Comparison of Multiple Bioactive Constituents in Different Parts of Eucommia ulmoides Based on UFLC-QTRAP-MS/MS Combined with PCA

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Plant Materials
2.3. Preparation of Standard Solution
2.4. Preparation of Sample Solutions
2.5. Chromatographic and Mass Spectrometric Conditions
2.6. Validation of The Method
2.6.1. Linearity and Range, LOD, and LOQ
2.6.2. Precision, Repeatability, Solution Stability, and Accuracy
2.7. Principal Component Analysis (PCA)
3. Results and Discussion
3.1. Optimization of Extraction Conditions
3.2. Optimization of UFLC Conditions
3.3. Optimization of MS Conditions
3.4. UFLC Method Validation
3.4.1. Linearity and Range, LOD, and LOQ
3.4.2. Precision, Repeatability, Solution Stability, and Accuracy
3.5. Sample Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ministry of Health of the People's Republic of China. Pharmacopoeia of the People’s Republic of China. Part I; Chemical Industry Press: Beijing, China, 2015; p. 165.
- Ding, Y.X.; Dou, D.Q.; Guo, Y.J.; Li, Q. Simultaneous quantification of eleven bioactive components of male flowers of Eucommia ulmoides oliver by HPLC and their quality evaluation by chemical fingerprint analysis with hierarchical clustering analysis. Pharmacogn. Mag. 2014, 10, 435–440. [Google Scholar] [PubMed]
- Zhang, K.J.; Dong, J.E.; Ma, B.L.; Gao, J.M.; Han, X.W. Study on the distribution differences of the secondary metabolites in Eucommia ulmoides. Sci. Silv. Sin. 2002, 6, 12–16. [Google Scholar]
- Dong, J.E.; Liang, Z.S.; Zhang, K.J.; Sun, S.H. Dynamic changes of secondary metabolites in Eucommia ulmoides male flower. J. Plant Resour. Environ. 2005, 14, 7–10. [Google Scholar]
- He, X.R.; Wang, J.H.; Li, M.X.; Hao, D.J.; Yang, Y.; Zhang, C.L.; He, R.; Tao, R. Eucommia ulmoides Oliv.: Ethnopharmacology, phytochemistry and pharmacology of an important traditional Chinese medicine. J. Ethnopharmacol. 2014, 151, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Zhu, W.X.; Bai, X.T.; Wang, N.; Liu, S.Y. Study advancement about chemical composition and pharmacological effects of Eucommia ulmoides Oliv. Sci. Technol. Food Ind. 2012, 33, 378–382. [Google Scholar]
- Feng, H.; Zhou, H.H.; Ouyang, D.S. Study advancement about chemical composition and pharmacological effects of Eucommia ulmoides Oliv. Chin. J. Clin. Pharmacol. Ther. 2015, 20, 713–720. [Google Scholar]
- Deyama, T.; Nishibe, S.; Nakazawa, Y. Constituents and pharmacological effects of Eucommia and Siberian ginseng. Acta Pharmacol. Sin. 2001, 22, 1057–1070. [Google Scholar] [PubMed]
- Hirata, T.; Kobayashi, T.; Wada, A.; Ueda, T.; Fujikawa, T.; Miyashita, H.; Ikeda, T.; Tsukamoto, S.; Nohara, T. Anti–obesity compounds in green leaves of Eucommia ulmoides. Bioorg. Med. Chem. Lett. 2011, 21, 1786–1791. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.N.; Lee, Y.H.; Park, J.S.; Jun, W.J.; Kim, H.K.; Hong, B.S.; Shin, D.H.; Cho, H.Y. Protective effects of aucubin isolated from Eucommia ulmoides against UVB–induced oxidative stress in human skin fibroblasts. Biol. Pharm. Bull. 2005, 28, 1244–1248. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.P.; Huang, Q.; Zhou, B.T.; Gong, Z.C.; Liu, Z.Q.; Shi, S.Y. Preparative isolation and purification of seven main antioxidants from Eucommia ulmoides Oliv.(Du–zhong) leaves using HSCCC guided by DPPH–HPLC experiment. Food Chem. 2013, 139, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Li, T.L.; Lu, Y.R.; Pan, J.P. Effects of Eucommia ulmoides Oliv. leaves tea on mouse immunity function. Chin. J. Public Health 2007, 23, 1221–1223. [Google Scholar]
- Chen, X.J.; Wang, F.C.; Yuan, Y.; Zhang, L.; Li, X.D.; Jin, S.A.; He, S.M.; Wang, J.Y. Comparative study of bark, leaf and male flower of Eucommia on pharmacodynamics. J. Gansu. Coll. Tradit. Chin. Med. 2016, 33, 5–8. [Google Scholar]
- Wang, J.L.; Liu, E.W.; Wu, S.; Yin, Z.Y.; Zhang, Y. Development of HPLC Method to Evaluate Drug-processing Technique of Eucommiae Cortex. Chin. Herb. Med. 2011, 3, 221–225. [Google Scholar]
- Weng, Z.B.; Yan, C.P.; Wu, Y.; Cai, B.C.; Li, W.D.; Chen, Z.P. Determination of Ten Components in Oliver by High-Performance Liquid Chromatography with Dual Detectors. Anal. Lett. 2014, 47, 1978–1986. [Google Scholar] [CrossRef]
- Fu, X.S.; Han, L.; Liu, X.H.; Xu, H.; Li, J.S.; Cai, B.C.; Yang, Y. Simultaneous Determination of Five Index Components in Eucommiae Cortex by HPCE. Chin. Pharm. J. 2012, 3, 378–382. [Google Scholar]
- Chai, X.; Wang, Y.F.; Su, Y.F.; Bah, A.J.; Hu, L.M.; Gao, Y.; Gao, X.M. A rapid ultra performance liquid chromatography-tandem mass spectrometric method for the qualitative and quantitative analysis of ten compounds in Eucommia ulmodies Oliv. J. Pharm. Biomed. Anal. 2012, 57, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhang, F.S.; Yang, N.Y.; Liu, X.H. Simultaneous Determination of 10 Nucleosides and Nucleobases in Antrodia camphorata Using QTRAP LC–MS/MS. J. Chromatogr. Sci. 2014, 52, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Bai, Y.L.; Shu, S.L.; Qian, D.W.; Ouyang, Z.; Liu, L.; Duan, J.A. Simultaneous quantitation of nucleosides, nucleobases, amino acids, and alkaloids in mulberry leaf by ultra high performance liquid chromatography with triple quadrupole tandem mass spectrometry. J. Sep. Sci. 2014, 37, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Masters, A.R.; McCoy, M.; Jones, D.R.; Desta, Z. Stereoselective method to quantify bupropion and its three major metabolites, hydroxybupropion, erythro-dihydrobupropion, and threo-dihydrobupropion using HPLC–MS/MS. J. Chromatogr. B 2016, 1015–1016, 201–208. [Google Scholar] [CrossRef] [PubMed]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Guidelines, Q2 (R1): Validation of Analytical Procedures (Text and Methodology); ICH: Geneva, Switherland, 1995. [Google Scholar]
- Bo, N.; Broberg, P.; Lindberg, C.; Plymoth, A. Analysis and understanding of high-dimensionality data by means of multivariate data analysis. Chem. Biodivers. 2005, 2, 1487–1494. [Google Scholar]
- Mediani, A.; Abas, F.; Maulidiani, M.; Khatib, A.; Tan, C.P.; Ismail, I.S.; Shaari, K.; Ismail, A. Characterization of Metabolite Profile in Phyllanthus niruri and Correlation with Bioactivity Elucidated by Nuclear Magnetic Resonance Based Metabolomics. Molecules 2017, 22, 902. [Google Scholar] [CrossRef] [PubMed]
- Han, D.Q.; Zhao, J.; Xu, J.; Peng, H.S.; Chen, X.J.; Li, S.P. Quality evaluation of Poly-gonum multiflorum in China based on HPLC analysis of hydrophilic bioactivecompounds and chemometrics. J. Pharm. Biomed. Anal. 2013, 72, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Da, J.; Wu, W.Y.; Hou, J.J.; Long, H.L.; Yao, S.; Yang, Z.; Cai, L.Y.; Yang, M.; Jiang, B.H.; Liu, X.; et al. Comparison of two officinal Chinese phar-macopoeia specie of Ganoderma based on chemical research with multipletechnologies and chemometric analysis. J. Chromatogr. A 1222, 1222, 59–70. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds (catalpol, aucubin, harpagide, geniposidic acid, neochlorogenic acid, protocatechuic acid, syringin, chlorogenic acid, pinoresinol-di-O-β-d-glucopyranoside, geniposide, cryptochlorogenic acid, caffeic acid, rutin, isoquercitrin, genipin, pinoresinol-4′-O-β-d-glucopyranoside, astragalin, isochlorogenic acid A, kaempferol, baicalein, and quercetin) are available from the authors. |
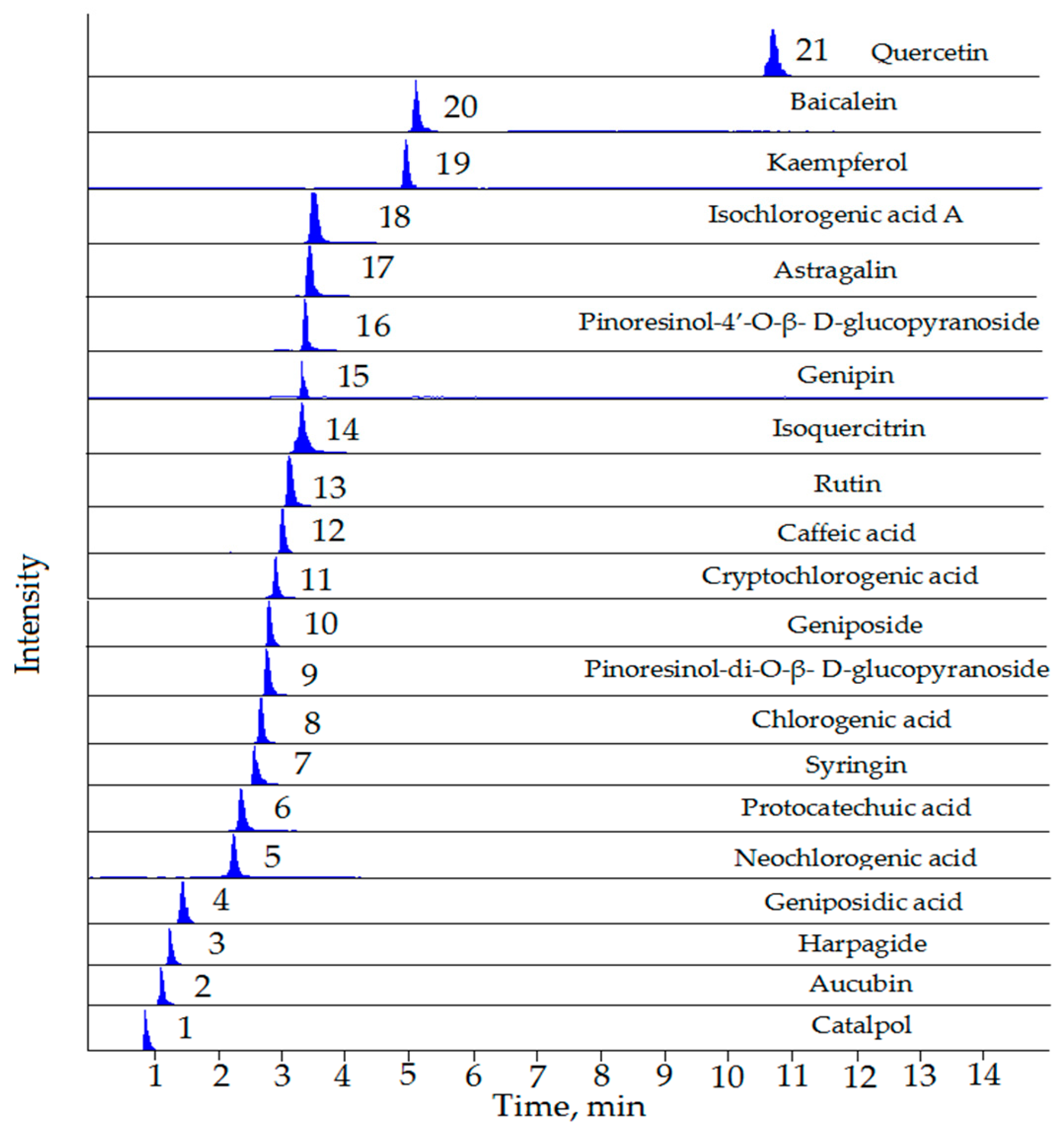


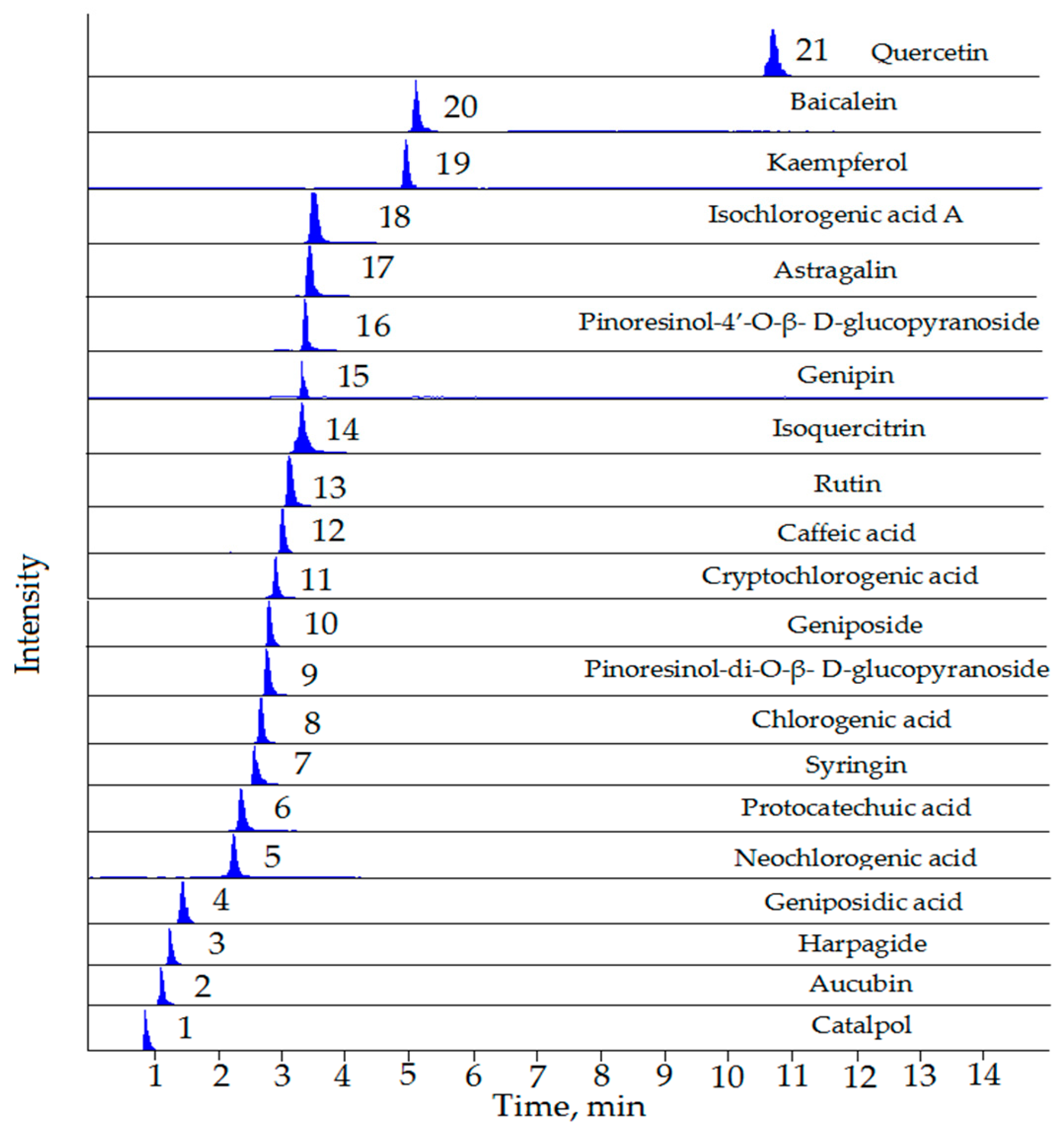{kind=link}
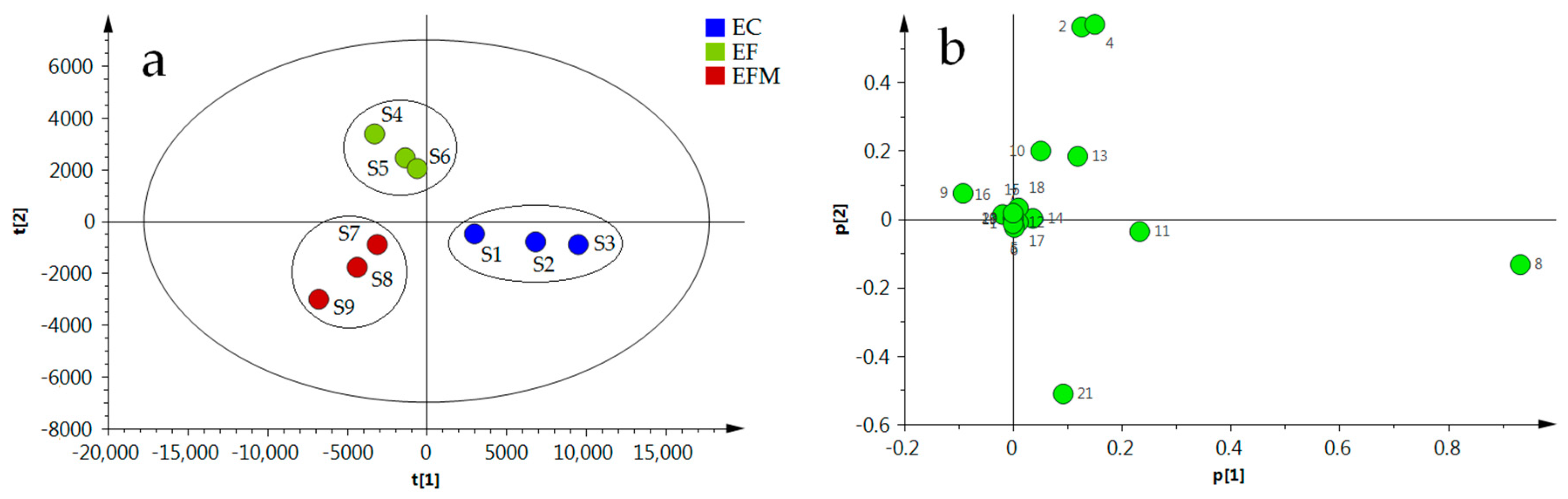{kind=link}
| No. | Compounds | RT (min) | Precursor Ion [M − H]− (m/z) | Product Ion (m/z) | Fragmentor (V) | CE (eV) |
|---|---|---|---|---|---|---|
| 1 | Catalpol | 0.90 | 361.2 | 169.0 | −60 | −18 |
| 2 | Aucubin | 1.18 | 345.1 | 183.0 | −50 | −22 |
| 3 | Harpagide | 1.26 | 363.0 | 201.0 | −135 | −18 |
| 4 | Geniposidic acid | 1.47 | 373.1 | 123.0 | −130 | −26 |
| 5 | Neochlorogenic acid | 2.24 | 353.0 | 191.0 | −95 | −20 |
| 6 | Protocatechuic acid | 2.37 | 152.9 | 109.0 | −85 | −16 |
| 7 * | Syringin | 2.59 | 395.2 | 232.1 | 136 | 31 |
| 8 | Chlorogenic acid | 2.71 | 353.0 | 191.0 | −95 | −20 |
| 9 | Pinoresinol-di-O-β- d-glucopyranoside | 2.79 | 681.2 | 357.3 | −155 | −34 |
| 10 | Geniposide | 2.82 | 387.1 | 225.0 | −180 | −12 |
| 11 | Cryptochlorogenic acid | 2.89 | 353.0 | 191.0 | −95 | −20 |
| 12 | Caffeic acid | 3.05 | 179.0 | 134.6 | −125 | −20 |
| 13 | Rutin | 3.15 | 609.1 | 300.0 | −245 | −46 |
| 14 | Isoquercitrin | 3.31 | 463.0 | 299.9 | −180 | −36 |
| 15 | Genipin | 3.33 | 225.0 | 101.1 | −30 | −16 |
| 16 | Pinoresinol-4’-O-β- d-glucopyranoside | 3.36 | 519.2 | 357.1 | −160 | −22 |
| 17 | Astragalin | 3.45 | 447.1 | 283.9 | −100 | −36 |
| 18 | Isochlorogenic acid A | 3.53 | 515.1 | 191.0 | −145 | −38 |
| 19 | Kaempferol | 4.98 | 285.0 | 116.9 | −180 | −48 |
| 20 | Baicalein | 5.15 | 269.0 | 139.0 | −130 | −40 |
| 21 | Quercetin | 10.78 | 301.1 | 255.1 | −45 | −10 |
| No. | Compounds | Regression Equation | r2 | Liner Range (μg/mL) | LOD (ng/mL) | LOQ (ng/mL) | Precision RSD (%) | Repeatability RSD (%) (n = 6) | Stability RSD (%) | Recovery (%) (n = 3) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intar-Day (n = 6) | Inter-Day (n = 6) | Low | Medium | High | ||||||||||||
| Mean | RSD | Mean | RSD | Mean | RSD | |||||||||||
| 1 | Catalpol | Y = 20.5X + 888 | 0.9997 | 0.015–7.600 | 1.52 | 5.32 | 2.22 | 3.70 | 3.68 | 1.44 | 98.67 | 1.62 | 98.34 | 2.74 | 99.26 | 3.35 |
| 2 | Aucubin | Y = 9.05X + 13,800 | 0.9997 | 0.130–195.000 | 3.90 | 13.65 | 3.45 | 2.10 | 2.87 | 1.16 | 99.00 | 2.27 | 99.16 | 1.78 | 98.77 | 2.10 |
| 3 | Harpagide | Y = 211X + 2990 | 0.9999 | 0.017–8.320 | 1.44 | 3.98 | 1.41 | 2.93 | 3.65 | 3.73 | 97.62 | 3.14 | 97.47 | 3.98 | 97.56 | 3.68 |
| 4 | Geniposidic acid | Y = 1060X + 48,400 | 0.9999 | 0.025–127.200 | 1.27 | 4.45 | 2.15 | 2.62 | 2.09 | 3.85 | 99.91 | 3.08 | 98.43 | 1.76 | 97.56 | 2.55 |
| 5 | Neochlorogenic acid | Y = 2130X + 26,000 | 0.9999 | 0.020–9.800 | 1.96 | 6.86 | 1.45 | 2.40 | 2.05 | 1.34 | 99.77 | 3.67 | 96.45 | 3.81 | 98.01 | 2.15 |
| 6 | Protocatechuic acid | Y = 3210X + 171,000 | 0.9996 | 0.013–12.520 | 1.25 | 4.38 | 2.08 | 3.39 | 3.23 | 1.86 | 95.83 | 3.09 | 99.05 | 3.40 | 97.01 | 3.55 |
| 7 | Syringin | Y = 115X + 7860 | 0.9997 | 0.019–19.440 | 1.94 | 6.80 | 2.96 | 2.23 | 3.56 | 1.24 | 98.38 | 2.29 | 100.16 | 2.82 | 98.81 | 2.82 |
| 8 | Chlorogenic acid | Y = 170X + 176,000 | 0.9996 | 0.238–476.000 | 4.76 | 16.66 | 1.88 | 2.61 | 1.57 | 2.76 | 98.20 | 2.64 | 98.24 | 2.19 | 97.59 | 2.85 |
| 9 | Pinoresinol-di-O-β- d-glucopyranoside | Y = 423X + 15,200 | 0.9998 | 0.150–75.000 | 3.75 | 13.13 | 1.09 | 3.07 | 2.83 | 3.20 | 98.29 | 1.20 | 98.01 | 2.92 | 96.72 | 3.88 |
| 10 | Geniposide | Y = 48.3X + 36,700 | 0.9991 | 0.080–40.200 | 1.47 | 4.94 | 1.20 | 2.16 | 1.45 | 2.64 | 98.20 | 3.85 | 96.45 | 1.17 | 96.99 | 3.52 |
| 11 | Cryptochlorogenic acid | Y = 350.5X + 506,010 | 0.9996 | 0.119–118.800 | 2.97 | 10.40 | 2.04 | 3.57 | 1.94 | 1.63 | 96.83 | 2.08 | 96.13 | 3.31 | 98.34 | 3.19 |
| 12 | Caffeic acid | Y = 2540X + 114,000 | 0.9998 | 0.024–11.920 | 1.19 | 4.17 | 1.87 | 2.91 | 2.74 | 3.84 | 98.38 | 3.68 | 96.95 | 3.37 | 100.43 | 3.07 |
| 13 | Rutin | Y = 652X + 238,000 | 0.9999 | 0.135–67.700 | 0.60 | 1.99 | 1.39 | 3.58 | 1.54 | 3.57 | 98.15 | 3.82 | 97.28 | 3.45 | 95.48 | 2.96 |
| 14 | Isoquercitrin | Y = 2640X + 216,000 | 0.9995 | 0.117–58.400 | 0.89 | 3.02 | 2.15 | 3.29 | 2.09 | 2.54 | 98.16 | 3.55 | 96.59 | 2.08 | 102.03 | 3.16 |
| 15 | Genipin | Y = 953X + 68,600 | 0.9991 | 0.034–33.500 | 0.67 | 2.35 | 3.26 | 3.14 | 2.68 | 3.30 | 98.41 | 2.29 | 96.34 | 3.44 | 98.09 | 3.55 |
| 16 | Pinoresinol-4’-O-β- d-glucopyranoside | Y = 0.89X +116 | 0.9991 | 0.022–11.180 | 1.12 | 3.91 | 2.70 | 2.40 | 3.60 | 2.99 | 102.35 | 3.29 | 97.18 | 2.95 | 97.43 | 2.13 |
| 17 | Astragalin | Y = 3050X + 130,000 | 0.9999 | 0.027–13.440 | 0.98 | 2.96 | 2.07 | 3.04 | 2.55 | 2.80 | 99.89 | 3.87 | 98.52 | 1.99 | 96.71 | 2.18 |
| 18 | Isochlorogenic acid A | Y = 1300X + 149,000 | 1.0000 | 0.090–44.800 | 2.24 | 7.84 | 2.99 | 3.07 | 3.31 | 3.59 | 97.34 | 3.49 | 97.69 | 2.58 | 97.07 | 2.98 |
| 19 | Kaempferol | Y = 429X + 1010 | 1.0000 | 0.026–13.000 | 2.36 | 5.90 | 1.87 | 2.16 | 3.77 | 3.36 | 96.13 | 3.15 | 96.85 | 2.93 | 97.92 | 2.04 |
| 20 | Baicalein | Y = 531X − 1080 | 0.9991 | 0.0015–7.740 | 0.32 | 1.15 | 2.68 | 3.08 | 3.04 | 3.57 | 97.62 | 3.52 | 103.30 | 2.38 | 98.64 | 3.38 |
| 21 | Quercetin | Y = 2.46X + 123,000 | 0.9991 | 0.126–62.950 | 1.45 | 4.69 | 2.14 | 3.76 | 1.02 | 3.38 | 95.91 | 3.41 | 98.65 | 3.73 | 97.45 | 2.03 |
| No. | Compounds | EC | EF | EFM | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | S9 | ||
| 1 | Catalpol | 26.33 ± 2.77 | 14.45 ± 1.82 | 18.80 ± 1.23 | 54.78 ± 3.60 | 91.33 ± 4.54 | 135.17 ± 4.80 | 18.80 ± 1.47 | 15.89 ± 0.60 | 21.07 ± 0.91 |
| 2 | Aucubin | 351.33 ± 34.59 | 557.00 ± 12.53 | 895.00 ± 13.23 | 27.70 ± 3.34 | 45.02 ± 3.11 | 17.71 ± 2.26 | 84.00 ± 7.21 | 2378.33 ± 12.58 | 4663.33 ± 132.04 |
| 3 | Harpagide | 2.43 ± 0.57 | 1.23 ± 0.05 | 4.15 ± 0.28 | 1.65 ± 0.04 | 2.06 ± 0.06 | 1.14 ± 0.10 | 2.66 ± 0.32 | 1.30 ± 0.05 | - |
| 4 | Geniposidic acid | 1630.00 ± 5.00 | 1040.00 ± 36.06 | 393.67 ± 7.51 | 129.17 ± 2.52 | 32.22 ± 2.06 | 67.11 ± 4.43 | 3285.00 ± 97.60 | 3541.67 ± 68.25 | 3628.33 ± 46.46 |
| 5 | Neochlorogenic acid | 31.52 ± 0.87 | 22.18 ± 0.28 | 11.20 ± 0.30 | 139.40 ± 0.85 | 83.80 ± 1.75 | 67.33 ± 5.69 | 28.94 ± 0.45 | 28.98 ± 0.81 | 30.21 ± 1.03 |
| 6 | Protocatechuic acid | 13.87 ± 0.39 | 4.85 ± 0.37 | 8.32 ± 0.69 | 43.25 ± 1.38 | 262.80 ± 8.78 | 153.17 ± 10.60 | 37.27 ± 1.81 | 31.78 ± 0.78 | 26.10 ± 0.48 |
| 7 | Syringin | 1.33 ± 0.20 | 5.40 ± 0.22 | 7.98 ± 0.50 | 37.37 ± 2.13 | 1.25 ± 0.26 | 6.29 ± 0.50 | 30.63 ± 0.74 | 25.12 ± 0.83 | 27.73 ± 1.52 |
| 8 | Chlorogenic acid | 8030.00 ± 62.45 | 4276.67 ± 102.14 | 1641.33 ± 116.57 | 14,086.67 ± 160.42 | 12,446.67 ± 291.43 | 11,593.33 ± 30.55 | 13,410.00 ± 284.78 | 14,543.33 ± 695.15 | 16,710.00 ± 608.52 |
| 9 | Pinoresinol-di-O-β-d-glucopyranoside | 888.33 ± 32.15 | 1295.00 ± 30.00 | 1125.00 ± 52.20 | 9.07 ± 0.55 | 15.91 ± 0.70 | 21.72 ± 1.01 | 16.88 ± 0.75 | 26.92 ± 2.54 | 74.17 ± 3.88 |
| 10 | Geniposide | 13.68 ± 0.98 | 231.33 ± 4.25 | 148.13 ± 6.79 | - | - | - | 111.83 ± 0.76 | 578.33 ± 20.82 | 1803.33 ± 35.12 |
| 11 | Cryptochlorogenic acid | 2013.33 ± 20.82 | 1073.33 ± 30.55 | 406.00 ± 27.22 | 3553.33 ± 70.24 | 3100.00 ± 70.00 | 2963.33 ± 118.46 | 3343.33 ± 75.72 | 3550.00 ± 90.00 | 4230.00 ± 115.33 |
| 12 | Caffeic acid | 31.32 ± 0.31 | 20.17 ± 0.81 | 14.79 ± 0.56 | 29.05 ± 1.13 | 182.50 ± 4.58 | 38.03 ± 1.95 | 51.28 ± 1.63 | 37.52 ± 1.93 | 20.62 ± 0.81 |
| 13 | Rutin | - | - | - | 541.00 ± 28.16 | 88.67 ± 2.52 | 343.00 ± 26.51 | 2561.67 ± 25.17 | 1509.33 ± 80.60 | 1685.00 ± 10.00 |
| 14 | Isoquercitrin | - | - | - | 475.00 ± 7.55 | 108.33 ± 1.53 | 369.67 ± 20.51 | 395.33 ± 11.56 | 441.00 ± 22.11 | 507.00 ± 14.73 |
| 15 | Genipin | 10.18 ± 0.83 | 92.83 ± 0.29 | 104.33 ± 6.03 | 11.50 ± 0.92 | 1.85 ± 0.18 | 18.22 ± 1.18 | 407.50 ± 15.55 | 36.38 ± 3.59 | 71.28 ± 4.08 |
| 16 | Pinoresinol-4′-O-β-d-glucopyranoside | 138.33 ± 1.26 | 202.00 ± 2.65 | 306.50 ± 17.26 | 7.30 ± 0.23 | 2.28 ± 0.01 | 6.19 ± 0.05 | 12.49 ± 0.94 | 11.22 ± 0.23 | 13.33 ± 0.45 |
| 17 | Astragalin | - | - | - | 165.50 ± 4.27 | 47.65 ± 0.23 | 160.73 ± 13.51 | 106.53 ± 4.00 | 116.33 ± 5.51 | 118.50 ± 9.00 |
| 18 | Isochlorogenic acid A | 100.17 ± 5.13 | 62.40 ± 0.85 | 47.02 ± 3.06 | 117.33 ± 2.52 | 11.07 ± 0.21 | 32.90 ± 0.90 | 95.17 ± 4.86 | 83.83 ± 1.04 | 403.40 ± 9.56 |
| 19 | Kaempferol | - | - | - | 22.13 ± 0.57 | 31.08 ± 0.58 | 20.07 ± 1.31 | 19.32 ± 1.09 | 5.99 ± 0.29 | 6.06 ± 0.23 |
| 20 | Baicalein | 0.21 ± 0.04 | 0.77 ± 0.09 | 0.43 ± 0.04 | 0.16 ± 0.01 | 0.15 ± 0.00 | 0.14 ± 0.01 | 0.14 ± 0.02 | 0.12 ± 0.00 | 0.14 ± 0.02 |
| 21 | Quercetin | - | - | - | 3740.67 ± 168.84 | 2085.00 ± 5.00 | 1651.67 ± 106.93 | 708.33 ± 5.77 | 637.83 ± 15.61 | 160.50 ± 5.29 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Y.; Zhao, H.; Chen, C.; Zou, L.; Liu, X.; Chai, C.; Wang, C.; Shi, J.; Chen, S. Comparison of Multiple Bioactive Constituents in Different Parts of Eucommia ulmoides Based on UFLC-QTRAP-MS/MS Combined with PCA. Molecules 2018, 23, 643. https://doi.org/10.3390/molecules23030643
Yan Y, Zhao H, Chen C, Zou L, Liu X, Chai C, Wang C, Shi J, Chen S. Comparison of Multiple Bioactive Constituents in Different Parts of Eucommia ulmoides Based on UFLC-QTRAP-MS/MS Combined with PCA. Molecules. 2018; 23(3):643. https://doi.org/10.3390/molecules23030643
Chicago/Turabian StyleYan, Ying, Hui Zhao, Cuihua Chen, Lisi Zou, Xunhong Liu, Chuan Chai, Chengcheng Wang, Jingjing Shi, and Shuyu Chen. 2018. "Comparison of Multiple Bioactive Constituents in Different Parts of Eucommia ulmoides Based on UFLC-QTRAP-MS/MS Combined with PCA" Molecules 23, no. 3: 643. https://doi.org/10.3390/molecules23030643