
Immunohistochemical Evidence from APP-Transgenic Mice for Glutaminyl Cyclase as Drug Target to Diminish pE-Abeta Formation

 ,
, 
Abstract
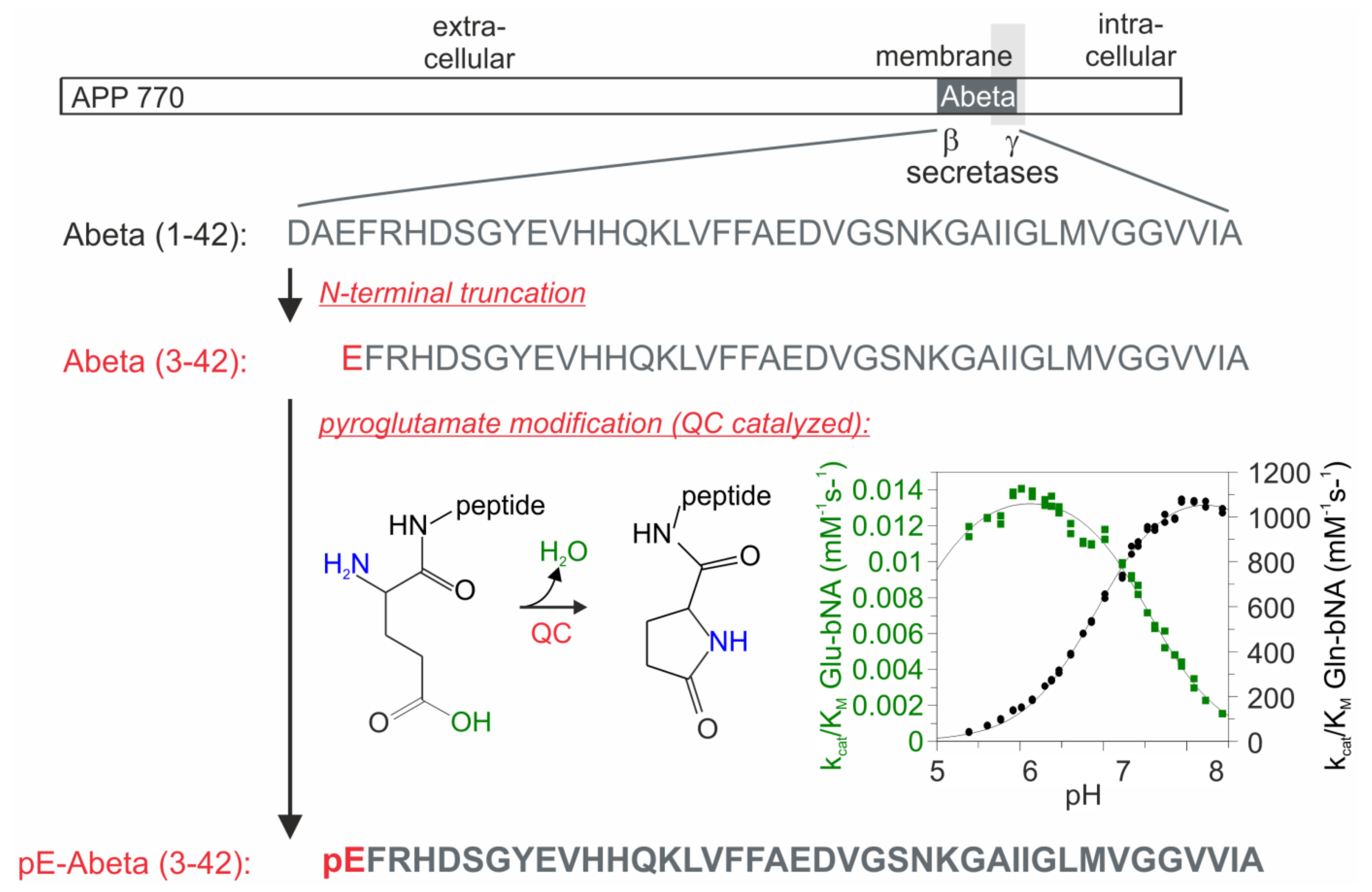
:1. Introduction
2. Results
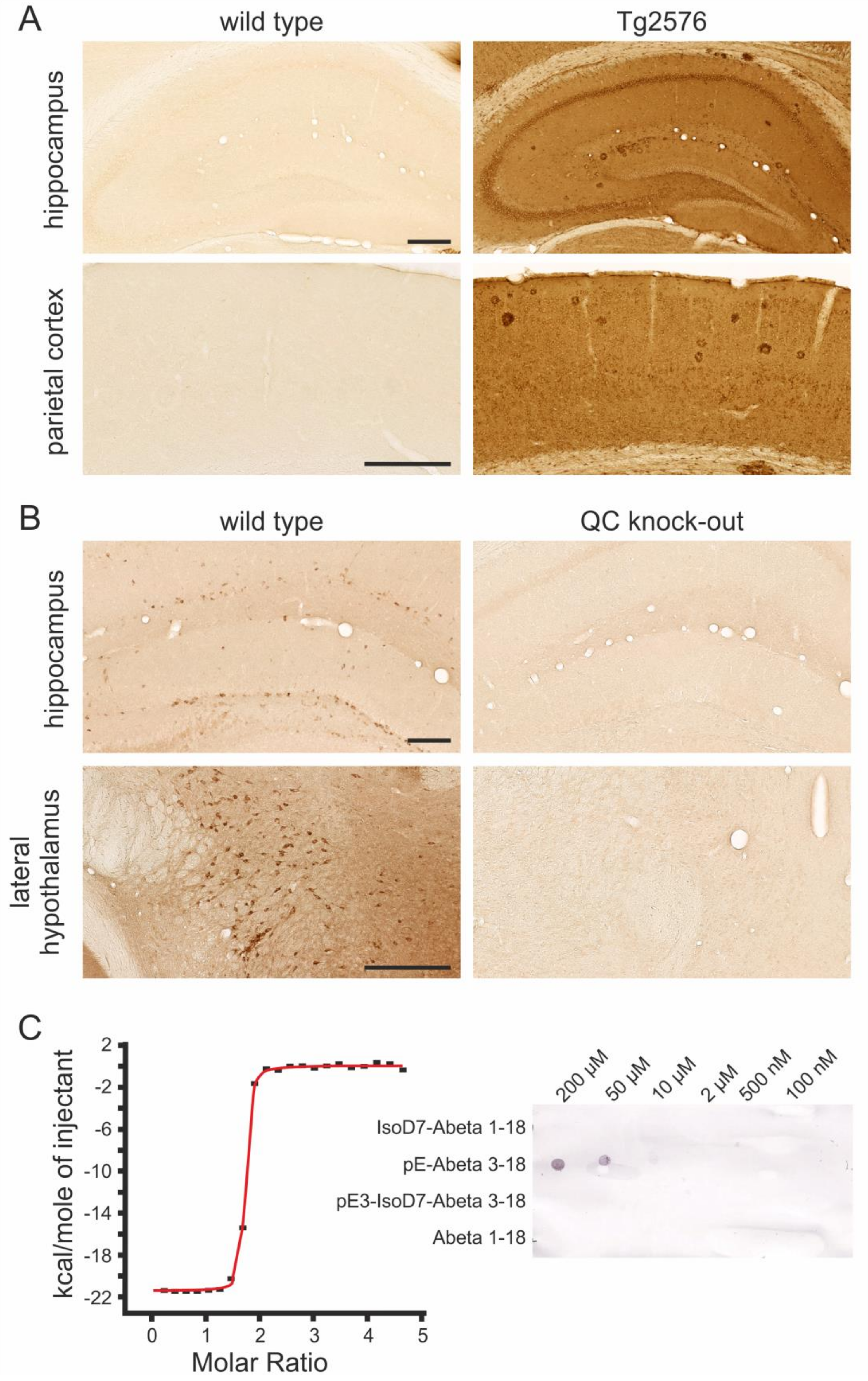
2.1. Specificity of the Rat Anti-hAPP, Goat Anti-Mouse QC and Mouse Anti-pE-Abeta Antibodies
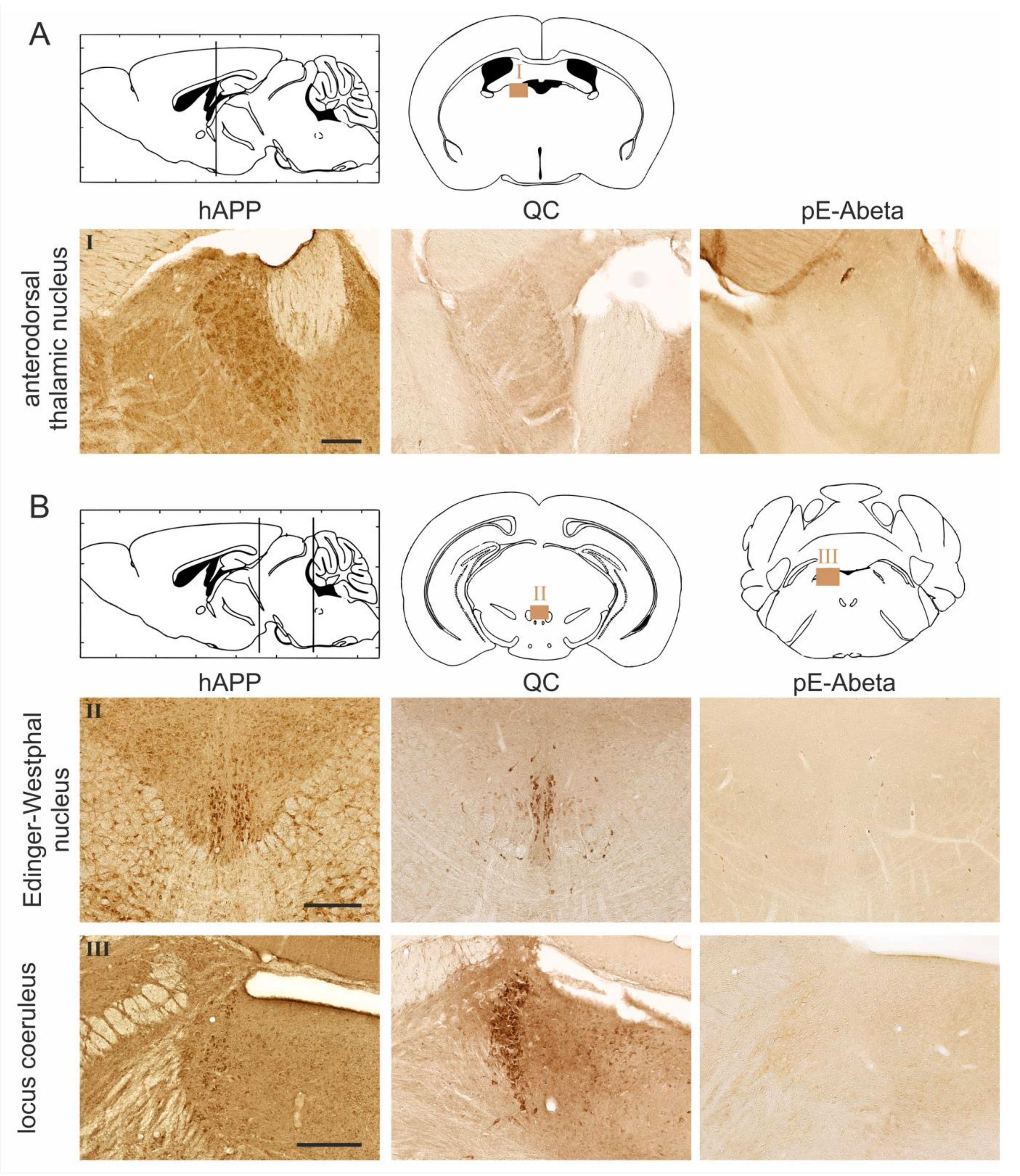
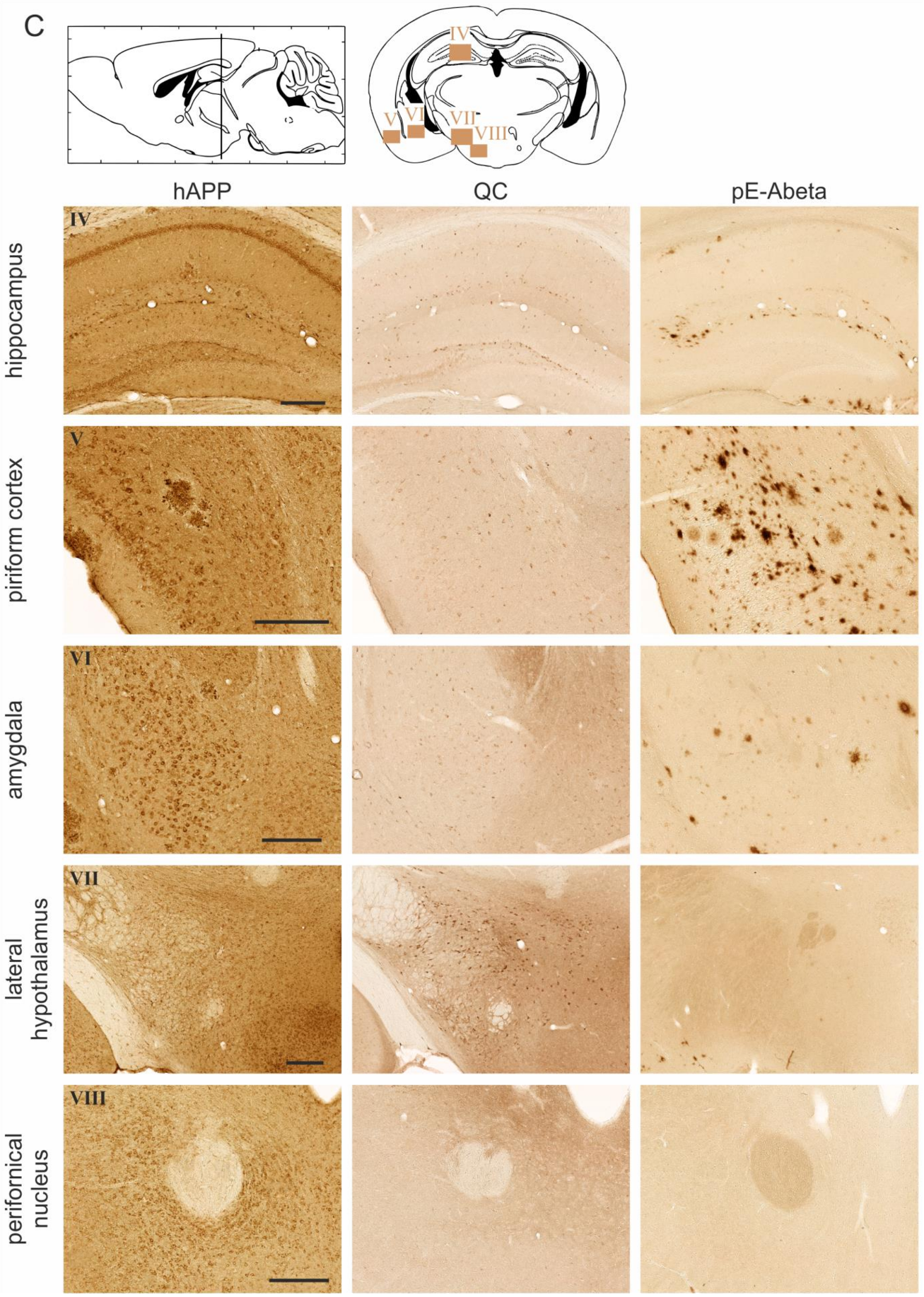
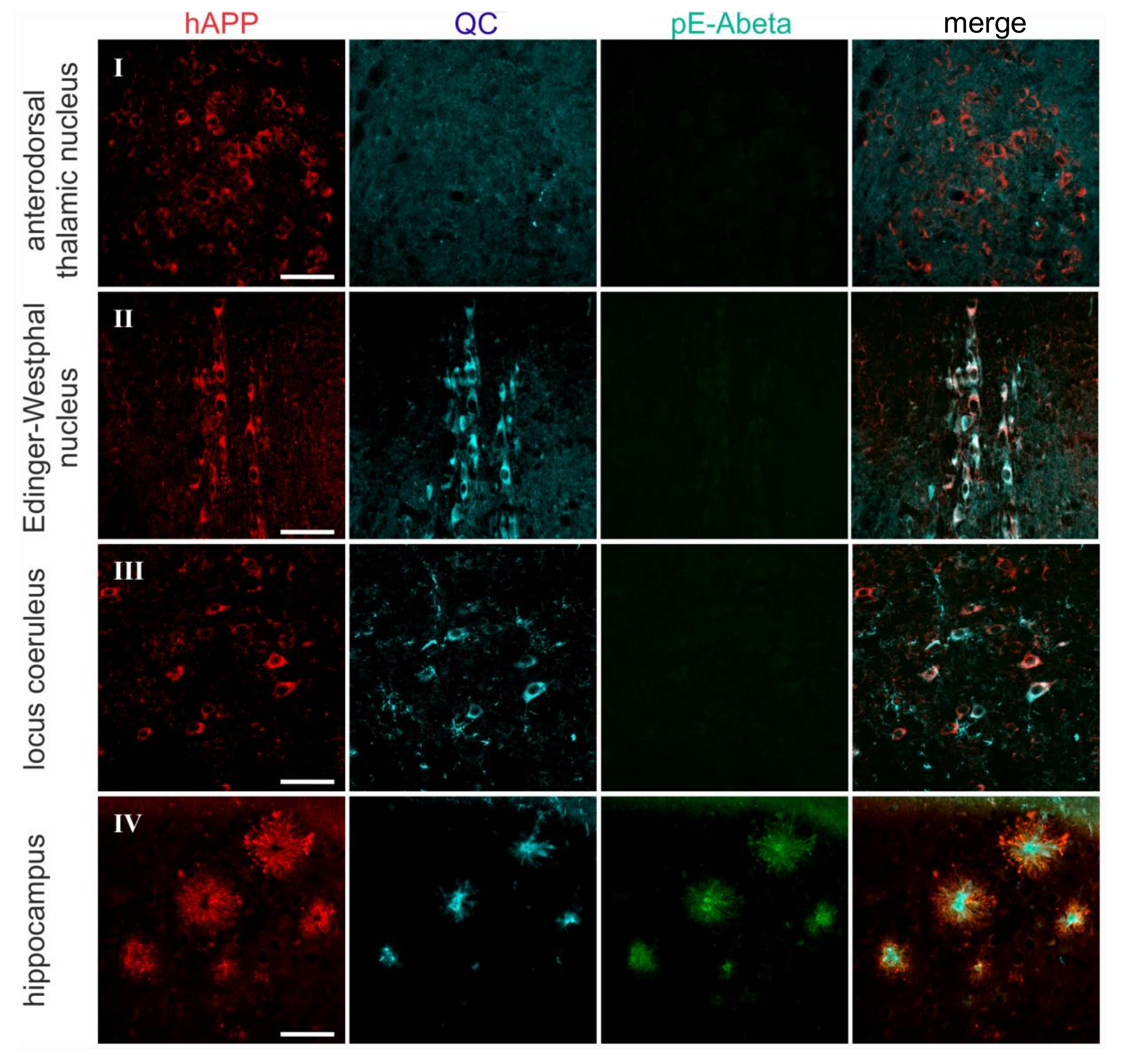
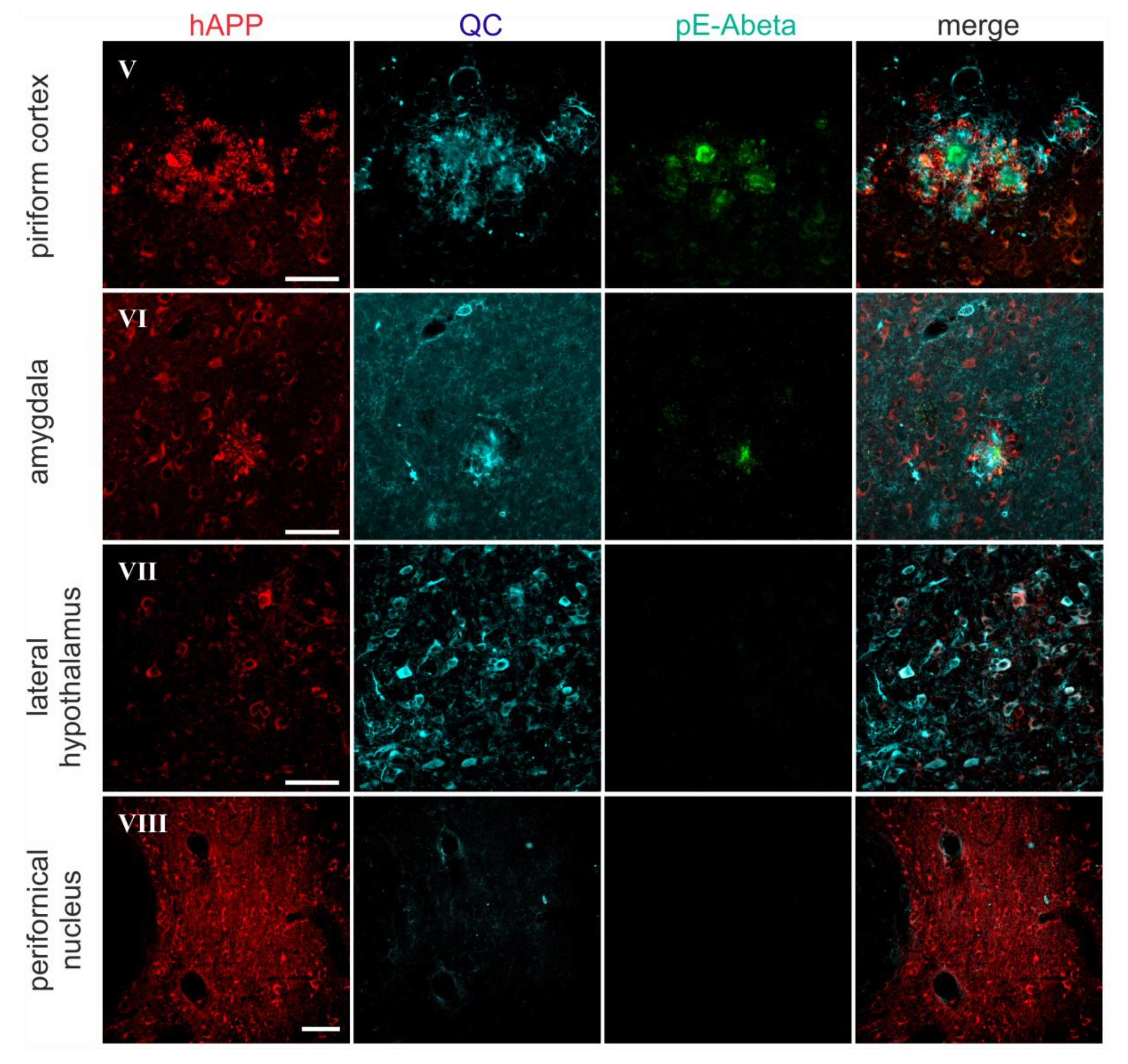
2.2. Spatial Relation of hAPP and QC Expression with pE-Abeta Deposition in Tg2576 Mouse Brain
3. Discussion
4. Materials and Methods
4.1. APP-Transgenic Tg2576 Mice
4.2. Antibodies against hAPP, QC, and pE-Abeta
4.3. Isothermal Titration Calorimetry
4.4. Dot Blot Analysis
4.5. Immunohistochemistry
4.5.1. Tissue Preparation
4.5.2. Single Labeling hAPP, QC, and pE-Abeta Immunohistochemistry
4.5.3. Staging of Immunohistochemical Labeling
4.5.4. Triple Immunofluorescent Labeling
4.5.5. Light Microscopy
4.5.6. Confocal Laser Scanning Microscopy
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AP | alkaline phosphatase |
| APP | amyloid precursor protein |
| BSA | bovine serum albumin |
| DAB | 3,3′-Diaminobenzidine |
| ITC | isothermal titration calorimetry |
| NOS | nitric oxide synthase |
| TBS | tris-buffered saline |
| pE | pyroglutamate |
| QC | glutaminyl cyclase |
References
- Kummer, M.P.; Heneka, M.T. Truncated and modified amyloid-beta species. Alzheimers Res. Ther. 2014, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Walter, J.; Saido, T.C.; Fändrich, M. Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015, 129, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.C.; König, S.; Roeber, S.; et al. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rezaei-Ghaleh, N.; Terwel, D.; Thal, D.R.; Richard, M.; Hoch, M.; Mc Donald, J.M.; Wüllner, U.; Glebov, K.; Heneka, M.T.; et al. Extracellular phosphorylation of the amyloid β-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J. 2011, 30, 2255–2265. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, S.; Hinze, D.; Josten, M.; Sahl, H.G.; Siepmann, M.; Walter, J. Phosphorylation of amyloid-β peptide at serine 8 attenuates its clearance via insulin-degrading and angiotensin-converting enzymes. J. Biol. Chem. 2012, 287, 8641–8651. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Wirths, O.; Stüber, K.; Wunderlich, P.; Koch, P.; Theil, S.; Rezaei-Ghaleh, N.; Zweckstetter, M.; Bayer, T.A.; Brüstle, O.; et al. Phosphorylation of the amyloid β-peptide at Ser26 stabilizes oligomeric assembly and increases neurotoxicity. Acta Neuropathol. 2016, 131, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Ashby, E.L.; Miners, J.S.; Kumar, S.; Walter, J.; Love, S.; Kehoe, P.G. Investigation of Aβ phosphorylated at serine 8 (pAβ) in Alzheimer’s disease, dementia with Lewy bodies and vascular dementia. Neuropathol. Appl. Neurobiol. 2015, 41, 428–444. [Google Scholar] [CrossRef] [PubMed]
- Wiltfang, J.; Esselmann, H.; Cupers, P.; Neumann, M.; Kretzschmar, H.; Beyermann, M.; Schleuder, D.; Jahn, H.; Rüther, E.; Kornhuber, J.; et al. Elevation of beta-amyloid peptide 2–42 in sporadic and familial Alzheimer’s disease and its generation in PS1 knockout cells. J. Biol. Chem. 2001, 276, 42645–42657. [Google Scholar] [CrossRef] [PubMed]
- Casas, C.; Sergeant, N.; Itier, J.M.; Blanchard, V.; Wirths, O.; van der Kolk, N.; Vingtdeux, V.; van de Steeg, E.; Ret, G.; Canton, T.; et al. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Aβ42 accumulation in a novel Alzheimer transgenic model. Am. J. Pathol. 2004, 165, 1289–1300. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, NA.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Bouter, Y.; Dietrich, K.; Wittnam, J.L.; Rezaei-Ghaleh, N.; Pillot, T.; Papot-Couturier, S.; Lefebvre, T.; Sprenger, F.; Wirths, O.; Zweckstetter, M.; et al. N-truncated amyloid β (Aβ) 4-42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 2013, 126, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Araki, W.; Akiyama, H.; Tabira, T. Amino-truncated amyloid beta-peptide (Aβ5-40/42) produced from caspase-cleaved amyloid precursor protein is deposited in Alzheimer’s disease brain. FASEB J. 2004, 18, 1755–1757. [Google Scholar] [CrossRef] [PubMed]
- Murayama, K.S.; Kametani, F.; Tabira, T.; Araki, W. A novel monoclonal antibody specific for the amino-truncated beta-amyloid Aβ5-40/42 produced from caspase-cleaved amyloid precursor protein. J. Neurosci. Methods 2007, 161, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, N.; Bombois, S.; Ghestem, A.; Drobecq, H.; Kostanjevecki, V.; Missiaen, C.; Wattez, A.; David, J.P.; Vanmechelen, E.; Sergheraert, C.; et al. Truncated beta-amyloid peptide species in pre-clinical Alzheimer’s disease as new targets for the vaccination approach. J. Neurochem. 2003, 85, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Schieb, H.; Kratzin, H.; Jahn, O.; Möbius, W.; Rabe, S.; Staufenbiel, M.; Wiltfang, J.; Klafki, H.W. Beta-amyloid peptide variants in brains and cerebrospinal fluid from amyloid precursor protein (APP) transgenic mice: Comparison with human Alzheimer amyloid. J. Biol. Chem. 2011, 286, 33747–33758. [Google Scholar] [CrossRef] [PubMed]
- Saido, T.C. Alzheimer’s disease as proteolytic disorders: Anabolism and catabolism of beta-amyloid. Neurobiol. Aging 1998, 19, S69–S75. [Google Scholar] [CrossRef]
- Saido, T.C.; Iwatsubo, T.; Mann, D.M.; Shimada, H.; Ihara, Y.; Kawashima, S. Dominant and differential deposition of distinct beta-amyloid peptide species, AβN3(pE), in senile plaques. Neuron 1995, 14, 457–466. [Google Scholar] [CrossRef]
- Schilling, S.; Hoffmann, T.; Manhart, S.; Hoffmann, M.; Demuth, H.U. Glutaminyl cyclases unfold glutamyl cyclase activity under mild acid conditions. FEBS Lett. 2004, 563, 191–196. [Google Scholar] [CrossRef]
- Morawski, M.; Hartlage-Rübsamen, M.; Jäger, C.; Waniek, A.; Schilling, S.; Schwab, C.; McGeer, P.; Arendt, T.; Demuth, H.U.; Roßner, S. Distinct glutaminyl cyclase expression in Edinger-Westphal nucleus, locus coeruleus and nucleus basalis Meynert contributes to pGlu-Aβ pathology in Alzheimer’s disease. Acta Neuropathol. 2010, 120, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Hartlage-Rübsamen, M.; Morawski, M.; Waniek, A.; Jäger, C.; Zeitschel, U.; Koch, B.; Cynis, H.; Schilling, S.; Schliebs, R.; Demuth, H.U.; et al. Glutaminyl cyclase contributes to the formation of focal and diffuse pyroglutamate (pGlu)-Aβ deposits in hippocampus via distinct cellular mechanisms. Acta Neuropathol. 2011, 121, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Morawski, M.; Schilling, S.; Kreuzberger, M.; Waniek, A.; Jäger, C.; Koch, B.; Cynis, H.; Kehlen, A.; Arendt, T.; Hartlage-Rübsamen, M.; et al. Glutaminyl cyclases in human cortex—Correlation with (pGlu)-Abeta load and cognitive decline in Alzheimer’s disease. J. Alzheimers Dis. 2014, 39, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Zeitschel, U.; Hoffmann, T.; Heiser, U.; Francke, M.; Kehlen, A.; Holzer, M.; Hutter-Paier, B.; Prokesch, M.; Windisch, M.; et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer’s disease-like pathology. Nat. Med. 2008, 14, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- Alexandru, A.; Jagla, W.; Graubner, S.; Becker, A.; Bäuscher, C.; Kohlmann, S.; Sedlmeier, R.; Raber, K.A.; Cynis, H.; Rönicke, R.; et al. Selective hippocampal neurodegeneration in transgenic mice expressing small amounts of truncated Aβ is induced by pyroglutamate-Aβ formation. J. Neurosci. 2011, 31, 12790–12801. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, S.; Wirths, O.; Schilling, S.; Graubner, S.; Demuth, H.U.; Bayer, T.A. Overexpression of glutaminyl cyclase, the enzyme responsible for pyroglutamate Aβ formation, induces behavioral deficits, and glutaminyl cyclase knock-out rescues the behavioral phenotype in 5XFAD mice. J. Biol. Chem. 2011, 286, 4454–4460. [Google Scholar] [CrossRef] [PubMed]
- Schlenzig, D.; Manhart, S.; Cinar, Y.; Kleinschmidt, M.; Hause, G.; Willbold, D.; Funke, S.A.; Schilling, S.; Demuth, H.U. Pyroglutamate formation influences solubility and amyloidogenicity of amyloid peptides. Biochemistry 2009, 48, 7072–7078. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, J.M.; Schilling, S.; Cynis, H.; Silva, A.; Swanson, E.; Wangsanut, T.; Tayler, K.; Wiltgen, B.; Hatami, A.; Rönicke, R.; et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 2012, 485, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Younkin, L.H.; Saido, T.C.; Shoji, M.; Ashe, K.H.; Younkin, S.G. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2001, 21, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Hartlage-Rübsamen, M.; Staffa, K.; Waniek, A.; Wermann, M.; Hoffmann, T.; Cynis, H.; Schilling, S.; Demuth, H.U.; Roßner, S. Developmental expression and subcellular localization of glutaminyl cyclase in mouse brain. Int. J. Dev. Neurosci. 2009, 27, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Höfling, C.; Indrischek, H.; Höpcke, T.; Waniek, A.; Cynis, H.; Koch, B.; Schilling, S.; Morawski, M.; Demuth, H.U.; Roßner, S.; et al. Mouse strain and brain region-specific expression of the glutaminyl cyclases QC and isoQC. Int. J. Dev. Neurosci. 2014, 36, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Höfling, C.; Morawski, M.; Zeitschel, U.; Zanier, E.R.; Moschke, K.; Serdaroglu, A.; Canneva, F.; von Hörsten, S.; De Simoni, M.G.; Forloni, G.; et al. Differential transgene expression patterns in Alzheimer mouse models revealed by novel human amyloid precursor protein-specific antibodies. Aging Cell 2016, 15, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Waniek, A.; Hartlage-Rübsamen, M.; Höfling, C.; Kehlen, A.; Schilling, S.; Demuth, H.U.; Roßner, S. Identification of thyrotropin-releasing hormone as hippocampal glutaminyl cyclase substrate in neurons and reactive astrocytes. Biochim. Biophys. Acta 2015, 1852, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Seifert, F.; Schulz, K.; Koch, B.; Manhart, S.; Demuth, H.U.; Schilling, S. Glutaminyl cyclases display significant catalytic proficiency for glutamyl substrates. Biochemistry 2009, 48, 11831–11833. [Google Scholar] [CrossRef] [PubMed]
- Cynis, H.; Scheel, E.; Saido, T.C.; Schilling, S.; Demuth, H.U. Amyloidogenic processing of amyloid precursor protein: Evidence of a pivotal role of glutaminyl cyclase in generation of pyroglutamate-modified amyloid-beta. Biochemistry 2008, 47, 7405–7413. [Google Scholar] [CrossRef] [PubMed]
- Cynis, H.; Schilling, S.; Bodnar, M.; Hoffmann, T.; Heiser, U.; Saido, T.C.; Demuth, H.U. Inhibition of glutaminyl cyclase alters pyroglutamate formation in mammalian cells. Biochim. Biophys. Acta 2006, 1764, 1618–1625. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.L.; Liu, B.; Kleinschmidt, M.; Schilling, S.; Demuth, H.U.; Lemere, C.A. Passive immunization against pyroglutamate-3 amyloid-β reduces plaque burden in Alzheimer-like transgenic mice: A pilot study. Neurodegener. Dis. 2012, 10, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.L.; Liu, B.; Rahfeld, J.U.; Kleinschmidt, M.; O’Nuallain, B.; Le, K.X.; Lues, I.; Caldarone, B.J.; Schilling, S.; Demuth, H.U.; et al. An anti-pyroglutamate-3 Aβ vaccine reduces plaques and improves cognition in APPswe/PS1ΔE9 mice. Neurobiol. Aging 2015, 36, 3187–3199. [Google Scholar] [CrossRef] [PubMed]
- Demattos, R.B.; Lu, J.; Tang, Y.; Racke, M.M.; Delong, C.A.; Tzaferis, J.A.; Hole, J.T.; Forster, B.M.; McDonnell, P.C.; Liu, F.; et al. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron 2012, 76, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Erck, C.; Martens, H.; Harmeier, A.; Geumann, C.; Jawhar, S.; Kumar, S.; Multhaup, G.; Walter, J.; Ingelsson, M.; et al. Identification of low molecular weight pyroglutamate Aβ oligomers in Alzheimer disease: A novel tool for therapy and diagnosis. J. Biol. Chem. 2010, 285, 41517–41524. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.H.; Spiess, J. Identification of a mammalian glutaminyl cyclase converting glutaminyl into pyroglutamyl peptides. Proc. Natl. Acad. Sci. USA 1987, 84, 3628–3632. [Google Scholar] [CrossRef] [PubMed]
- Busby, W.H.; Quackenbush, G.E.; Humm, J.; Youngblood, W.W.; Kizer, J.S. An enzyme(s) that converts glutaminyl-peptides into pyroglutamyl-peptides. J. Biol. Chem. 1987, 262, 8532–8536. [Google Scholar] [PubMed]
- Schilling, S.; Kohlmann, S.; Bäuscher, C.; Sedlmeier, R.; Koch, B.; Eichentopf, R.; Becker, A.; Cynis, H.; Hoffmann, T.; Berg, S.; et al. Glutaminyl cyclase knock-out mice exhibit slight hypothyroidism but no hypogonadism: Implications for enzyme function and drug development. J. Biol. Chem. 2011, 286, 14199–14208. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Eichentopf, R.; Sedlmeier, R.; Waniek, A.; Cynis, H.; Koch, B.; Stephan, A.; Bäuscher, C.; Kohlmann, S.; Hoffmann, T.; et al. IsoQC (QPCTL) knock-out mice suggest differential substrate conversion by glutaminyl cyclase isoenzymes. Biol. Chem. 2016, 397, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.; Meyer, A.; Heiser, U.; Kurat, S.; Böhme, L.; Kleinschmidt, M.; Bühring, K.U.; Hutter-Paier, B.; Farcher, M.; Demuth, H.U.; et al. Glutaminyl cyclase inhibitor PQ912 improves cognition in mouse models of Alzheimer’s disease-studies on relation to effective target occupancy. J. Pharmacol. Exp. Ther. 2017, 362, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, M.; Heiser, U.; Schilling, S.; Niestroj, A.J.; Zunkel, K.; Demuth, H.U. The first potent inhibitors for human glutaminyl cyclase: Synthesis and structure-activity relationship. J. Med. Chem. 2006, 49, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Koch, B.; Buchholz, M.; Wermann, M.; Heiser, U.; Schilling, S.; Demuth, H.U. Probing secondary glutaminyl cyclase (QC) inhibitor interactions applying an in silico-modeling/site-directed mutagenesis approach: Implications for drug development. Chem. Biol. Drug Des. 2012, 80, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Lues, I.; Weber, F.; Meyer, A.; Bühring, U.; Hoffmann, T.; Kühn-Wache, K.; Manhart, S.; Heiser, U.; Pokorny, R.; Chiesa, J.; et al. A phase 1 study to evaluate the safety and pharmacokinetics of PQ912, a glutaminyl cyclase inhibitor, in healthy subjects. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2015, 1, 182–195. [Google Scholar] [CrossRef]
- Alzforum. 2017. Available online: https://www.alzforum.org/news/research-news/new-alzheimers-drug-shows-safety-hints-efficacy-phase-2 (accessed on 20 March 2018).
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the antibodies rat anti-human APP, mouse anti-pE-Abeta and goat anti-QC are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Brain Region | hAPP | Endogenous QC | pE-Abeta |
|---|---|---|---|
| anterodorsal thalamic nucleus | 4–5 | 0 | 0 |
| Edinger-Westphal nucleus | 5 | 5 | 0 |
| locus coeruleus | 3 | 5 | 0 |
| hippocampus | |||
| granule cells | 0 | 0 | 0 |
| interneurons | 4 | 3 | 3 |
| pyramidal cells | 1 | 1 | 1 |
| piriform cortex | 4–5 | 3 | 5 |
| amygdala | 5 | 2 | 3 |
| lateral hypothalamus | 2 | 4–5 | 0 |
| perifornical nucleus | 4 | 0 | 0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartlage-Rübsamen, M.; Bluhm, A.; Piechotta, A.; Linnert, M.; Rahfeld, J.-U.; Demuth, H.-U.; Lues, I.; Kuhn, P.-H.; Lichtenthaler, S.F.; Roßner, S.; et al. Immunohistochemical Evidence from APP-Transgenic Mice for Glutaminyl Cyclase as Drug Target to Diminish pE-Abeta Formation. Molecules 2018, 23, 924. https://doi.org/10.3390/molecules23040924
Hartlage-Rübsamen M, Bluhm A, Piechotta A, Linnert M, Rahfeld J-U, Demuth H-U, Lues I, Kuhn P-H, Lichtenthaler SF, Roßner S, et al. Immunohistochemical Evidence from APP-Transgenic Mice for Glutaminyl Cyclase as Drug Target to Diminish pE-Abeta Formation. Molecules. 2018; 23(4):924. https://doi.org/10.3390/molecules23040924
Chicago/Turabian StyleHartlage-Rübsamen, Maike, Alexandra Bluhm, Anke Piechotta, Miriam Linnert, Jens-Ulrich Rahfeld, Hans-Ulrich Demuth, Inge Lues, Peer-Hendrik Kuhn, Stefan F. Lichtenthaler, Steffen Roßner, and et al. 2018. "Immunohistochemical Evidence from APP-Transgenic Mice for Glutaminyl Cyclase as Drug Target to Diminish pE-Abeta Formation" Molecules 23, no. 4: 924. https://doi.org/10.3390/molecules23040924
APA StyleHartlage-Rübsamen, M., Bluhm, A., Piechotta, A., Linnert, M., Rahfeld, J.-U., Demuth, H.-U., Lues, I., Kuhn, P.-H., Lichtenthaler, S. F., Roßner, S., & Höfling, C. (2018). Immunohistochemical Evidence from APP-Transgenic Mice for Glutaminyl Cyclase as Drug Target to Diminish pE-Abeta Formation. Molecules, 23(4), 924. https://doi.org/10.3390/molecules23040924