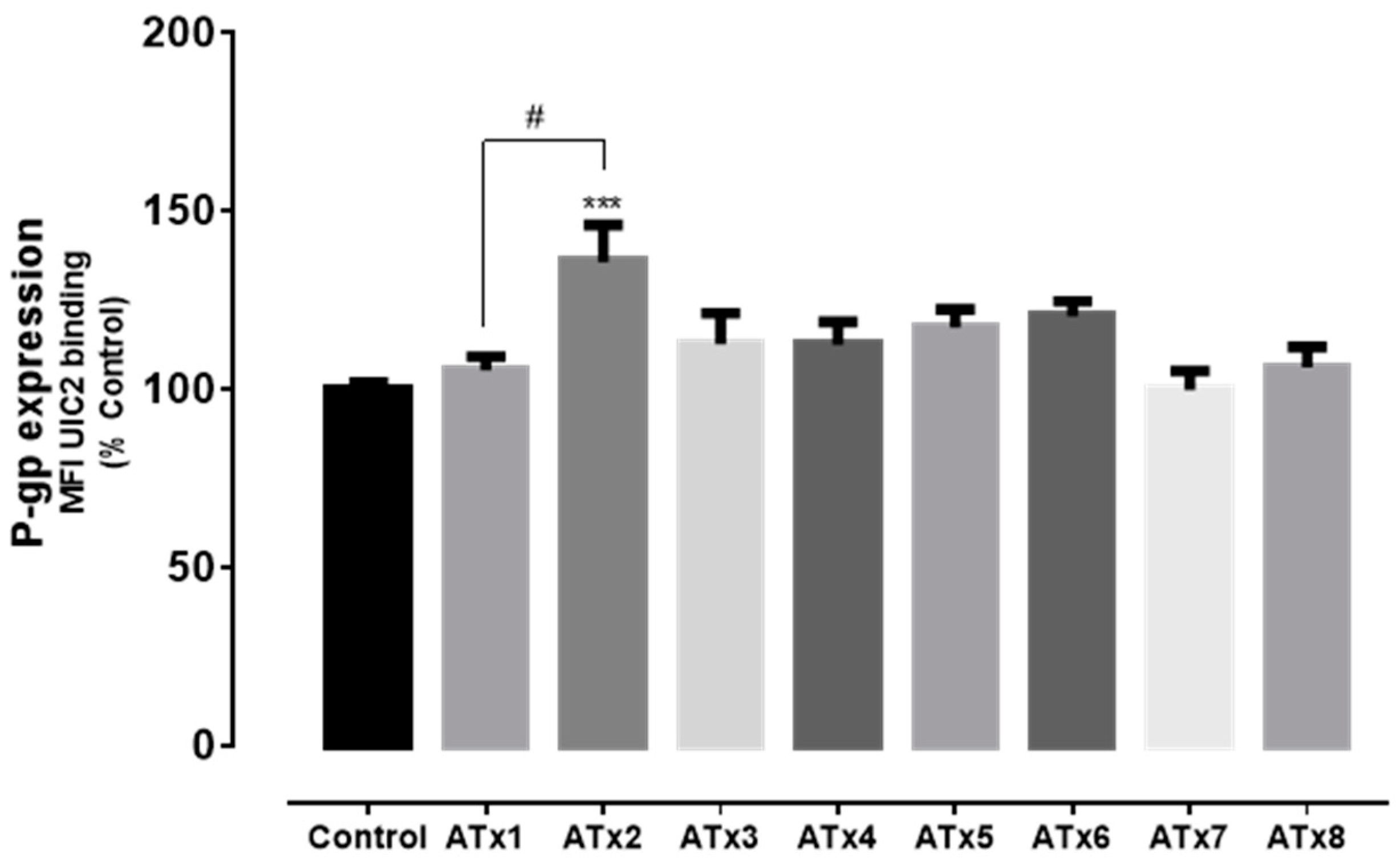
2.3. Flow Cytometry Analysis of P-gp Expression
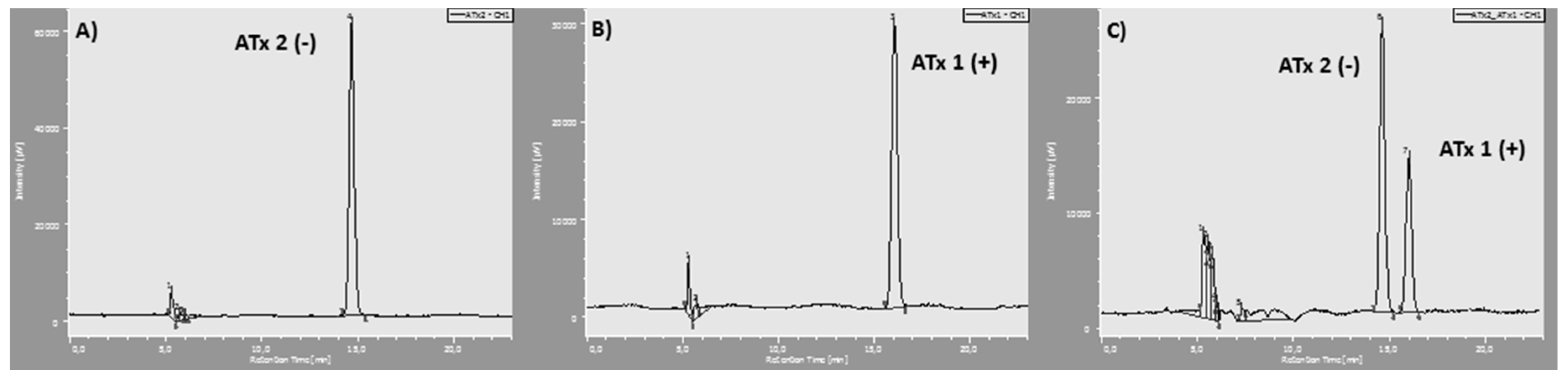
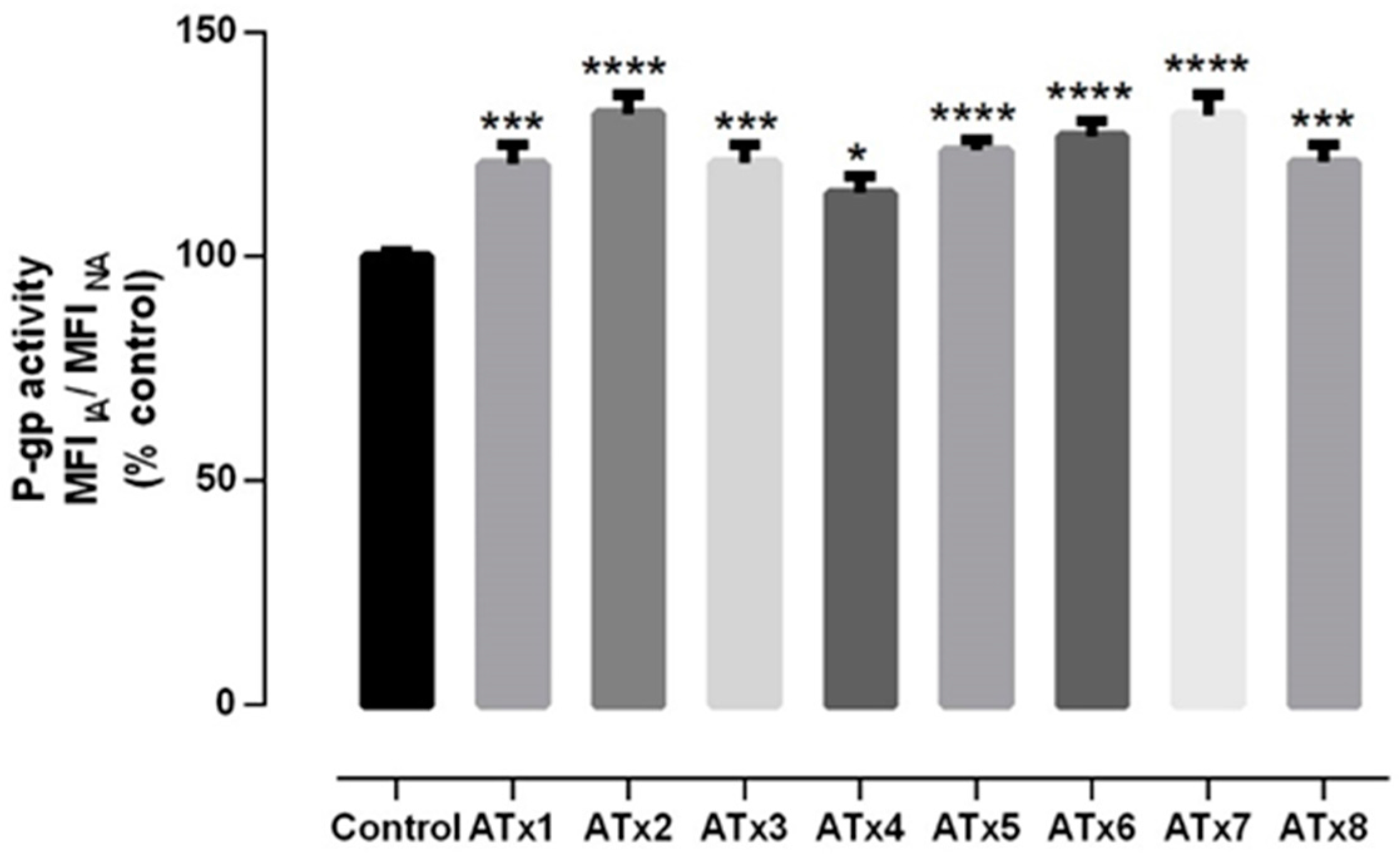
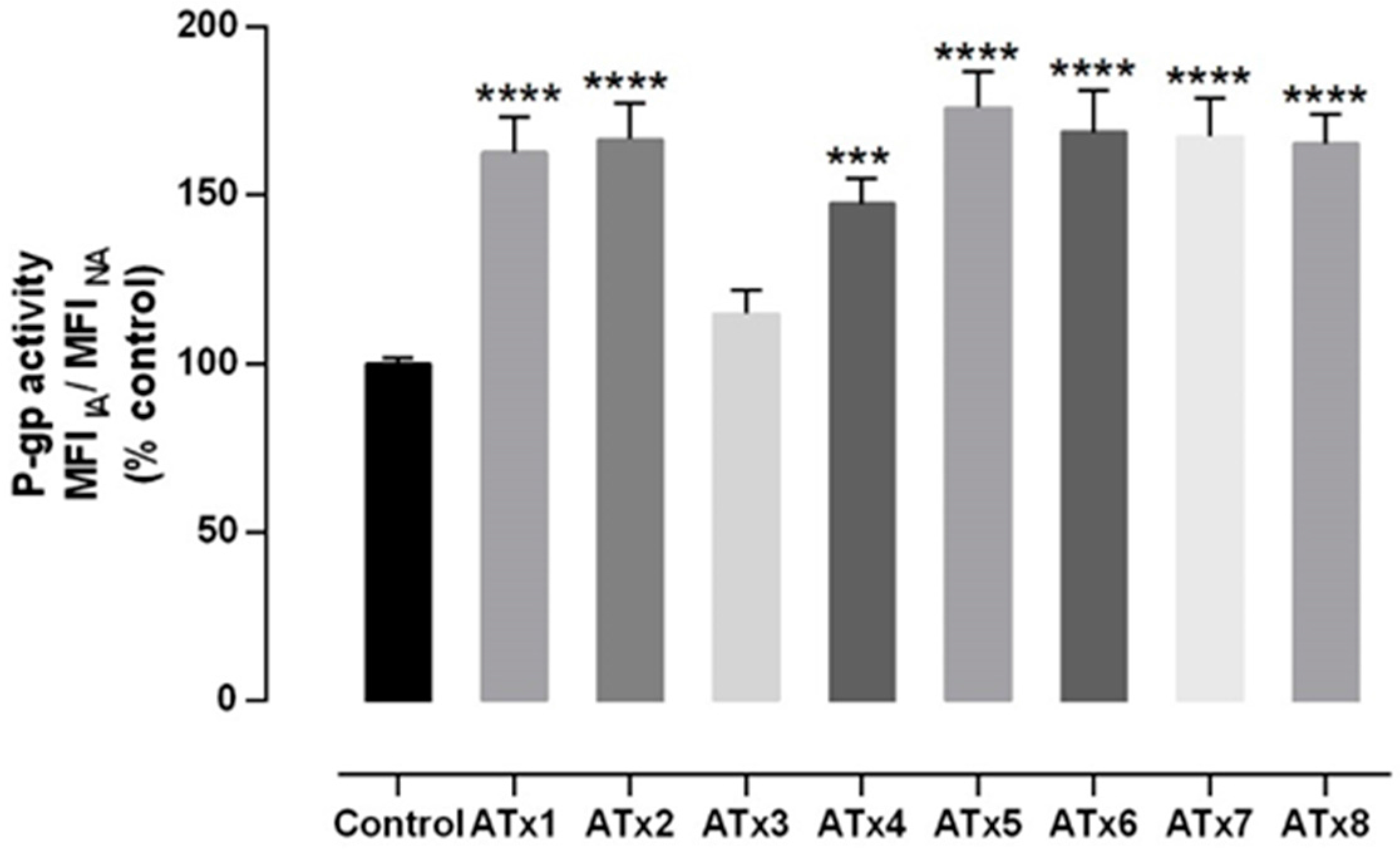
2.4. Evaluation of P-gp Transport Activity
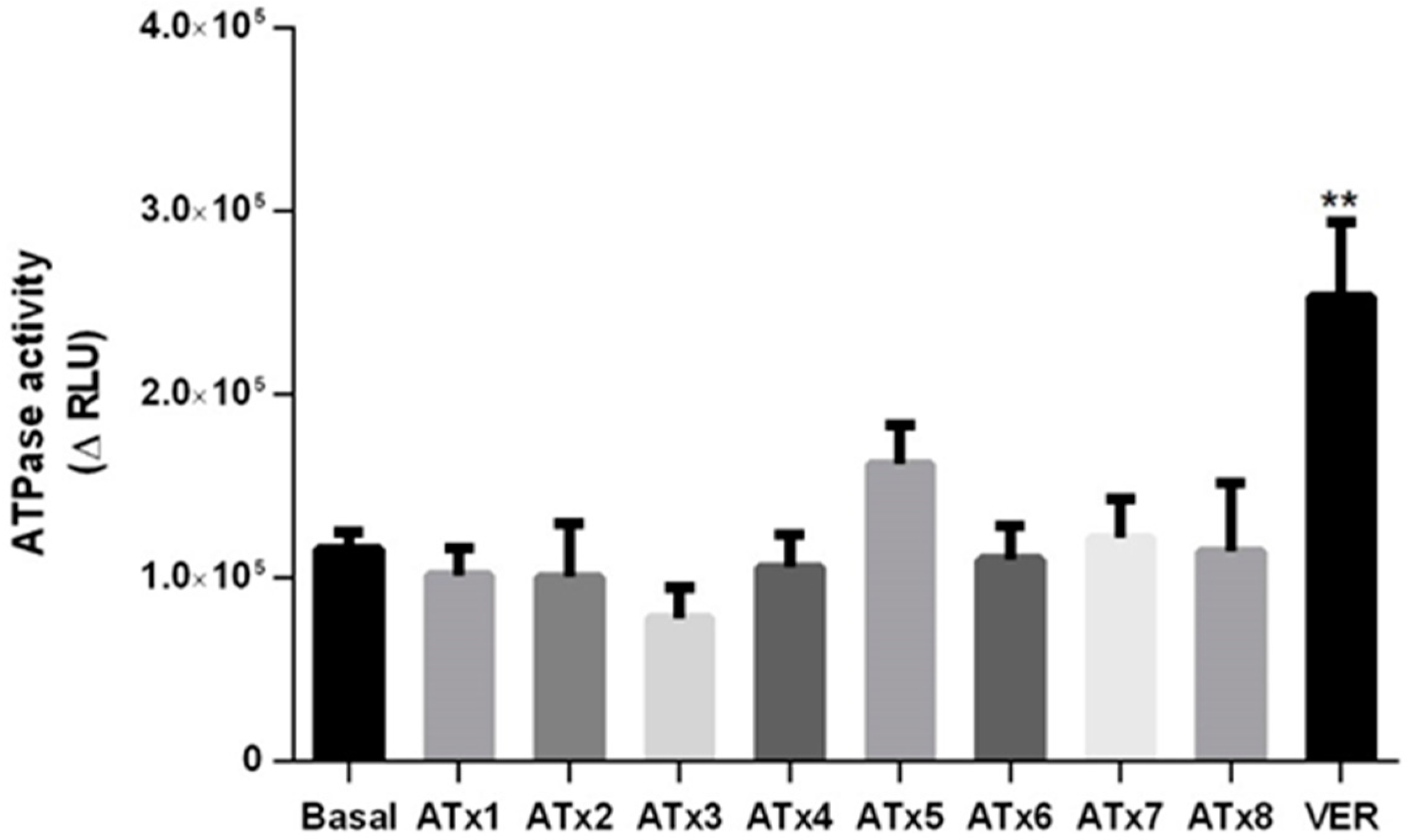
2.5. Determination of ATPase Activity
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Synthesis of Chiral Thioxanthones (ATxs 1–8)
4.3. Caco-2 Cell Culture
4.4. Compounds Cytotoxicity Assays
4.5. Flow Cytometry Analysis of P-gp Expression
4.6. Evaluation of P-gp Transport Activity
4.6.1. RHO 123 Efflux Assay in the Presence of ATxs 1–8
4.6.2. RHO 123 Efflux Assay in Caco-2 Cells Pre-Expose to ATxs 1–8 for 24 h
4.7. Evaluation of P-gp ATPase Activity
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Capparelli, E.; Zinzi, L.; Cantore, M.; Contino, M.; Perrone, M.G.; Luurtsema, G.; Berardi, F.; Perrone, R.; Colabufo, N.A. Sar studies on tetrahydroisoquinoline derivatives: The role of flexibility and bioisosterism to raise potency and selectivity toward P-glycoprotein. J. Med. Chem. 2014, 57, 9983–9994. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735–7738. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.S.; Ashokraj, Y.; Dey, C.S.; Panchagnula, R. P-Glycoprotein inhibitors and their screening: A perspective from bioavailability enhancement. Pharmacol. Res. 2003, 48, 347–359. [Google Scholar] [CrossRef]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; de Lourdes Bastos, M.; Remião, F. Modulation of P-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, M.; Spiers, J.P. A primer on the mechanics of P-glycoprotein the multidrug transporter. Pharmacol. Res. 2007, 55, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. The P-glycoprotein multidrug transporter: Interactions with membrane lipids, and their modulation of activity. Biochem. Soc. Trans. 1997, 25, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, M.; Silva, R.; Rocha-Pereira, C.; Carmo, H.; Carvalho, F.; Bastos, M.L.; Remiao, F. Cellular models and in vitro assays for the screening of modulators of P-gp, MRP1 and BCRP. Molecules 2017, 22, 600. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.M.; Deeley, R.G.; Cole, S.P.C. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 2005, 204, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Fojo, A.T.; Ueda, K.; Slamon, D.J.; Poplack, D.G.; Gottesman, M.M.; Pastan, I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Kobori, T.; Harada, S.; Nakamoto, K.; Tokuyama, S. Mechanisms of P-glycoprotein alteration during anticancer treatment: Role in the pharmacokinetic and pharmacological effects of various substrate drugs. J. Pharmacol. Sci. 2014, 125, 242–254. [Google Scholar] [CrossRef] [PubMed]
- DeGorter, M.K.; Xia, C.Q.; Yang, J.J.; Kim, R.B. Drug transporters in drug efficacy and toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–273. [Google Scholar] [CrossRef] [PubMed]
- Couture, L.; Nash, J.A.; Turgeon, J. The ATP-binding cassette transporters and their implication in drug disposition: A special look at the heart. Pharmacol. Rev. 2006, 58, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Estudante, M.; Morais, J.G.; Soveral, G.; Benet, L.Z. Intestinal drug transporters: An overview. Adv. Drug Deliv. Rev. 2013, 65, 1340–1356. [Google Scholar] [CrossRef] [PubMed]
- Doring, B.; Petzinger, E. Phase 0 and phase iii transport in various organs: Combined concept of phases in xenobiotic transport and metabolism. Drug Metab. Rev. 2014, 46, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 2008, 38, 802–832. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.S.; Bauer, B.; Hartz, A.M. Modulation of P-glycoprotein at the blood-brain barrier: Opportunities to improve central nervous system pharmacotherapy. Pharmacol. Rev. 2008, 60, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Limtrakul, P.; Chearwae, W.; Shukla, S.; Phisalphong, C.; Ambudkar, S.V. Modulation of function of three abc drug transporters, P-glycoprotein (ABCB1), mitoxantrone resistance protein (ABCG2) and multidrug resistance protein 1 (ABCC1) by tetrahydrocurcumin, a major metabolite of curcumin. Mol. Cell. Biochem. 2007, 296, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Krishna, R.; Mayer, L.D. Multidrug resistance (MDR) in cancer: Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharm. Sci. 2000, 11, 265–283. [Google Scholar] [CrossRef]
- Coley, H.M. Overcoming multidrug resistance in cancer: Clinical studies of P-glycoprotein inhibitors. Methods Mol. Biol. 2010, 596, 341–358. [Google Scholar] [PubMed]
- Choong, E.; Dobrinas, M.; Carrupt, P.A.; Eap, C.B. The permeability P-glycoprotein: A focus on enantioselectivity and brain distribution. Expert Opin. Drug Metab. Toxicol. 2010, 6, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Bansal, T.; Jaggi, M.; Khar, R.K.; Talegaonkar, S. Emerging significance of flavonoids as P-glycoprotein inhibitors in cancer chemotherapy. J. Pharm. Pharm. Sci. 2009, 12, 46–78. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Rodrigues, F.; Sousa, E.; Pinto, M.; Vasconcelos, M.H.; Fernandes, M.X. New uses for old drugs: Pharmacophore-based screening for the discovery of P-glycoprotein inhibitors. Chem. Biol. Drug Des. 2011, 78, 57–72. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, C.A.; Callaghan, R. How can we best use structural information on P-glycoprotein to design inhibitors? Pharm. Ther. 2007, 113, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.B.; Kuo, C.L.; Lien, L.L.; Lien, E.J. Structure-activity relationship: Analyses of P-glycoprotein substrates and inhibitors. J. Clin. Pharm. Ther. 2003, 28, 203–228. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Palmeira, A.; Carmo, H.; Barbosa, D.J.; Gameiro, M.; Gomes, A.; Paiva, A.M.; Sousa, E.; Pinto, M.; Bastos Mde, L.; et al. P-glycoprotein induction in caco-2 cells by newly synthetized thioxanthones prevents paraquat cytotoxicity. Arch. Toxicol. 2015, 89, 1783–1800. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Carmo, H.; Dinis-Oliveira, R.; Cordeiro-da-Silva, A.; Lima, S.C.; Carvalho, F.; Bastos Mde, L.; Remiao, F. In vitro study of P-glycoprotein induction as an antidotal pathway to prevent cytotoxicity in caco-2 cells. Arch. Toxicol. 2011, 85, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Sousa, E.; Carmo, H.; Palmeira, A.; Barbosa, D.J.; Gameiro, M.; Pinto, M.; Bastos Mde, L.; Remiao, F. Induction and activation of P-glycoprotein by dihydroxylated xanthones protect against the cytotoxicity of the P-glycoprotein substrate paraquat. Arch. Toxicol. 2014, 88, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, V.; Silva, R.; Palmeira, A.; Sousa, E.; Ferreira, L.M.; Branco, P.S.; Carvalho, F.; Bastos Mde, L.; Remiao, F. Development of novel rifampicin-derived P-glycoprotein activators/inducers. Synthesis, in silico analysis and application in the rbe4 cell model, using paraquat as substrate. PLoS ONE 2013, 8, e74425. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Sugiyama, Y. Drug transporters: Their role and importance in the selection and development of new drugs. Drug Metab. Pharm. 2002, 17, 93–108. [Google Scholar] [CrossRef]
- Rosi, D.; Peruzzotti, G.; Dennis, E.W.; Berberian, D.A.; Freele, H.; Archer, S. A new active metabolite of “Miracil D”. Nature 1965, 208, 1005–1006. [Google Scholar] [CrossRef] [PubMed]
- Cioli, D.; Pica-Mattoccia, L.; Archer, S. Antischistosomal drugs: Past, present … and future? Pharmacol. Ther. 1995, 68, 35–85. [Google Scholar] [CrossRef]
- Palmeira, A.; Vasconcelos, M.H.; Paiva, A.; Fernandes, M.X.; Pinto, M.; Sousa, E. Dual inhibitors of P-glycoprotein and tumor cell growth: (Re)discovering thioxanthones. Biochem. Pharmacol. 2012, 83, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Bases, R.E.; Mendez, F. Topoisomerase inhibition by lucanthone, an adjuvant in radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 1997, 37, 1133–1137. [Google Scholar] [CrossRef]
- Woo, S.; Kang, D.; Kim, J.; Lee, C.-S.; Lee, E.-S.; Jahng, Y.; Kwon, Y.; Na, Y. Synthesis, cytotoxicity and topoisomerase ii inhibition study of new thioxanthone analogues. Bull. Korean Chem. Soc. 2008, 29, 471–474. [Google Scholar]
- Kostakis, I.K.; Pouli, N.; Marakos, P.; Mikros, E.; Skaltsounis, A.-L.; Leonce, S.; Atassi, G.; Renard, P. Synthesis, cytotoxic activity, nmr study and stereochemical effects of some new pyrano[3,2-b]thioxanthen-6-ones and pyrano[2,3-c]thioxanthen-7-ones. Bioorg. Med. Chem. 2001, 9, 2793–2802. [Google Scholar] [CrossRef]
- Chen, C.-L.; Chen, T.-C.; Lee, C.-C.; Shih, L.-C.; Lin, C.-Y.; Hsieh, Y.-Y.; Ali, A.A.A.; Huang, H.-S. Synthesis and evaluation of new 3-substituted-4-chloro-thioxanthone derivatives as potent anti-breast cancer agents. Arab. J. Chem. 2015. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Fernandes, M.X.; Pinto, M.M.; Vasconcelos, M.H. Multidrug resistance reversal effects of aminated thioxanthones and interaction with cytochrome P450 3A4. J. Pharm. Pharm. Sci. 2012, 15, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, J.P.; Massova, I.; Wiese, T.E.; Besler, B.H.; Corbett, T.H. Comparative molecular field analysis of the antitumor activity of 9h-thioxanthen-9-one derivatives against pancreatic ductal carcinoma 03. J. Med. Chem. 1994, 37, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Corbett, T.H.; Panchapor, C.; Polin, L.; Lowichik, N.; Pugh, S.; White, K.; Kushner, J.; Meyer, J.; Czarnecki, J.; Chinnukroh, S.; et al. Preclinical efficacy of thioxanthone SR271425 against transplanted solid tumors of mouse and human origin. Investig. New Drugs 1999, 17, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Varvaresou, A.; Tsotinis, A.; Papadaki-Valiraki, A.; Siatra-Papastaikoudi, T. New aza-thioxanthones: Synthesis and cytotoxicity. Bioorg. Med. Chem. Lett. 1996, 6, 861–864. [Google Scholar] [CrossRef]
- Neumann, M.G.; Gehlen, M.H.; Encinas, M.V.; Allen, N.S.; Corrales, T.; Peinado, C.; Catalina, F. Photophysics and photoreactivity of substituted thioxanthones. J. Chem. Soc. Faraday Trans. 1997, 93, 1517–1521. [Google Scholar] [CrossRef]
- Paiva, A.M.; Pinto, M.M.; Sousa, E. A century of thioxanthones: Through synthesis and biological applications. Curr. Med. Chem. 2013, 20, 2438–2457. [Google Scholar] [CrossRef] [PubMed]
- Belal, F.; Hefnawy, M.M.; Aly, F.A. Analysis of pharmaceutically-important thioxanthene derivatives. J. Pharm. Biomed. Anal. 1997, 16, 369–376. [Google Scholar] [CrossRef]
- Lory, P.M.J.; Estrella-Jimenez, M.E.; Shashack, M.J.; Lokesh, G.L.; Natarajan, A.; Gilbertson, S.R. Synthesis and screening of 3-substituted thioxanthen-9-one-10,10-dioxides. Bioorg. Med. Chem. Lett. 2007, 17, 5940–5943. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the atp binding cassette (abc) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Pousinho, S.; Fortuna, A.; Falcão, A.; Alves, G. Flavonoid compounds as reversal agents of the P-glycoprotein-mediated multidrug resistance: Biology, chemistry and pharmacology. Phytochem. Rev. 2015, 14, 233–272. [Google Scholar] [CrossRef]
- Silva, R.; Carmo, H.; Vilas-Boas, V.; de Pinho, P.G.; Dinis-Oliveira, R.J.; Carvalho, F.; Silva, I.; Correia-de-Sa, P.; Bastos Mde, L.; Remiao, F. Doxorubicin decreases paraquat accumulation and toxicity in caco-2 cells. Toxicol. Lett. 2013, 217, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yu, L.S.; Zeng, S. Stereoselectivity of chiral drug transport: A focus on enantiomer- transporter interaction. Drug Metab. Rev. 2014, 46, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; He, Y.; Zeng, S. Stereoselective regulation of mdr1 expression in caco-2 cells by cetirizine enantiomers. Chirality 2007, 19, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Pham, Y.-T.; Régina, A.; Farinotti, R.; Couraud, P.-O.; Wainer, I.W.; Roux, F.; Gimenez, F. Interactions of racemic mefloquine and its enantiomers with P-glycoprotein in an immortalised rat brain capillary endothelial cell line, GPNT. Biochim. Biophys. Acta 2000, 1524, 212–219. [Google Scholar] [CrossRef]
- Sousa, M.E.; Tiritan, M.E.; Belaz, K.R.A.; Pedro, M.; Nascimento, M.S.J.; Cass, Q.B.; Pinto, M.M.M. Multimilligram enantioresolution of low-solubility xanthonolignoids on polysaccharide chiral stationary phases using a solid-phase injection system. J. Chromatogr. A 2006, 1120, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Palmeira, A.; Ramos, I.; Carneiro, C.; Afonso, C.; Tiritan, M.; Cidade, H.; Pinto, P.; Saraiva, M.; Reis, S.; et al. Chiral derivatives of xanthones: Investigation of the effect of enantioselectivity on inhibition of cyclooxygenases (COX-1 and COX-2) and binding interaction with human serum albumin. Pharmaceuticals 2017, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Masawang, K.; Tiritan, M.E.; Sousa, E.; Lima, V.; Afonso, C.; Bousbaa, H.; Sudprasert, W.; Pedro, M.; Pinto, M.M. New chiral derivatives of xanthones: Synthesis and investigation of enantioselectivity as inhibitors of growth of human tumor cell lines. Bioorg. Med. Chem. 2014, 22, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Takara, K.; Hayashi, R.; Kokufu, M.; Yamamoto, K.; Kitada, N.; Ohnishi, N.; Yokoyama, T. Effects of nonsteroidal anti-inflammatory drugs on the expression and function of P-glycoprotein/mdr1 in caco-2 cells. Drug Chem. Toxicol. 2009, 32, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Carmo, H.; Vilas-Boas, V.; Barbosa, D.J.; Palmeira, A.; Sousa, E.; Carvalho, F.; Bastos Mde, L.; Remiao, F. Colchicine effect on P-glycoprotein expression and activity: In silico and in vitro studies. Chem. Biol. Interact. 2014, 218, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, V.; Silva, R.; Gaio, A.R.; Martins, A.M.; Lima, S.C.; Cordeiro-da-Silva, A.; de Lourdes Bastos, M.; Remiao, F. P-Glycoprotein activity in human caucasian male lymphocytes does not follow its increased expression during aging. Cytom. Part A J. Int. Soc. Anal. Cytol. 2011, 79, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Taipalensuu, J.; Tornblom, H.; Lindberg, G.; Einarsson, C.; Sjoqvist, F.; Melhus, H.; Garberg, P.; Sjostrom, B.; Lundgren, B.; Artursson, P. Correlation of gene expression of ten drug efflux proteins of the atp-binding cassette transporter family in normal human jejunum and in human intestinal epithelial caco-2 cell monolayers. J. Pharmacol. Exp. Ther. 2001, 299, 164–170. [Google Scholar] [PubMed]
- Chang, K.L.; Pee, H.N.; Yang, S.; Ho, P.C. Influence of drug transporters and stereoselectivity on the brain penetration of pioglitazone as a potential medicine against alzheimer’s disease. Sci. Rep. 2015, 5, 9000. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.-J.; Hua, F.; Zhu, X.-L.; Li, M.; Wang, H.-X.; Yu, X.-M.; Li, Y. Stereoselective regulation of P-gp activity by clausenamide enantiomers in caco-2, kb/kbv and brain microvessel endothelial cells. PLoS ONE 2015, 10, e0135866. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.W. Chiral toxicology: It’s the same thing...only different. Toxicol. Sci. 2009, 110, 4–30. [Google Scholar] [CrossRef] [PubMed]
- Maglich, J.M.; Stoltz, C.M.; Goodwin, B.; Hawkins-Brown, D.; Moore, J.T.; Kliewer, S.A. Nuclear pregnane x receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol. Pharmacol. 2002, 62, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tang, Y.; Guo, C.; Wang, J.; Boral, D.; Nie, D. Nuclear receptors in the multidrug resistance through the regulation of drug-metabolizing enzymes and drug transporters. Biochem. Pharmacol. 2012, 83, 1112–1126. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Huang, H.; Sundarababu, S.; Liu, W.; Kalpana, G.; Smith, A.B.; Horwitz, S.B. Activation of the steroid and xenobiotic receptor (human pregnane x receptor) by nontaxane microtubule-stabilizing agents. Clin. Cancer Res. 2005, 11, 6359–6369. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.N.; Nolan, G.T.; Hood, S.R. Lignans, bacteriocides and organochlorine compounds activate the human pregnane x receptor (PXR). Toxicol. Appl. Pharmacol. 2005, 209, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Synold, T.W.; Dussault, I.; Forman, B.M. The orphan nuclear receptor sxr coordinately regulates drug metabolism and efflux. Nat. Med. 2001, 7, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Murry, D.J.; Kolwankar, D.; Hall, S.D.; Foster, D.R. Vincristine transcriptional regulation of efflux drug transporters in carcinoma cell lines. Biochem. Pharmacol. 2006, 71, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, H.; Suwaki, N.; Tateishi, Y.; Nakatsukasa, H.; Segawa, T.; Hiramatsu, Y. The pregnane X receptor regulates gene expression in a ligand- and promoter-selective fashion. Mol. Endocrinol. 2005, 19, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds ATxs 1–8 are available from the authors. |








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes, A.; Martins, E.; Silva, R.; Pinto, M.M.M.; Remião, F.; Sousa, E.; Fernandes, C. Chiral Thioxanthones as Modulators of P-glycoprotein: Synthesis and Enantioselectivity Studies. Molecules 2018, 23, 626. https://doi.org/10.3390/molecules23030626
Lopes A, Martins E, Silva R, Pinto MMM, Remião F, Sousa E, Fernandes C. Chiral Thioxanthones as Modulators of P-glycoprotein: Synthesis and Enantioselectivity Studies. Molecules. 2018; 23(3):626. https://doi.org/10.3390/molecules23030626
Chicago/Turabian StyleLopes, Ana, Eva Martins, Renata Silva, Madalena M. M. Pinto, Fernando Remião, Emília Sousa, and Carla Fernandes. 2018. "Chiral Thioxanthones as Modulators of P-glycoprotein: Synthesis and Enantioselectivity Studies" Molecules 23, no. 3: 626. https://doi.org/10.3390/molecules23030626
APA StyleLopes, A., Martins, E., Silva, R., Pinto, M. M. M., Remião, F., Sousa, E., & Fernandes, C. (2018). Chiral Thioxanthones as Modulators of P-glycoprotein: Synthesis and Enantioselectivity Studies. Molecules, 23(3), 626. https://doi.org/10.3390/molecules23030626