
Synaptic Alterations in Mouse Models for Alzheimer Disease—A Special Focus on N-Truncated Abeta 4-42

1
Division of Molecular Psychiatry, Department of Psychiatry and Psychotherapy, University Medical Center (UMG), Georg-August-University, von-Siebold-Strasse 5, 37075 Göttingen, Germany
2
Center for Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB), Humboldtallee 23, 37073 Göttingen, Germany
3
Center for Physiology and Pathophysiology, Institute for Neuro- and Sense Physiology, University Medical Center (UMG), Georg-August-University, Humboldtallee 23, 37073 Göttingen, Germany
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(4), 718; https://doi.org/10.3390/molecules23040718
Submission received: 21 February 2018
/
Revised: 16 March 2018
/
Accepted: 19 March 2018
/
Published: 21 March 2018
(This article belongs to the Special Issue 25th Anniversary of the Amyloid Hypothesis and Alzheimer Disease)
Abstract
:This commentary reviews the role of the Alzheimer amyloid peptide Aβ on basal synaptic transmission, synaptic short-term plasticity, as well as short- and long-term potentiation in transgenic mice, with a special focus on N-terminal truncated Aβ4-42. Aβ4-42 is highly abundant in the brain of Alzheimer’s disease (AD) patients. It demonstrates increased neurotoxicity compared to full length Aβ, suggesting an important role in the pathogenesis of AD. Transgenic Tg4-42 mice, a model for sporadic AD, express human Aβ4-42 in Cornu Ammonis (CA1) neurons, and develop age-dependent hippocampal neuron loss and neurological deficits. In contrast to other transgenic AD mouse models, the Tg4-42 model exhibits synaptic hyperexcitability, altered synaptic short-term plasticity with no alterations in short- and long-term potentiation. The outcomes of this study are discussed in comparison with controversial results from other AD mouse models.
1. N-Terminally Truncated Amyloid-β Variants in Alzheimer’s Disease
It is generally well accepted that Alzheimer’s disease (AD) is neuropathologically characterized by extracellular beta-amyloid plaques (Aβ) and neurofibrillary tangles. These pathologies, although typical and important for neuropathological diagnosis of AD, do not convincingly explain synaptic deficits and neuron loss, which are the basis for clinical AD [1]. The amyloid hypothesis was originally based on the discovery that inherited forms of AD can be induced by an enhanced production of full length Aβ peptides [2]. Aβ is released by proteolytic processing of the amyloid precursor protein (APP) [3]. Of interest for the current review is that N-truncated Aβ peptides are major constituents of AD plaques. It was discovered already in 1985 that Aβ (Phe-4; Aβ4–x), beginning with phenylalanine at position 4, is a main component of amyloid plaques [4]. Other studies supported the initial findings, and added pyroglutamate Aβ as an additional N-truncated amyloid species [5,6,7,8,9,10], which was previously reviewed in detail [11].
Our group has recently developed a transgenic mouse model for sporadic AD [12]. The Tg4-42 mice express human Aβ4-42 and develop an age-dependent massive CA1 pyramidal neuron loss in the hippocampus. The hippocampus-specific expression of Aβ4-42 correlated well with spatial reference memory deficits assessed by the Morris water maze test [12,13]. These findings indicate that N-truncated Aβ4-42 triggers behavioral deficits comparable to AD-typical memory dysfunction, even without plaque formation and appearance of neurofibrillary tangles.
In order to demonstrate that the Tg4-42 mouse model is a unique AD mouse model, we compared the Aβ pathology to 5XFAD mice [14], a widely used mouse model with typical amyloid plaques. Both models were analyzed with a pan-Aβ antibody in order to visualize intraneuronal Aβ and plaque deposits in CA1 neurons of the hippocampus of 3-month-old transgenic mice. Immunostaining demonstrates strong intraneuronal Aβ accumulation only in the Tg4-42 model (Figure 1A–C), but not in 5XFAD (Figure 1D–F). As expected, 5XFAD mice showed significant extracellular plaque deposition throughout the hippocampus and cortex [14].
2. Synaptic Alterations in the Tg4-42 Mouse Model
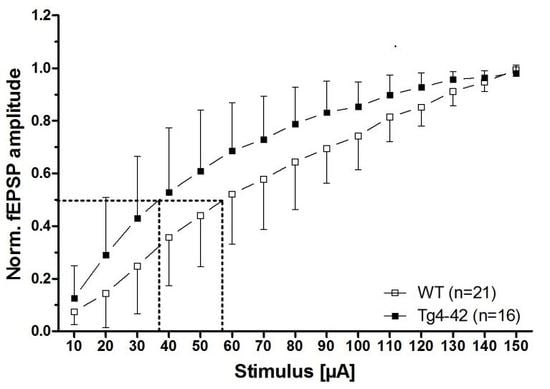
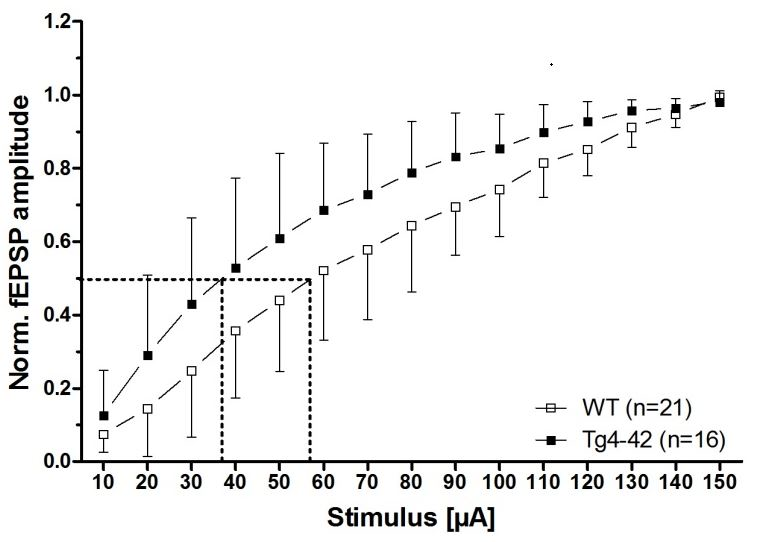
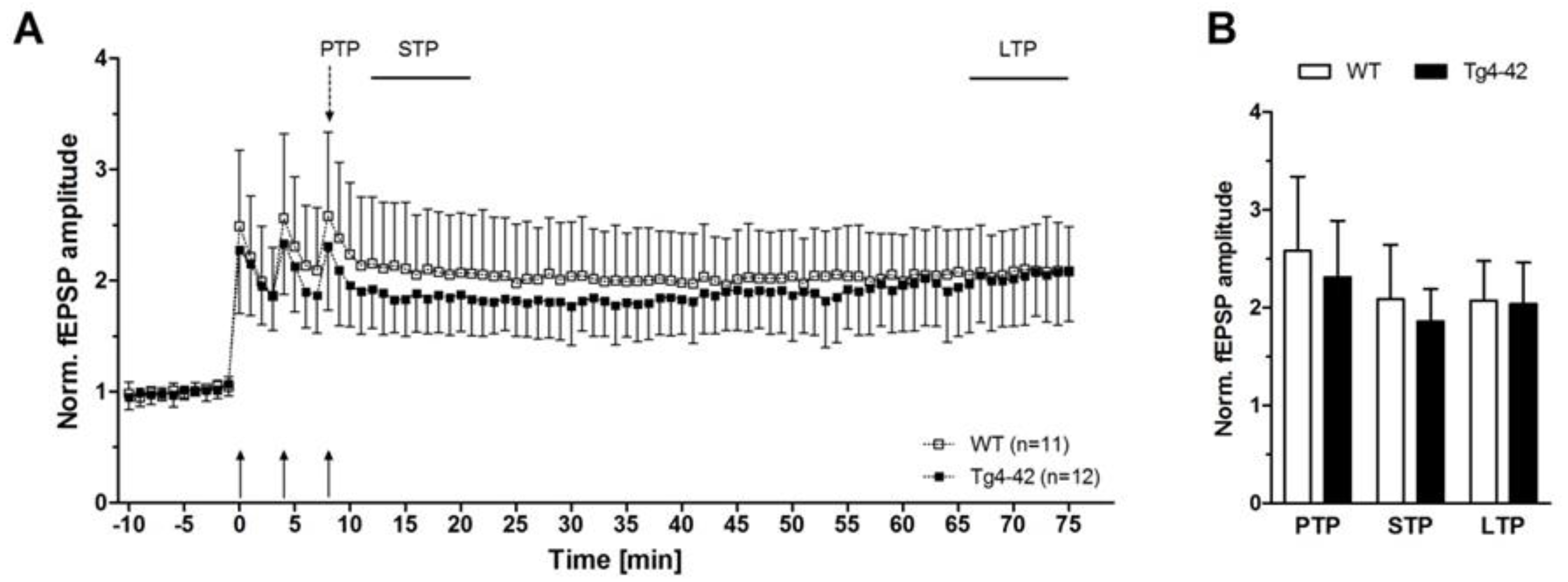
The decline in synaptic function is an early event in AD pathology. It is mainly related to pathological alterations in the hippocampal formation of AD patients, and correlates well with the clinical symptoms and cognitive dysfunction [15]. Interestingly, expression of Aβ4-42 in the CA1 area of the hippocampus in Tg4-42 mice induced certain aspects in synaptic dysfunction and plasticity at a time point prior to neuron death in this model [16]. The main outcomes of this study are summarized as follows. In order to study the possible chronic neurotoxic effects of N-truncated Aβ4-42 on synaptic function and plasticity orthodromically evoked field potentials were recorded in hippocampal slices. Field excitatory postsynaptic potentials were evoked at the CA3/CA1 region and orthodromic responses were recorded in the stratum radiatum of the CA1 region [16]. Details on the recording conditions were previously published [16]. In short, two slices from each brain of male hemizygous Tg4-42 and wildtype littermate controls (3 months of age) were used (6–8 animals per group). Field excitatory postsynaptic potential (fEPSPs) were evoked by 0.1 ms unipolar stimuli using a steel wire microelectrode. Responses were recorded using glass electrodes [17]. Sampling rate was 20 kHz. The acute hippocampal tissue slices were subjected to three different test paradigms, i.e., input–output curves, paired-pulse facilitation (PPF), as well as recording for short-term (PTP, STP) and long-term potentiation (LTP). Input–output curves were recorded for stimulation intensities of 10–150 µA. fEPSP amplitudes were normalized to their absolute minimum. Four consecutive stimulus trains were pooled and averaged for each stimulus intensity. Remarkably, a left shift of the input–output curve was observed. This is an indication for altered basal excitatory synaptic transmission (Figure 2A). The increased neuronal excitability corroborated this finding with the half-maximal stimulus intensity in 3-month-old Tg4-42 mice (Figure 2B). It has been shown that PPF is a paradigm for synaptic short-term plasticity [18] with a mostly presynaptic origin [19]. Using the half maximal stimulus intensity obtained from input–output recordings, this twin-pulse stimulation was measured at eight different interstimulus intervals (25–200 ms), and calculated as the ratio of the second fEPSP to the first fEPSP amplitude. Recordings revealed characteristic PPF amplitudes declining fast with increasing interstimulus interval duration (Figure 2C). Noticeably, Tg4-42 mice showed lower output intensities—a sign for a decline in short-term plasticity (Figure 2C). Furthermore, the effect of N-truncated Aβ4-42 on post-tetanic potentiation (PTP), short-term potentiation (STP), as well as long-term potentiation (LTP) at the Schaffer collateral CA1 pathway was examined. Post-tetanic potentiation is mostly considered to be of presynaptic origin [20], and lasts between 30 s and several minutes. When applying brief high-frequency stimuli trains to the Schaffer collaterals, three different phases can be distinguished: PTP, STP, and LTP. Presynaptic accumulation of Ca2+ causes PTP that readily decreases after Ca2+ clearance. In this phase, PTP is N-methyl-d-aspartate (NMDA) receptor-independent. By contrast, the following two phases (STP, LTP) are of postsynaptic origin, and NMDA receptor-dependent forms of potentiation [21]. Baseline fEPSPs were determined using the half-maximal stimulus intensity and a low stimulation frequency (measured every 15 s; 4× averaged for 1 min) and recorded for 10 min. Different forms of synaptic potentiation were induced by applying three tetanic stimuli, and trains of 100 Hz for 1 s every 5 min. After the third tetanic stimulus, recordings were continued for additional 65 min. Absolute fEPSP amplitudes were normalized to the average of pre-tetanus baseline fEPSP amplitudes. Post-tetanic potentiation was defined as the maximal response within 1 min after the third tetanic stimulus. STP and LTP were defined as the period between 12th and 21st min, and 65th and 75th min after induction, respectively. Induction of synaptic potentiation induced PTP of fEPSP amplitudes with no significant difference between wildtype and Tg4-42 mice (Figure 3A,B). The same was true for STP, which remained stable (Figure 3A,B). Tg4-42 mice showed stable LTP even after 65 min after the high-frequency stimulation (Figure 3A,B). The extent of LTP in Tg4-42 mice was not different compared to wildtype mice.
Therefore, one can conclude that the expression of N-truncated Aβ4-42 in the hippocampus of Tg4-42 mice leads to neuronal hyperexcitability, and affects synaptic short-term plasticity, while no significant changes in STP or LTP were observed [16]. This is partially in contrast to previous studies in other AD mouse models. As it is now well established that Aβ4-42 oligomers are highly soluble in comparison to full length Aβ1–42, we believe that the controversial lack of changes in STP and LTP are due to the different biophysical characteristics of both peptides.
Additionally, distinct Aβ levels might determine synaptic activity in young Tg4-42 mice, as described above. The expression levels of amyloid peptides may influence synaptic activity at the presynaptic site [22]. It is likely that enhanced synaptic excitability could be triggered by the oligomerization state of Aβ. A change in basal synaptic function was recently detected in a mouse model that harbors two FAD-linked mutations. Megill and colleagues found an increase in fEPSP slope and fiber volley amplitude in 2-month-old transgenic mice [23]. Another transgenic mouse model overexpressing mutated APP and mutated Presenilin-1 in neurons also showed hippocampal hyperactivity, as seen in the Tg4-42 model [24]. Previously, Kamenetz et al. observed that activity-dependent Aβ secretion induces a negative feedback loop, thereby influencing neuronal hyperactivity [25]. In good agreement with the outcomes of the Tg4-42 study [16], treating hippocampal CA1 neurons of wildtype mice with nanomolar concentrations of Aβ dimers induced hyperactivity as well [24]. The authors hypothesized that Aβ dimers may induce inward currents, leading to increased firing rate of action potentials and an increase in intracellular Ca2+ concentrations [24]. Hippocampal synaptic hyperactivity influences compensatory mechanisms that may be part of network dysfunctions in the hippocampus [26]. These in vitro studies are supported by a report demonstrating that patients with mild cognitive impairment exhibited hyperactivity in the hippocampus/parahippocampal region [27].
3. Neurophysiological Alterations in Mouse Models for AD
Numerous other groups found neurophysiological alterations in various mouse models for AD. Progressively more and more studies tried to analyze how amyloid Aβ can affect neuronal and synaptic functioning. For example, several signaling pathways are impaired after receptor binding of Aβ peptides [28]. In the course of this, cellular dysfunction or cell death has been associated with binding of Aβ oligomers to the Frizzled receptor and the low-affinity nerve growth factor. Alternatively, Aβ might be involved in the loss of insulin receptors, bind to prion protein, or interact with cell surface APP, impair kinase activity, impair Ca2+ currents at glutamatergic and GABAergic synapses, or directly form pores for Ca2+ in the synaptic membrane [28]. Aβ also might affect NMDA receptor functioning like Ca2+ homeostasis, oxidative stress, and synapse loss [29,30,31,32], and affect mGluR5 receptor clustering, diffusion properties of mGluR5, and elevated intracellular Ca2+ [33]. Aβ might interact with α7 nicotinic acetylcholine receptors, receptor for advanced glycation endproducts, and Ephrin type-B receptor 2 [22]. How Aβ might impair synaptic plasticity is still a matter of scientific debate [34,35].
An overview of neurophysiological alterations in the hippocampus of different mouse models is presented in Table 1, including Tg4-42 and 5XFAD as examples for models with abundant intraneuronal Aβ and/or plaques, respectively.
As the focus of the current review is on similar experimental settings in the hippocampal CA1 region, we did not include other studies on altered synapse functions in other AD mouse models, e.g., PS2APP [66], SAMP8 [67], and PLB1Triple [68].
There are obvious discrepancies in the outcomes of the reports. The comparability of studies using mouse models is hampered by several factors: different transgenic expression vectors and promotors, expression levels of transgenes, genetic background of the strains, gender and age of the mice. Many of the AD mouse models develop amyloid plaques, but no or minor neuron loss. The presence of low Aβ levels facilitates the generation of LTP [28,69]. As discussed in detail by K. Dietrich [16], the variations in different methodologies will impact the results of such assays: interface versus submission style recording chambers, differences in the stimulus protocols, variations in the definition of parameters to be analyzed, like the type of normalization of fEPSP, conditions of in vitro preparations, etc.
Most of the listed studies reported a decline of synaptic function, in contrast to the observations in the Tg4-42 mouse model, with a significant increase in synaptic excitability and no impairment in LTP. As mentioned already, the design of the experiments may influence their outcome, which may account for diverging results of studies with the same AD models (Table 1). Reduced synaptic function was often observed in young mice prior to plaque development.
Soluble (and/or intraneuronal) Aβ is a critical key player in AD-related synaptic deficits [70]: The APP E693∆ transgenic model shows oligomerized, accumulated, and intraneuronal Aβ in an age-dependent manner. Aβ plaques develop late, at the age of 24 months. A significant reduction of PPF and LTP was reported in the granular cells of the dentate gyrus, with no effect on basal synaptic transmission. This important report clearly demonstrated that Aβ oligomers trigger synaptic deficits prior to plaque formation [70]. This correlation was further substantiated by studies using other transgenic mouse models, e.g., APPSLPS1 KI [1], 3xTg-AD [64], acrAβ [71], and PD-APP [43]. Terry et al. have reported that only weak correlations exist between psychological values and plaques and tangles, but the density of synaptic markers correlated well in the neocortex of patients with Alzheimer’s disease [72]. The expression levels of synaptic proteins correlated well in AD cases clinically classified by the Clinical Dementia Rating score with more severe cases having a progressive decline [73]. Other studies corroborated these findings, and discuss that intracellular accumulation of amyloid-β is a predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease (reviewed by [1,74,75]). The current study is in good agreement with these observations.
In addition to the amyloid hypothesis-driven research, there is an increasing interest in gene–environmental interactions. For example, exposure to early life stress has recently been shown to influence synaptic plasticity after induction of epileptic activity [76], and more specifically, early life stress may alter amyloid-β processing and cognition in transgenic Alzheimer mice [77].
In summary, the Tg4-42 mice develop early synaptic deficits and neuron loss in the hippocampus, which correlates well with learning and memory dysfunction [12,78]. This is likely due to soluble oligomers of Aβ4-42. Of interest, these oligomers are derived from wildtype Aβ sequence and are not mutated as in other studies (cf. [70]). Finally, besides pyroglutamate Aβ3–42, Aβ1–42, and Aβ1–40, Aβ4-42 is a major species in the brain of AD patients, and is therefore an important player in the etiology of AD (reviewed in [11]).
Acknowledgments
We acknowledge support by the Open Access Publication Funds of the Göttingen University and the CNMPB. We are deeply grateful to Oliwia A. Janc and Belinda Kempkes for extraordinary technical help when performing the experiments.
Author Contributions
K.D. performed experiments, analyzed data and wrote the manuscript; Y.B. performed experiments; M.M. provided the electrophysiological setup, supervised the work and proofread the manuscript; T.A.B. contributed in writing the manuscript and supervised the work.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in the manuscript:
| AD | Alzheimer’s disease |
| Aβ | Abeta |
| APP | β-amyloid precursor protein |
| MALDI-TOF | Matrix-assisted-laser-desorption ionization time-of-flight |
| pyroGlu-3, AβpE3-x | pyroglutamate Abeta starting with position 3 |
| Asp-1 | Abeta starting with aspartate at position 1 |
| Ala-2 | Abeta starting with alanine at position 2 |
| Phe-4 | Abeta starting with phenylalanine at position 4 |
| Arg-5 | Abeta starting with arginine at position 5 |
| fEPSP | field excitatory postsynaptic potential |
| PPF | paired-pulse facilitation |
| Ca2+ | calcium 2+ |
| PTP | post-tetanic potentiation |
| STP | short-term potentiation |
| LTP | long-term potentiation |
| NMDA | N-methyl-D-aspartate |
| RM-ANOVA | repeated measures ANOVA |
| WT | wildtype |
| IO curve | input–output curve |
| PS1 | presenilin-1 |
| Swe | Swedish |
| Flo | Florida |
| Lon | London |
| Ind | Indiana |
| m | age in months |
| n.a. | not analyzed |
| ↓ | decreased |
| ↑ | increased |
| sec | seconds |
| min | minutes |
References
- Bayer, T.A.; Wirths, O. Intracellular accumulation of amyloid-Beta—A predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease. Front. Aging Neurosci. 2010, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 1998, 8, 447–453. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Papayannopoulos, I.A.; Styles, J.; Bobin, S.A.; Lin, Y.Y.; Biemann, K.; Iqbal, K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch. Biochem. Biophys. 1993, 301, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Saido, T.C.; Iwatsubo, T.; Mann, D.M.; Shimada, H.; Ihara, Y.; Kawashima, S. Dominant and differential deposition of distinct beta-amyloid peptide species, Abeta N3(pE), in senile plaques. Neuron 1995, 14, 457–466. [Google Scholar] [CrossRef]
- Russo, C.; Saido, T.C.; DeBusk, L.M.; Tabaton, M.; Gambetti, P.; Teller, J.K. Heterogeneity of water-soluble amyloid beta-peptide in Alzheimer’s disease and Down’s syndrome brains. FEBS Lett. 1997, 409, 411–416. [Google Scholar] [CrossRef]
- Sergeant, N.; Bombois, S.; Ghestem, A.; Drobecq, H.; Kostanjevecki, V.; Missiaen, C.; Wattez, A.; David, J.P.; Vanmechelen, E.; Sergheraert, C.; et al. Truncated beta-amyloid peptide species in pre-clinical Alzheimer’s disease as new targets for the vaccination approach. J. Neurochem. 2003, 85, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.; Beher, D.; Cookson, N.; Oakley, A.; Piggott, M.; Morris, C.M.; Jaros, E.; Perry, R.; Ince, P.; Kenny, R.A.; et al. Quantification of Alzheimer pathology in ageing and dementia: Age-related accumulation of amyloid-β (42) peptide in vascular dementia. Neuropathol. Appl. Neurobiol. 2006, 32, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Portelius, E.; Bogdanovic, N.; Gustavsson, M.K.; Volkmann, I.; Brinkmalm, G.; Zetterberg, H.; Winblad, B.; Blennow, K. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010, 120, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Bayer, T.; Wirths, O. Focusing the amyloid cascade hypothesis on N-truncated Abeta peptides as drug targets against Alzheimer’s disease. Acta Neuropathol. 2014, 127, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Bouter, Y.; Dietrich, K.; Wittnam, J.L.; Rezaei-Ghaleh, N.; Pillot, T.; Papot-Couturier, S.; Lefebvre, T.; Sprenger, F.; Wirths, O.; Zweckstetter, M.; et al. N-truncated amyloid beta (Abeta) 4-42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 2013, 126, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Antonios, G.; Borgers, H.; Richard, B.C.; Brauß, A.; Meißner, J.; Weggen, S.; Pena, V.; Pillot, T.; Davies, S.L.; Bakrania, P.; et al. Alzheimer therapy with an antibody against N-terminal Abeta 4-X and pyroglutamate Abeta 3-X. Sci. Rep. 2015, 5, 17338. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Marcello, E.; Epis, R.; Saraceno, C.; Luca, M. Synaptic dysfunction in Alzheimer’s disease. In Synaptic Plasticity; Kreutz, M.R., Sala, C., Eds.; Springer: Vienna, Austria, 2012; pp. 573–601. [Google Scholar]
- Dietrich, K. Impact of N-Terminally Truncated Aβ4-42 on Memory and Synaptic Plasticity—Tg4-42 a New Mouse Model of Alzheimer’s Disease. Ph.D. Thesis, Georg-August University Göttingen, Göttingen, Germany, 2015. [Google Scholar]
- Fischer, M.; Reuter, J.; Gerich, F.J.; Hildebrandt, B.; Hagele, S.; Katschinski, D.; Muller, M. Enhanced hypoxia susceptibility in hippocampal slices from a mouse model of rett syndrome. J. Neurophysiol. 2009, 101, 1016–1032. [Google Scholar] [CrossRef] [PubMed]
- Zucker, R.S. Short-term synaptic plasticity. Annu. Rev. Neurosci. 1989, 12, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Kuhnt, U.; Voronin, L.L. Interaction between paired-pulse facilitation and long-term potentiation in area CA1 of guinea-pig hippocampal slices: Application of quantal analysis. Neuroscience 1994, 62, 391–397. [Google Scholar] [CrossRef]
- Zucker, R.S.; Regehr, W.G. Short-term synaptic plasticity. Annu. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef] [PubMed]
- Volianskis, A.; Collingridge, G.L.; Jensen, M.S. The roles of STP and LTP in synaptic encoding. PeerJ 2013, 1, e3. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of Amyloid β-Protein: Synaptic and Network Dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Megill, A.; Tran, T.; Eldred, K.; Lee, N.J.; Wong, P.C.; Hoe, H.S.; Kirkwood, A.; Lee, H.K. Defective Age-Dependent Metaplasticity in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 11346–11357. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Chen, X.; Henning, H.A.; Reichwald, J.; Staufenbiel, M.; Sakmann, B.; Konnerth, A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2012, 109, 8740–8745. [Google Scholar] [CrossRef] [PubMed]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP processing and synaptic function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Maruszak, A.; Thuret, S. Why looking at the whole hippocampus is not enough-a critical role for anteroposterior axis, subfield and activation analyses to enhance predictive value of hippocampal changes for Alzheimer’s disease diagnosis. Front. Cell. Neurosci. 2014, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Pozueta, J.; Lefort, R.; Shelanski, M.L. Synaptic changes in Alzheimer’s disease and its models. Neuroscience 2013, 251, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural Oligomers of the Alzheimer Amyloid-{beta} Protein Induce Reversible Synapse Loss by Modulating an NMDA-Type Glutamate Receptor-Dependent Signaling Pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Koffie, R.M.; Hyman, B.T.; Spires-Jones, T.L. Alzheimer’s disease: Synapses gone cold. Mol. Neurodegener. 2011, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Dewachter, I.; Filipkowski, R.K.; Priller, C.; Ris, L.; Neyton, J.; Croes, S.; Terwel, D.; Gysemans, M.; Devijver, H.; Borghgraef, P.; et al. Deregulation of NMDA-receptor function and down-stream signaling in APP[V717I] transgenic mice. Neurobiol. Aging 2009, 30, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Ronicke, R.; Mikhaylova, M.; Ronicke, S.; Meinhardt, J.; Schroder, U.H.; Fandrich, M.; Reiser, G.; Kreutz, M.R.; Reymann, K.G. Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol. Aging 2011, 32, 2219–2228. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; Lacor, P.N.; Velasco, P.T.; Xu, J.; Contractor, A.; Klein, W.L.; Triller, A. Deleterious Effects of Amyloid [beta] Oligomers Acting as an Extracellular Scaffold for mGluR5. Neuron 2010, 66, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Small, D.H.; Mok, S.S.; Bornstein, J.C. Alzheimer’s disease and Abeta toxicity: From top to bottom. Nat. Rev. Neurosci. 2001, 2, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Abeta oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 29, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Alexandru, A.; Jagla, W.; Graubner, S.; Becker, A.; Bäuscher, C.; Kohlmann, S.; Sedlmeier, R.; Raber, K.A.; Cynis, H.; Rönicke, R.; et al. Selective hippocampal neurodegeneration in transgenic mice expressing small amounts of truncated Aβ is induced by pyroglutamate–Aβ formation. J. Neurosci. 2011, 31, 12790–12801. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Aß elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Capetillo-Zarate, E.; Lin, M.T.; Milner, T.A.; Gouras, G.K. Accumulation of intraneuronal beta-amyloid 42 peptides is associated with early changes in microtubule-associated protein 2 in neurites and synapses. PLoS ONE 2013, 8, e51965. [Google Scholar]
- D’Amelio, M.; Cavallucci, V.; Middei, S.; Marchetti, C.; Pacioni, S.; Ferri, A.; Diamantini, A.; De Zio, D.; Carrara, P.; Battistini, L.; et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2011, 14, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.F.; White, G.L.; Jones, M.W.; Cooper-Blacketer, D.; Marshall, V.J.; Irizarry, M.; Younkin, L.; Good, M.A.; Bliss, T.V.; Hyman, B.T.; et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat. Neurosci. 1999, 2, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Fitzjohn, S.M.; Morton, R.A.; Kuenzi, F.; Rosahl, T.W.; Shearman, M.; Lewis, H.; Smith, D.; Reynolds, D.S.; Davies, C.H.; Collingridge, G.L.; et al. Age-related impairment of synaptic transmission but normal long-term potentiation in transgenic mice that overexpress the human APP695SWE mutant form of amyloid precursor protein. J. Neurosci. 2001, 21, 4691–4698. [Google Scholar] [PubMed]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.; Lynch, G.; Games, D.; Seubert, P. Alterations in synaptic transmission and long-term potentiation in hippocampal slices from young and aged PDAPP mice. Brain Res. 1999, 840, 23–35. [Google Scholar] [CrossRef]
- Saganich, M.J.; Schroeder, B.E.; Galvan, V.; Bredesen, D.E.; Koo, E.H.; Heinemann, S.F. Deficits in Synaptic Transmission and Learning in Amyloid Precursor Protein (APP) Transgenic Mice Require C-Terminal Cleavage of APP. J. Neurosci. 2006, 26, 13428–13436. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [PubMed]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Burki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.M.; Beach, T.G.; Sue, L.I.; Scott, S.; Layne, K.J.; Kokjohn, T.A.; Kalback, W.M.; Luehrs, D.C.; Vishnivetskaya, T.A.; Abramowski, D.; et al. The evolution of A beta peptide burden in the APP23 transgenic mice: Implications for A beta deposition in Alzheimer disease. Mol. Med. 2001, 7, 609–618. [Google Scholar] [PubMed]
- Roder, S.; Danober, L.; Pozza, M.F.; Lingenhoehl, K.; Wiederhold, K.H.; Olpe, H.R. Electrophysiological studies on the hippocampus and prefrontal cortex assessing the effects of amyloidosis in amyloid precursor protein 23 transgenic mice. Neuroscience 2003, 120, 705–720. [Google Scholar] [CrossRef]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33. [Google Scholar] [CrossRef] [PubMed]
- Wittnam, J.L.; Portelius, E.; Zetterberg, H.; Gustavsson, M.K.; Schilling, S.; Koch, B.; Demuth, H.-U.; Blennow, K.; Wirths, O.; Bayer, T.A. Pyroglutamate amyloid β (Aβ) aggravates behavioral deficits in transgenic amyloid mouse model for Alzheimer disease. J. Biol. Chem. 2012, 287, 8154–8162. [Google Scholar] [CrossRef] [PubMed]
- Kimura, R.; Ohno, M. Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol. Dis. 2009, 33, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Crouzin, N.; Baranger, K.; Cavalier, M.; Marchalant, Y.; Cohen-Solal, C.; Roman, F.S.; Khrestchatisky, M.; Rivera, S.; Feron, F.; Vignes, M. Area-specific alterations of synaptic plasticity in the 5XFAD mouse model of Alzheimer’s disease: Dissociation between somatosensory cortex and hippocampus. PLoS ONE 2013, 8, e74667. [Google Scholar] [CrossRef] [PubMed]
- Casas, C.; Sergeant, N.; Itier, J.M.; Blanchard, V.; Wirths, O.; van der Kolk, N.; Vingtdeux, V.; van de Steeg, E.; Ret, G.; Canton, T.; et al. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta 42 accumulation in a novel Alzheimer transgenic model. Am. J. Pathol. 2004, 165, 1289–1300. [Google Scholar] [CrossRef]
- Christensen, D.Z.; Kraus, S.L.; Flohr, A.; Cotel, M.C.; Wirths, O.; Bayer, T.A. Transient intraneuronal Abeta rather than extracellular plaque pathology correlates with neuron loss in the frontal cortex of APP/PS1KI mice. Acta Neuropathol. 2008, 116, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Breyhan, H.; Wirths, O.; Duan, K.; Marcello, A.; Rettig, J.; Bayer, T.A. APP/PS1KI bigenic mice develop early synaptic deficits and hippocampus atrophy. Acta Neuropathol. 2009, 117, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: Evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, K.S.; Edwards, G., III; Levites, Y.; Kumar, A.; Myers, C.E.; Gluck, M.A.; Setlow, B.; Bizon, J.L. Deficits in hippocampal-dependent transfer generalization learning accompany synaptic dysfunction in a mouse model of amyloidosis. Hippocampus 2016, 26, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Savonenko, A.; Xu, G.M.; Melnikova, T.; Morton, J.L.; Gonzales, V.; Wong, M.P.; Price, D.L.; Tang, F.; Markowska, A.L.; Borchelt, D.R. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: Relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol. Dis. 2005, 18, 602–617. [Google Scholar] [CrossRef] [PubMed]
- Chishti, M.A.; Yang, D.S.; Janus, C.; Phinney, A.L.; Horne, P.; Pearson, J.; Strome, R.; Zuker, N.; Loukides, J.; French, J.; et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 2001, 276, 21562–21570. [Google Scholar] [CrossRef] [PubMed]
- Jolas, T.; Zhang, X.S.; Zhang, Q.; Wong, G.; Del Vecchio, R.; Gold, L.; Priestley, T. Long-term potentiation is increased in the CA1 area of the hippocampus of APP(swe/ind) CRND8 mice. Neurobiol. Dis. 2002, 11, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Kimura, R.; MacTavish, D.; Yang, J.; Westaway, D.; Jhamandas, J.H. Beta amyloid-induced depression of hippocampal long-term potentiation is mediated through the amylin receptor. J. Neurosci. 2012, 32, 17401–17406. [Google Scholar] [CrossRef] [PubMed]
- Tozzi, A.; Sclip, A.; Tantucci, M.; de Iure, A.; Ghiglieri, V.; Costa, C.; Di Filippo, M.; Borsello, T.; Calabresi, P. Region- and age-dependent reductions of hippocampal long-term potentiation and NMDA to AMPA ratio in a genetic model of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Clark, J.K.; Furgerson, M.; Crystal, J.D.; Fechheimer, M.; Furukawa, R.; Wagner, J.J. Alterations in synaptic plasticity coincide with deficits in spatial working memory in presymptomatic 3xTg-AD mice. Neurobiol. Learn. Mem. 2015, 125, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.G.; Higgins, G.A.; Ouagazzal, A.M.; Ozmen, L.; Kew, J.N.; Bohrmann, B.; Malherbe, P.; Brockhaus, M.; Loetscher, H.; Czech, C.; et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J. Neurosci. 2003, 23, 8989–9003. [Google Scholar] [PubMed]
- Lin, N.; Pan, X.D.; Chen, A.Q.; Zhu, Y.G.; Wu, M.; Zhang, J.; Chen, X.C. Tripchlorolide improves age-associated cognitive deficits by reversing hippocampal synaptic plasticity impairment and NMDA receptor dysfunction in SAMP8 mice. Behav. Brain Res. 2014, 258, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Koss, D.J.; Drever, B.D.; Stoppelkamp, S.; Riedel, G.; Platt, B. Age-dependent changes in hippocampal synaptic transmission and plasticity in the PLB1Triple Alzheimer mouse. Cell. Mol. Life Sci. 2013, 70, 2585–2601. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Privitera, L.; Leznik, E.; Fa, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 2008, 28, 14537–14545. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Matsuyama, S.; Iso, H.; Umeda, T.; Takuma, H.; Ohnishi, K.; Ishibashi, K.; Teraoka, R.; Sakama, N.; Yamashita, T.; et al. A Mouse Model of Amyloid {beta} Oligomers: Their Contribution to Synaptic Alteration, Abnormal Tau Phosphorylation, Glial Activation, and Neuronal Loss In Vivo. J. Neurosci. 2010, 30, 4845–4856. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Farinelli, M.; Konietzko, U.; Nitsch, R.M.; Mansuy, I.M. Abeta oligomer-mediated long-term potentiation impairment involves protein phosphatase 1-dependent mechanisms. J. Neurosci. 2007, 27, 7648–7653. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Mallory, M.; Alford, M.; DeTeresa, R.; Hansen, L.A.; McKeel, D.W., Jr.; Morris, J.C. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 2001, 56, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Bayer, T.A. Intraneuronal Aβ accumulation and neurodegeneration: Lessons from transgenic models. Life Sci. 2012, 91, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Multhaup, G.; Bayer, T.A. A modified beta-amyloid hypothesis: Intraneuronal accumulation of the beta-amyloid peptide - the first step of a fatal cascade. J. Neurochem. 2004, 91, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Van Campen, J.S.; Hessel, E.V.S.; Bohmbach, K.; Rizzi, G.; Lucassen, P.J.; Lakshmi Turimella, S.; Umeoka, E.H.L.; Meerhoff, G.F.; Braun, K.P.J.; de Graan, P.N.E.; et al. Stress and Corticosteroids Aggravate Morphological Changes in the Dentate Gyrus after Early-Life Experimental Febrile Seizures in Mice. Front. Endocrinol. 2018, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Lesuis, S.L.; Weggen, S.; Baches, S.; Lucassen, P.J.; Krugers, H.J. Targeting glucocorticoid receptors prevents the effects of early life stress on amyloid pathology and cognitive performance in APP/PS1 mice. Transl. Psychiatry 2018, 8, 53. [Google Scholar] [CrossRef] [PubMed]
- Bouter, Y.; Kacprowski, T.; Weissmann, R.; Dietrich, K.; Borgers, H.; Brauß, A.; Sperling, C.; Wirths, O.; Albrecht, M.; Jensen, L.R.; et al. Deciphering the molecular profile of plaques, memory decline and neuron-loss in two mouse models for Alzheimer’s disease by deep sequencing. Front. Aging Neurosci. 2014, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: The mouse model Tg4-42 is available from the authors. |
Figure 1.
Amyloid pathology in 3-month-old Tg4-42 and 5XFAD. Immunohistochemical staining showing Tg4-42 as an example for an Alzheimer’s disease (AD) mouse model with intraneuronal Aβ and 5XFAD as an example for abundant plaque pathology. Significant intraneuronal Aβ was only detected in CA1 in Tg4-42 mice (A–C), but not in 5XFAD (D–F), whereas plaques were only found in the hippocampus of 5XFAD mice. Immunohistochemistry was performed on 4 µm paraffin sections, as previously described [12]. The polyclonal antibody 24311 recognizes pan-Aβ (1:500; rabbit [12]). Biotinylated secondary anti-rabbit and anti-mouse antibodies (1:200) were purchased from DAKO (Glostrup, Denmark). Staining was visualized using the ABC method, with a Vectastain kit (Vector Laboratories, Burlingame, CA, USA) and diaminobenzidine as chromogen. Counterstaining was carried out with hematoxylin (Merck, Darmstadt, Germany). Scale bar: (A,D) 200 μm; (B,E) 100 μm; (C,F) 50 µm.
Figure 1.
Amyloid pathology in 3-month-old Tg4-42 and 5XFAD. Immunohistochemical staining showing Tg4-42 as an example for an Alzheimer’s disease (AD) mouse model with intraneuronal Aβ and 5XFAD as an example for abundant plaque pathology. Significant intraneuronal Aβ was only detected in CA1 in Tg4-42 mice (A–C), but not in 5XFAD (D–F), whereas plaques were only found in the hippocampus of 5XFAD mice. Immunohistochemistry was performed on 4 µm paraffin sections, as previously described [12]. The polyclonal antibody 24311 recognizes pan-Aβ (1:500; rabbit [12]). Biotinylated secondary anti-rabbit and anti-mouse antibodies (1:200) were purchased from DAKO (Glostrup, Denmark). Staining was visualized using the ABC method, with a Vectastain kit (Vector Laboratories, Burlingame, CA, USA) and diaminobenzidine as chromogen. Counterstaining was carried out with hematoxylin (Merck, Darmstadt, Germany). Scale bar: (A,D) 200 μm; (B,E) 100 μm; (C,F) 50 µm.

Figure 2.
Aβ4-42 induced neuronal hyperexcitability and affects short-term plasticity in 3-month-old Tg4-42 mice (taken from [16]). Impact of Aβ4-42 on basal synaptic function and short-term plasticity in acute hippocampal tissue slices of Tg4-42 and controls at 3 months of age. (A) An altered basal excitatory synaptic transmission was demonstrated by a left shift of the input–output curve; (B) The half-maximal stimulus intensity (dashed lines in A) corroborated this observation; (C) Paired-pulse facilitation (PPF), quantified as a paradigm for synaptic short-term plasticity, was affected in Tg4-42 mice in comparison to wildtype control mice. (A + C) Mean ± SD. n = number of slices per group (B) Mean ± SD. The number of slices analyzed is indicated at the bottom of the bars. Half-maximal stimulus intensity: unpaired t-test, ** p < 0.01. Amplitude fEPSP2/fEPSP1: unpaired t-test, * p < 0.05.
Figure 2.
Aβ4-42 induced neuronal hyperexcitability and affects short-term plasticity in 3-month-old Tg4-42 mice (taken from [16]). Impact of Aβ4-42 on basal synaptic function and short-term plasticity in acute hippocampal tissue slices of Tg4-42 and controls at 3 months of age. (A) An altered basal excitatory synaptic transmission was demonstrated by a left shift of the input–output curve; (B) The half-maximal stimulus intensity (dashed lines in A) corroborated this observation; (C) Paired-pulse facilitation (PPF), quantified as a paradigm for synaptic short-term plasticity, was affected in Tg4-42 mice in comparison to wildtype control mice. (A + C) Mean ± SD. n = number of slices per group (B) Mean ± SD. The number of slices analyzed is indicated at the bottom of the bars. Half-maximal stimulus intensity: unpaired t-test, ** p < 0.01. Amplitude fEPSP2/fEPSP1: unpaired t-test, * p < 0.05.

Figure 3.
N-truncated Aβ4-42 did not alter short-term and long-term plasticity in 3-month-old Tg4-42 mice (taken from [16]). Effects of Aβ4-42 on synaptic plasticity were assessed in hippocampal slices of Tg4-42 and wildtype (WT) littermate controls. Post-tetanic potentiation (PTP = was defined as the maximal response within 1 min after the third tetanic stimulus. Short-term potentiation (STP) and long-term potentiation (LTP) were defined as the period between 12th and 21st min, and 65th and 75th min after induction, respectively. (A + B) Induction of potentiation by trains of high-frequency stimuli triggered PTP, STP, and LTP in both Tg4-42 and control mice. Recordings of STP and LTP revealed stable amplitudes in hippocampal slices of Tg4-42 and WT. Mean ± SD. n = number of slices per group.
Figure 3.
N-truncated Aβ4-42 did not alter short-term and long-term plasticity in 3-month-old Tg4-42 mice (taken from [16]). Effects of Aβ4-42 on synaptic plasticity were assessed in hippocampal slices of Tg4-42 and wildtype (WT) littermate controls. Post-tetanic potentiation (PTP = was defined as the maximal response within 1 min after the third tetanic stimulus. Short-term potentiation (STP) and long-term potentiation (LTP) were defined as the period between 12th and 21st min, and 65th and 75th min after induction, respectively. (A + B) Induction of potentiation by trains of high-frequency stimuli triggered PTP, STP, and LTP in both Tg4-42 and control mice. Recordings of STP and LTP revealed stable amplitudes in hippocampal slices of Tg4-42 and WT. Mean ± SD. n = number of slices per group.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Overview of neurophysiological alterations in hippocampal slices from transgenic Alzheimer mouse models. Electrophysiological recordings of fEPSPs in CA1 subfield (adapted from [16]).
Table 1.
Overview of neurophysiological alterations in hippocampal slices from transgenic Alzheimer mouse models. Electrophysiological recordings of fEPSPs in CA1 subfield (adapted from [16]).
| Mouse Line (Mutations) (Promoter) | Intra-Neuronal Aβ | Plaques | Input-Output Curve (IO) | PPF | PTP/STP | LTP |
|---|---|---|---|---|---|---|
| Tg4-42 [12] (none) (Thy-1) | >2 m: yes | none | 3 m: yes ↑ >12 m: none | 3 m: yes ↓ >12 m: none | >3 m: none/ >3 m: none | >3 m: none |
| TBA2.1hom [36] (Aβ3E-42 → Aβ3Q-42) (Thy-1.2) | >1 m: yes | >1 m: yes | 2 m: none 5 m: yes ↓ | n.a. | n.a./ n.a. | 2 m: none 5 m: yes ↓ |
| Tg2576 [37,38,39,40,41] (APP: Swe) (PrP) | >2 m: yes | >6 m: yes | 2–8 m: none [39,40] 15–17 m: none [40] 12 m: none/yes [41] ↓ 18 m: yes [41] ↓ | 3 m: none [39] <17 m: none [40] <18 m: none [41] | n.a./ n.a. | 3 m: none [39] 2–8 m: none [40] 15–17 m: yes [40] ↓ <18 m: none [41] |
| PD-APP [42,43] line H6 (APP: Ind) (PDGF-β) | n.a. | 2–5 m: none 8–10 m: yes | 1–4 m: yes ↓ 8–10 m: yes ↓ | 1–4 m: n.a. 8–10 m: none | n.a./ n.a. | 1–4 m: n.a. 8–10 m: none |
| PD-APP [42,44] line 109 (APP: Ind) (PDGF-β) | n.a. | 27 m: yes | 4–5 m: none 27–29 m: yes ↓ | 4–5 m: yes ↑ 27–29 m: yes ↓ | n.a./ n.a. | 4–5 m: yes ↓ 27–29 m: none |
| PD-APP [42,44] line J9 (APP: Ind, Swe) (PDGF-β) | n.a. | 2–4 m: none 8–10 m: yes | 2–4 m: yes ↓ | n.a. | n.a./ n.a. | n.a. |
| PD-APP [26,45,46] line J20 (APP: Ind, Swe) (PDGF-β) | n.a. | >2 m: yes | 3–6 m: yes [45] ↓ 4–7 m: yes [26] ↓ | 3–6 m: none [45] 4–7 m: none [26] | n.a./ n.a. | 3–6 m: yes [45] ↓ 4–7 m: none [26] |
| APP23 [47,48,49] (APP: Swe) (Thy-1.2) | 4 m: yes | >9 m: yes | 3–9 m: none 12–18 m: yes ↓ 24 m: none | n.a. | n.a./ n.a. | 3 m: none 6 m: yes ↓ 9–12 m: none 18 m: yes ↑ 24 m: none |
| 5XFAD [14,50,51,52,53] (APP: Swe, Flo, Lon, PS1: M146L, L286V) (Thy-1) | >1.5 m: yes | >2 m: yes | 4 m: none 5.5 m: yes ↓ | <6 m: none | n.a./ n.a. | 4 m: none 5.5 m: yes ↓ |
| APPSLPS1KI [54,55,56] (APP: Lon, Swe, PS1: M233T/ L235P) (Thy-1 (APP), PS1 knock-in) | >1.5 m: yes | >2 m: yes | n.a. | 2–4 m: n.a. 6 m: yes ↓ | n.a./ n.a.. | 2–4 m: none 6 m: yes ↓ |
| APPswe; PS1∆E9 [23,57,58,59] (APP: Swe, PS1: deltaE9) (PrP) | n.a. | >6 m: yes | 6 m: yes [58] ↓ 1 m: yes [23] ↑ 6 m: none [23] | 6 m: none [58] 1 m: none [23] 6 m: yes [23] ↓ | n.a./ n.a. | 6 m: yes [58] ↓ 1 m: yes [23] ↑ 6 m: yes [23] ↓ |
| TgCRND8 [60,61,62,63] (APP: Swe, Ind) (PrP) | n.a. | >3 m: yes | 2 m: none [61] 5 m: yes [61] ↓ 6–12 m: yes [62] ↓ | 2 m: none [61,63] 5–6 m: none [61,63] | n.a./ n.a. | 2–5 m: yes [61] ↑ 6–12 m: yes [62] ↓ 2 m: yes [63] ↓ 6 m: yes [63] ↓ |
| 3xTg-AD [64,65] (APP: Swe, tau: P301L, PS1: M146V) (Thy-1.2 (APP, tau), PS1 knock-in) | >3 m: yes | >6 m: yes | 1 m: none [64] 6 m: yes [64] ↓ 3 m: yes [65] ↓ 8 m: yes [65]↓ | 1–6 m: none [64] 3 m: yes [65] ↓ 8 m: yes [65] ↓ | n.a./ 1–6 m: none [64] | 1 m: none [64] 6 m: yes [64] ↓ 3 m: none [65] 8 m: yes [65] ↓ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dietrich, K.; Bouter, Y.; Müller, M.; Bayer, T.A. Synaptic Alterations in Mouse Models for Alzheimer Disease—A Special Focus on N-Truncated Abeta 4-42. Molecules 2018, 23, 718. https://doi.org/10.3390/molecules23040718
AMA Style
Dietrich K, Bouter Y, Müller M, Bayer TA. Synaptic Alterations in Mouse Models for Alzheimer Disease—A Special Focus on N-Truncated Abeta 4-42. Molecules. 2018; 23(4):718. https://doi.org/10.3390/molecules23040718
Chicago/Turabian StyleDietrich, Katharina, Yvonne Bouter, Michael Müller, and Thomas A. Bayer. 2018. "Synaptic Alterations in Mouse Models for Alzheimer Disease—A Special Focus on N-Truncated Abeta 4-42" Molecules 23, no. 4: 718. https://doi.org/10.3390/molecules23040718