Carbene Transfer Reactions Catalysed by Dyes of the Metalloporphyrin Group

 , , , , , and
, , , , , and
Abstract
:1. Introduction
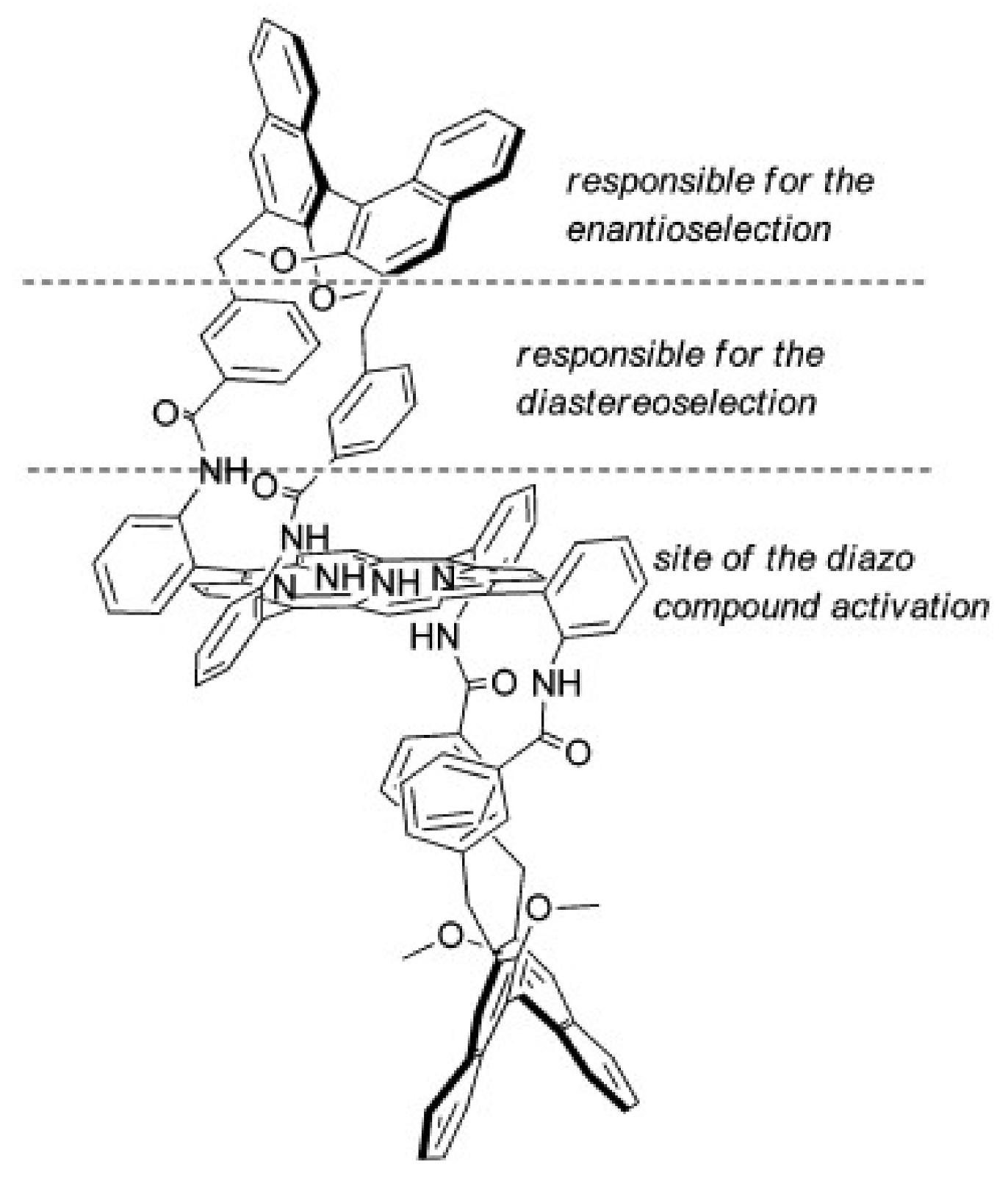
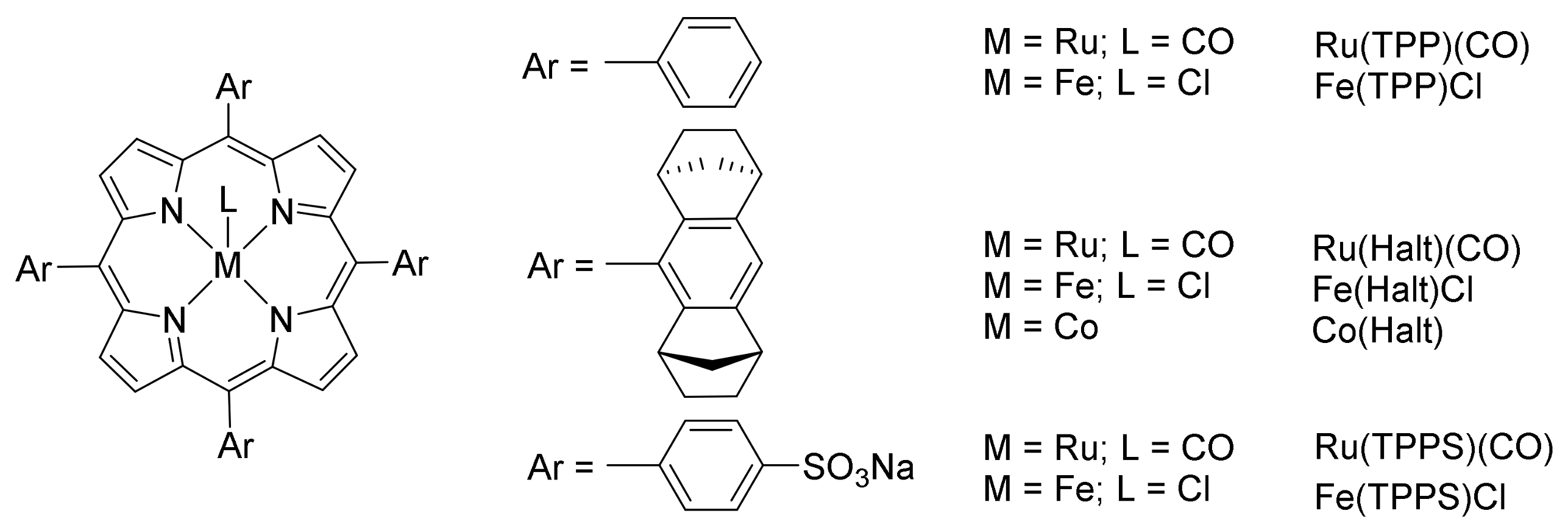
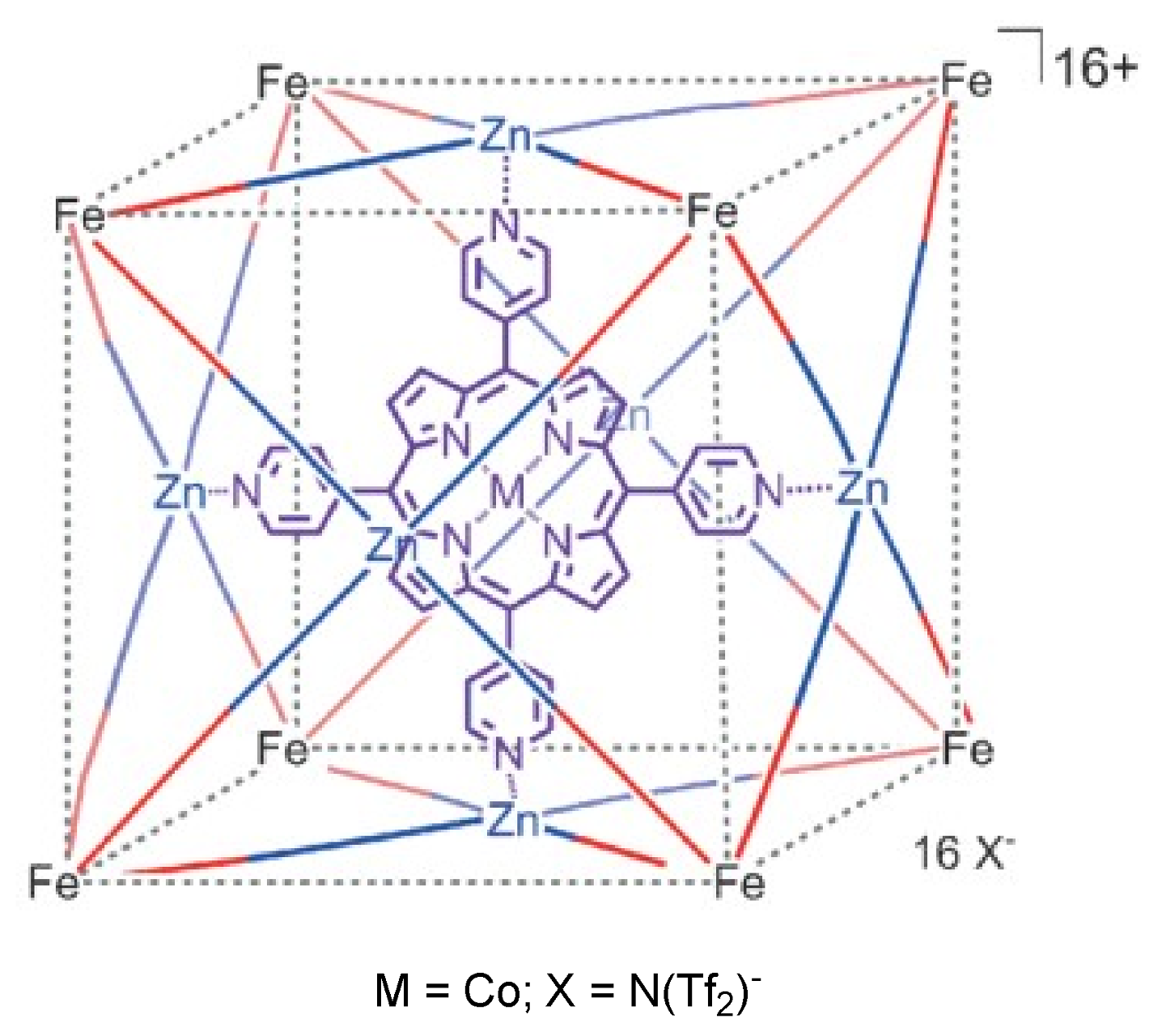
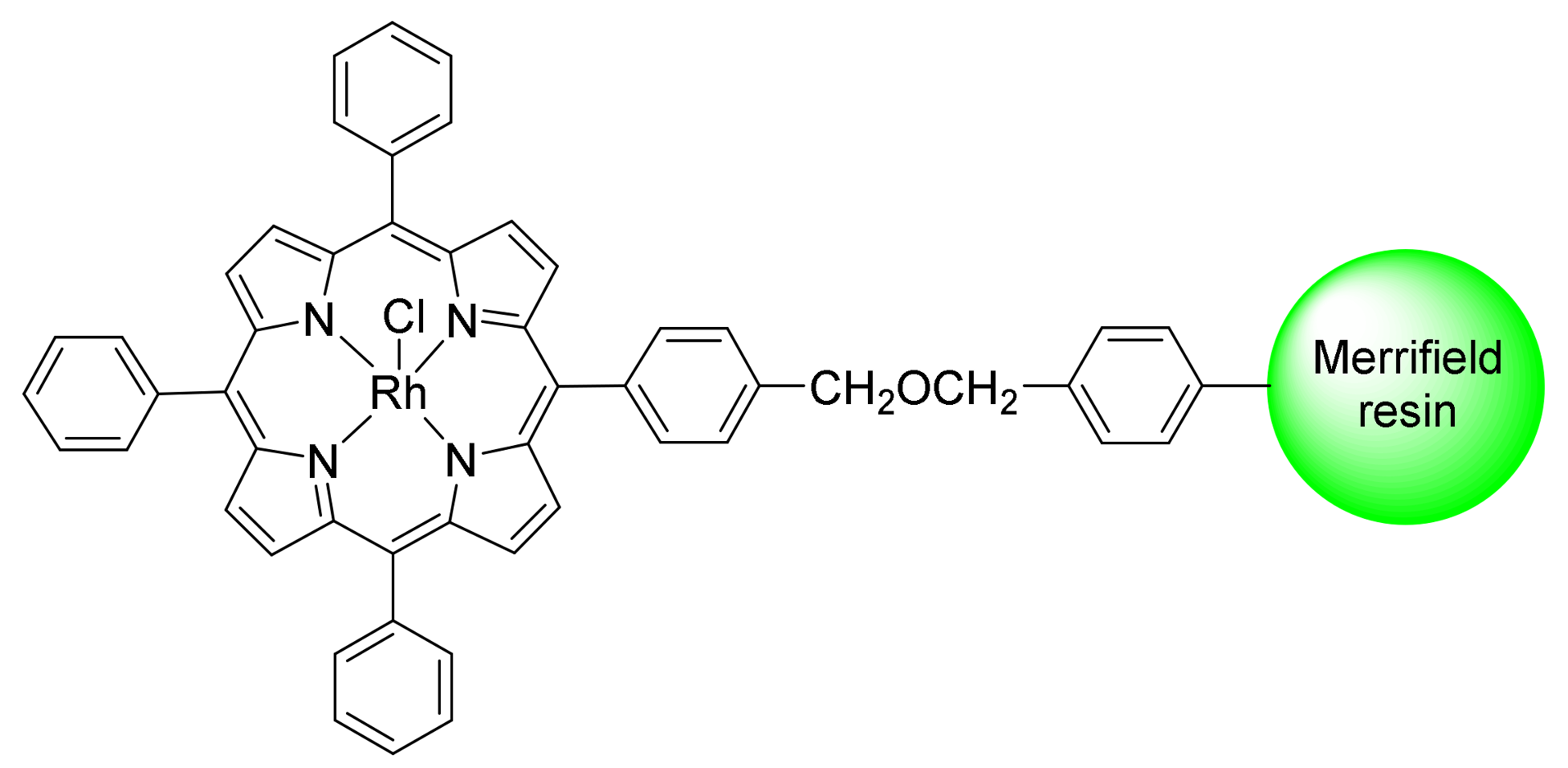
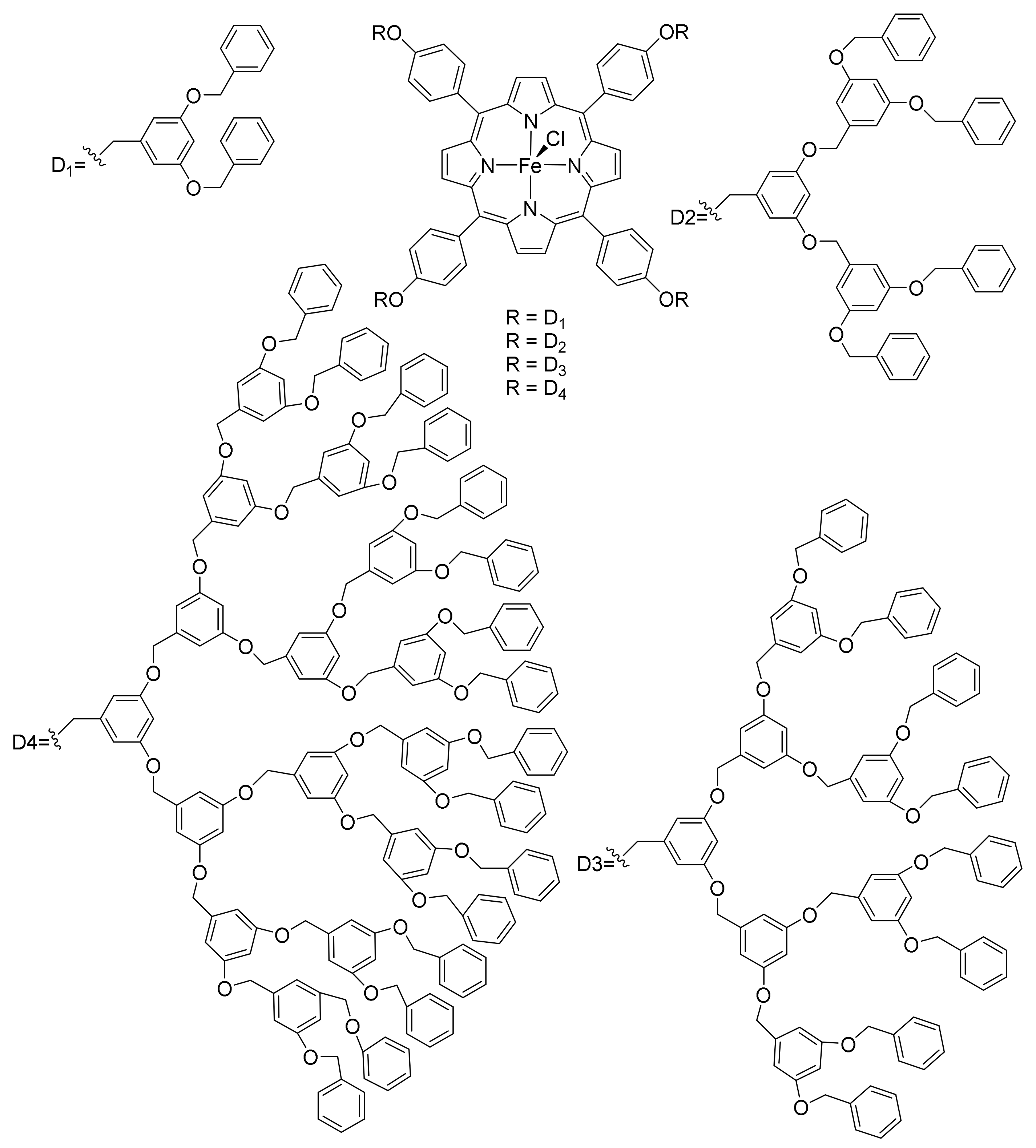
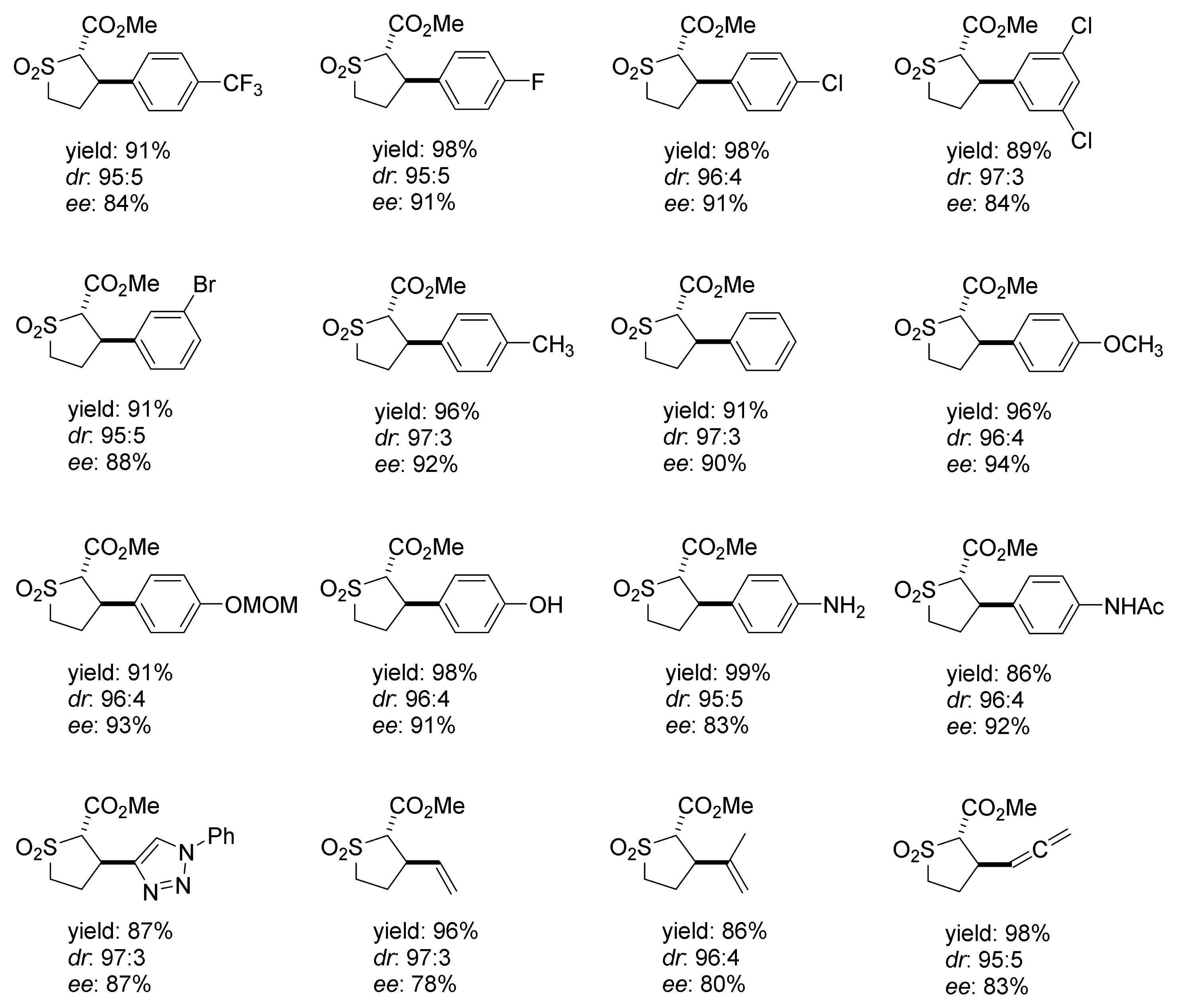
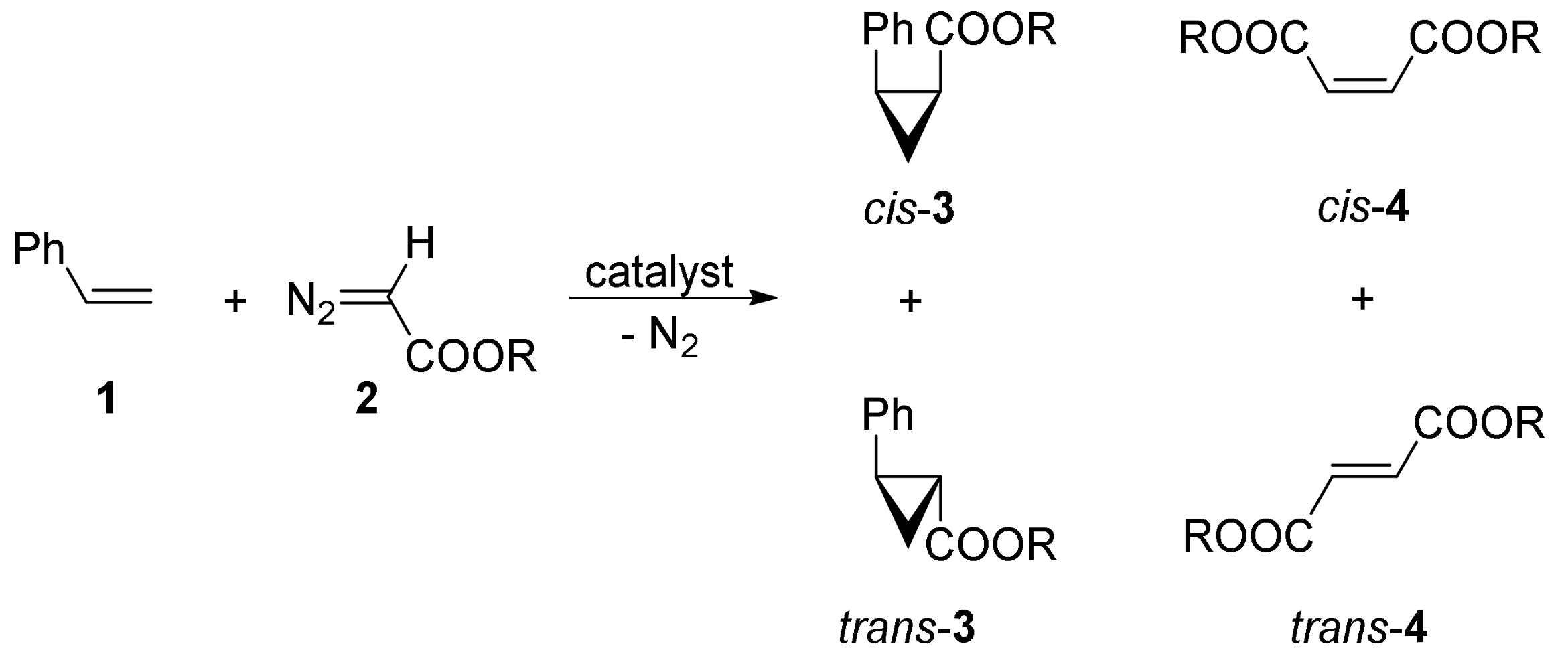
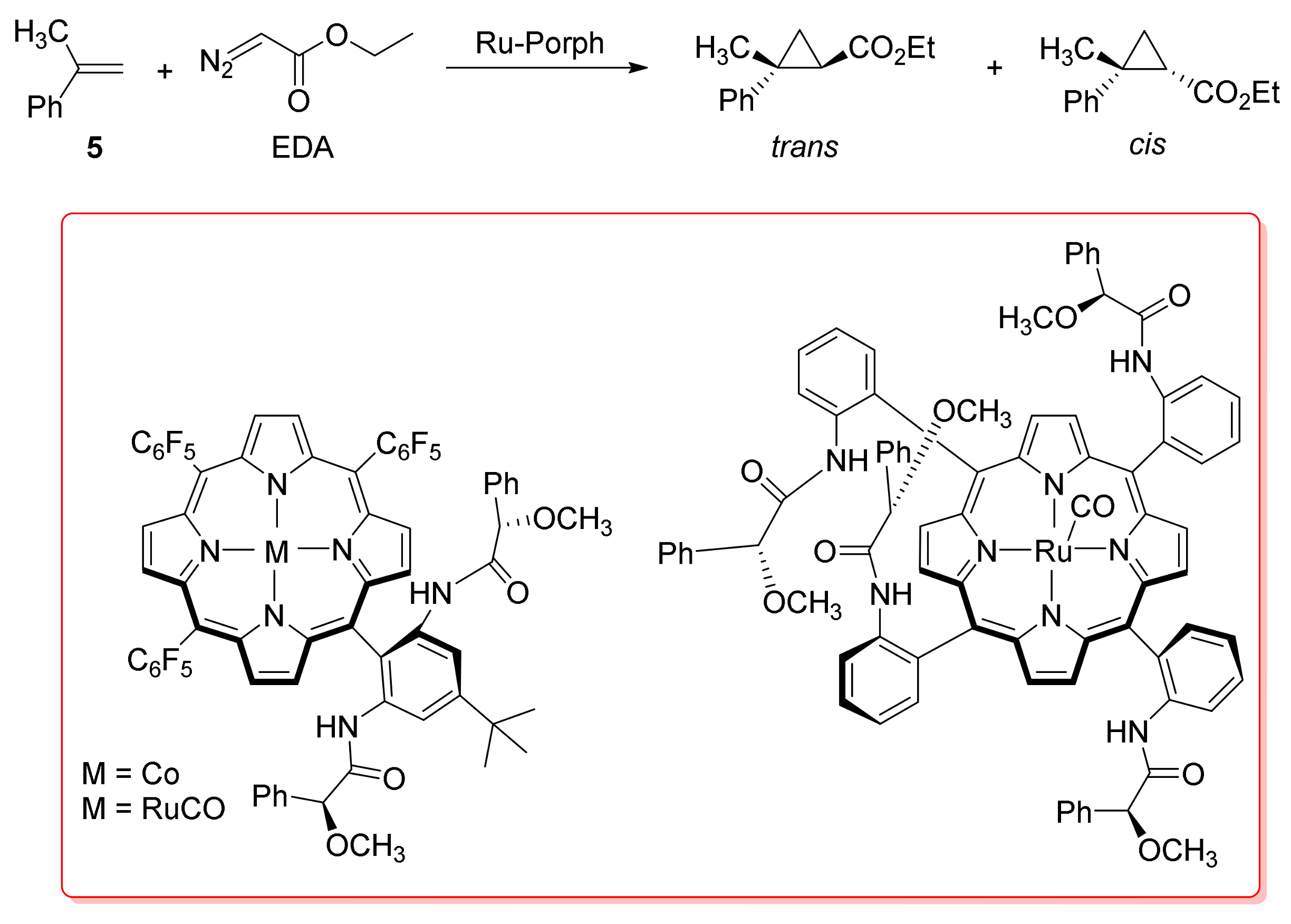
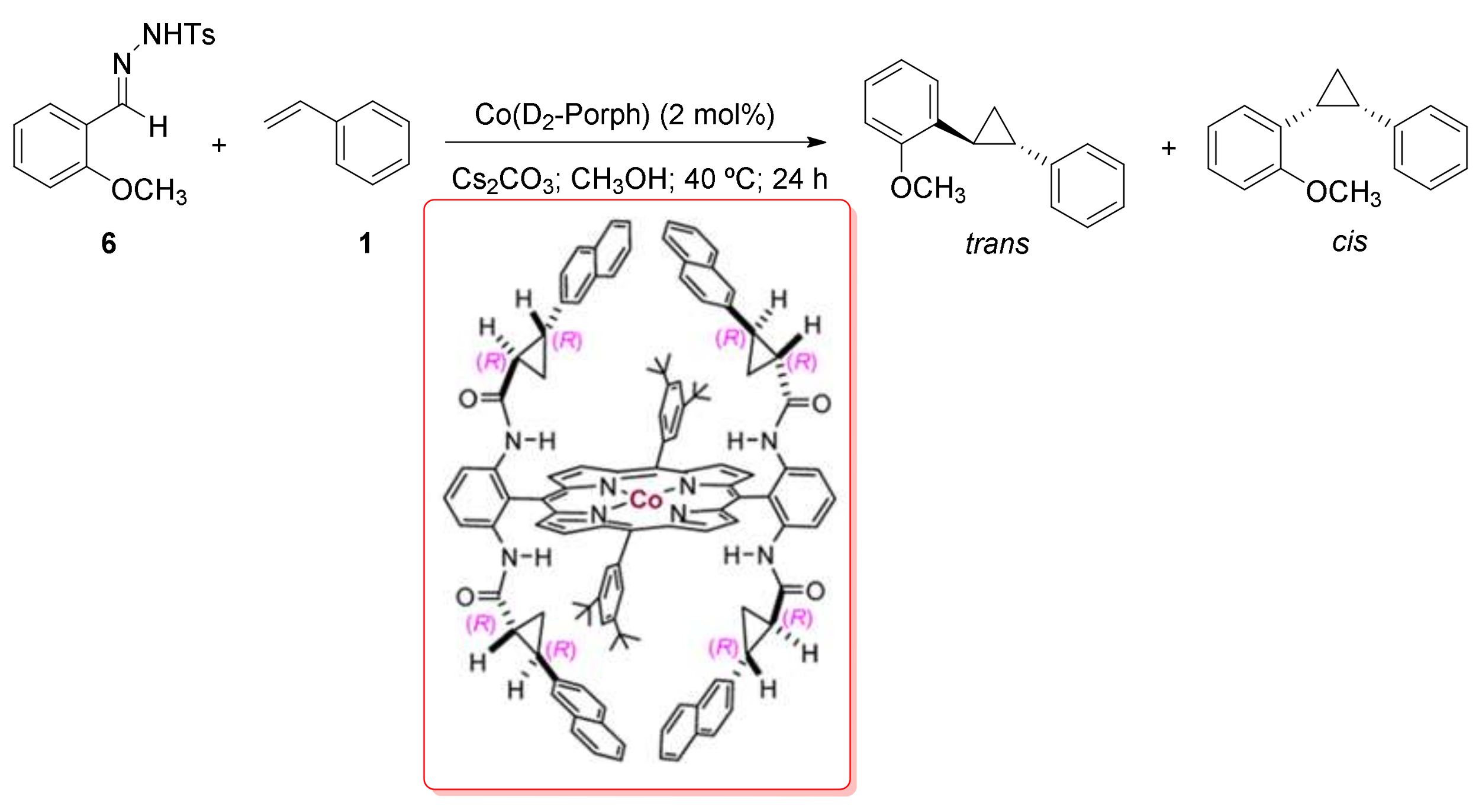
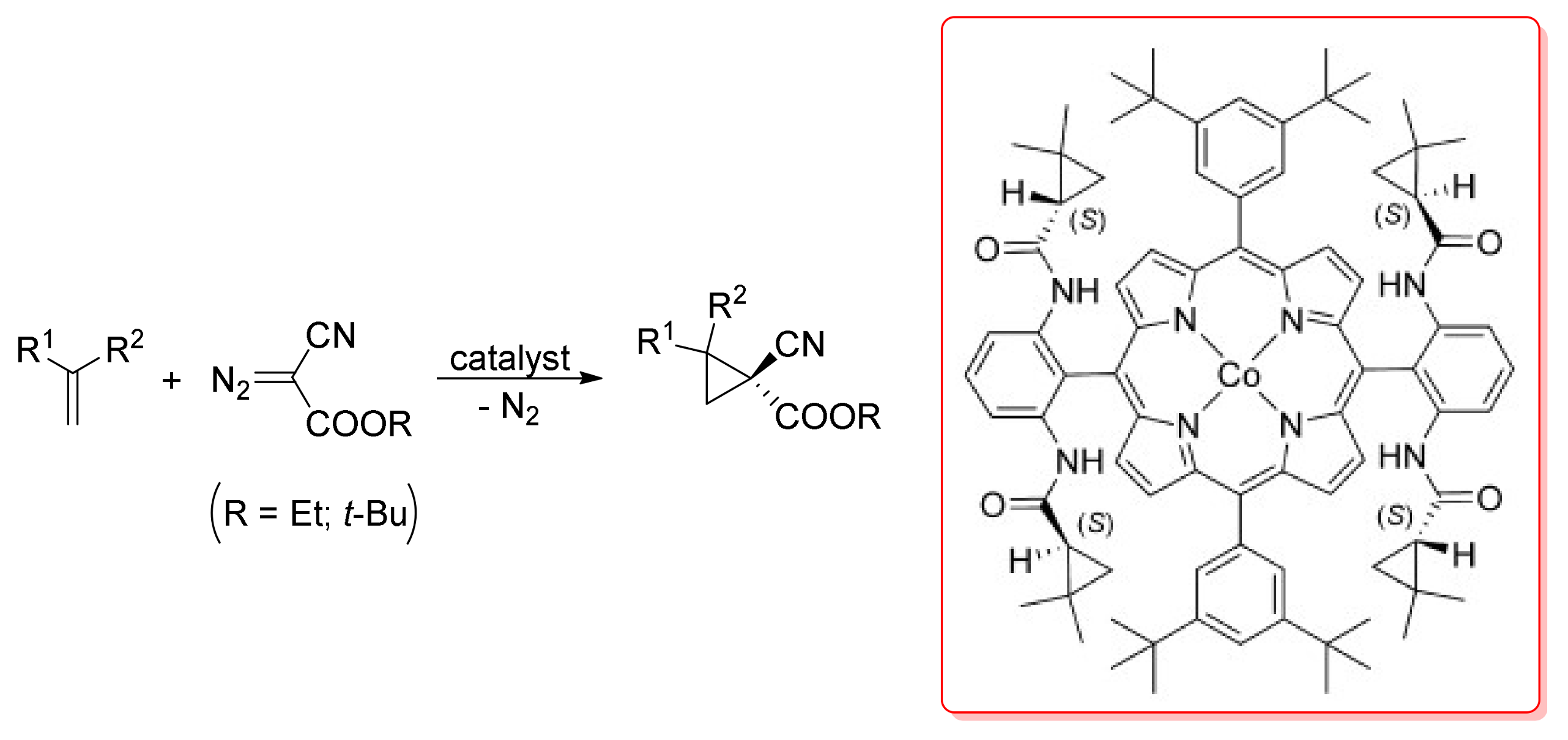
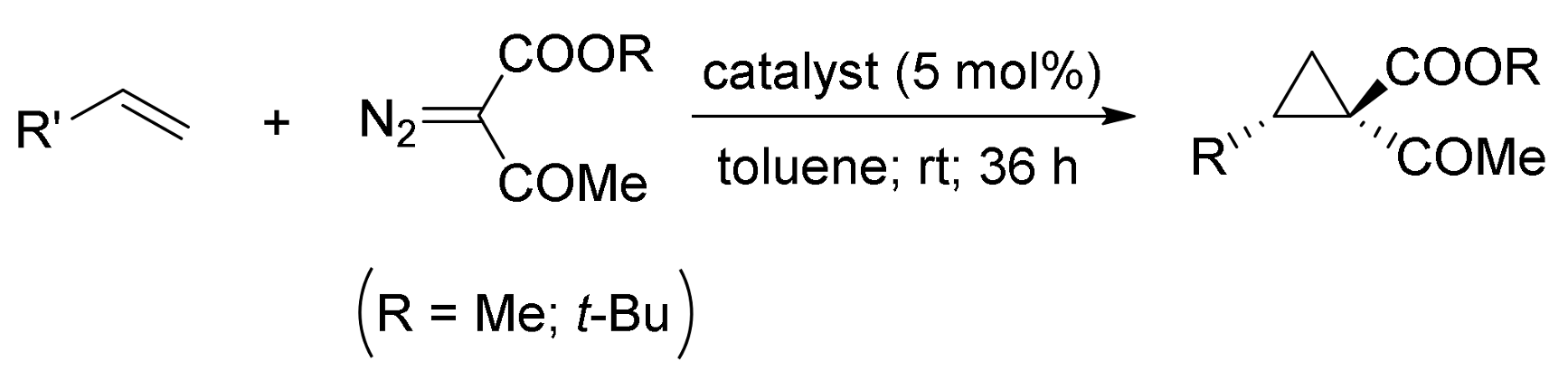
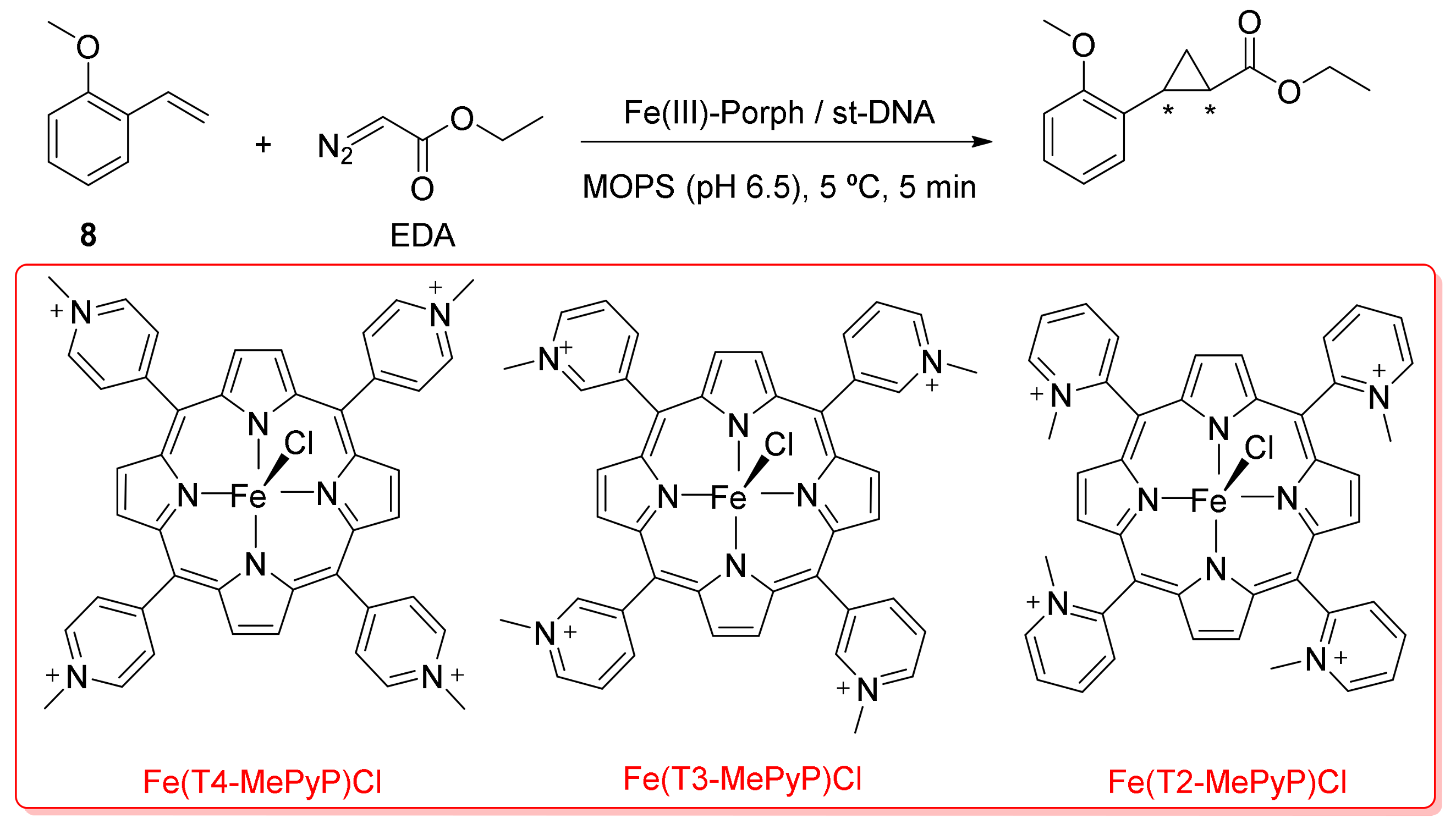
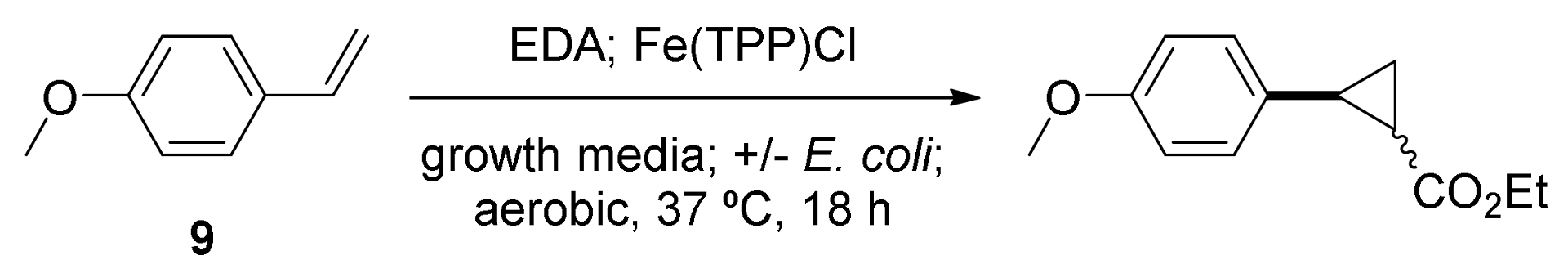
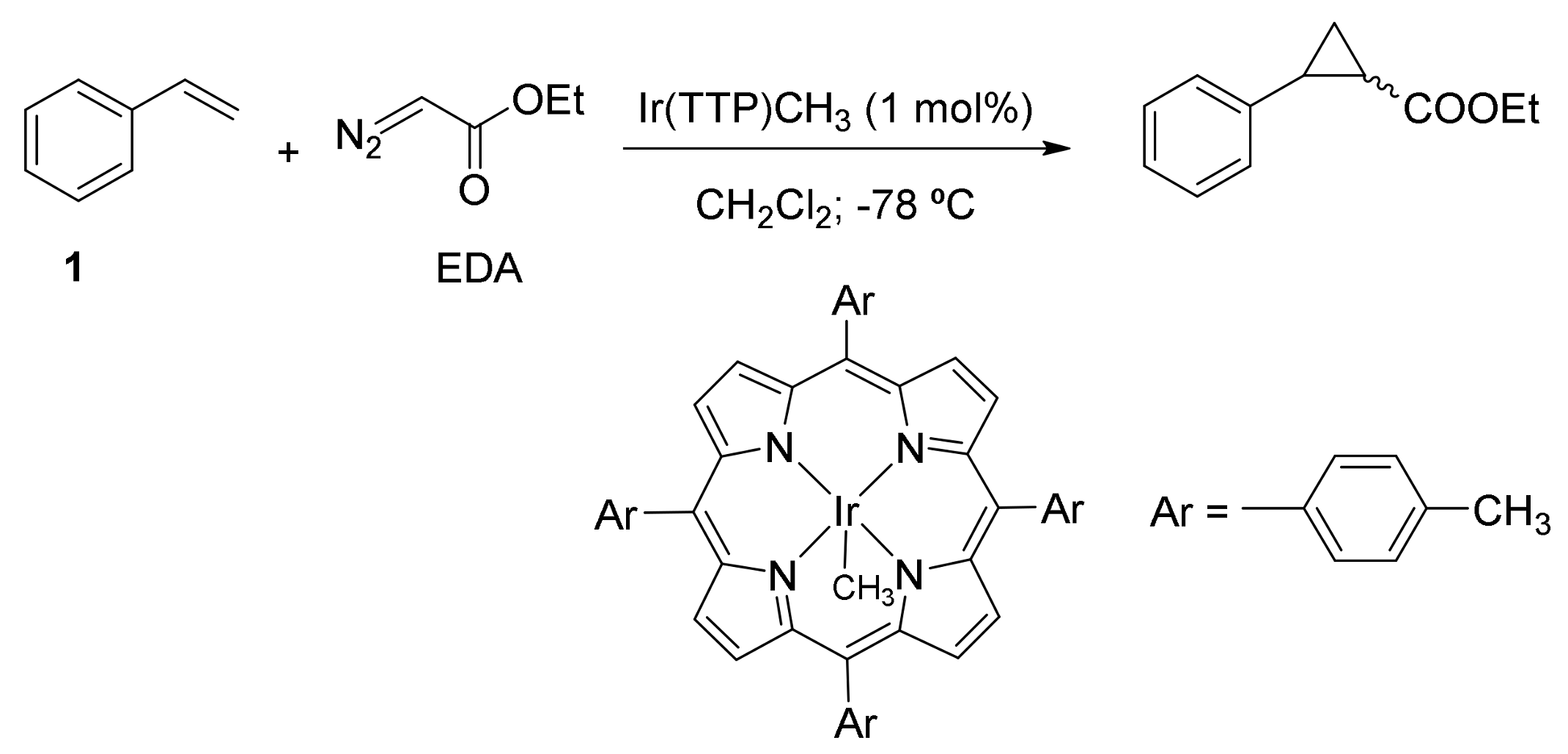
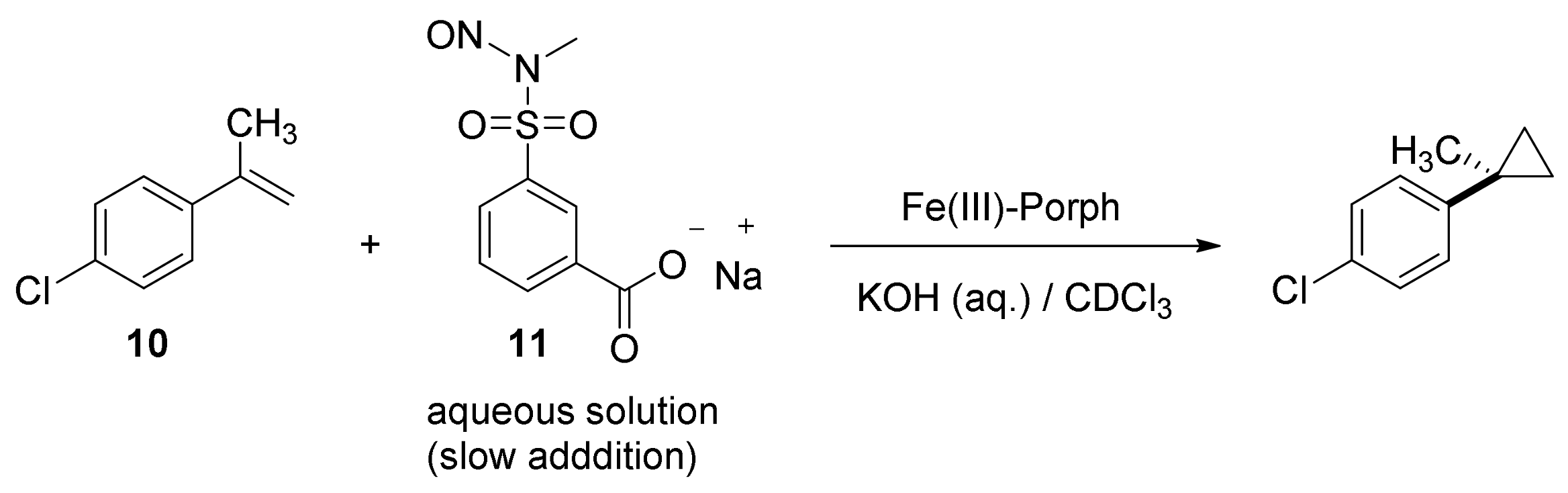
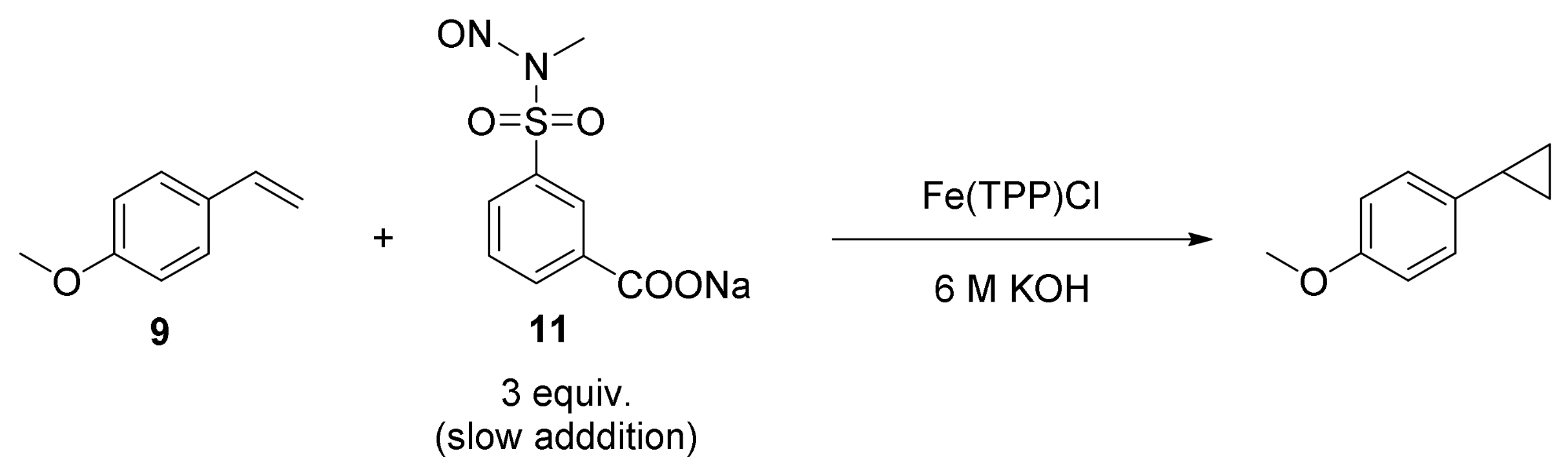
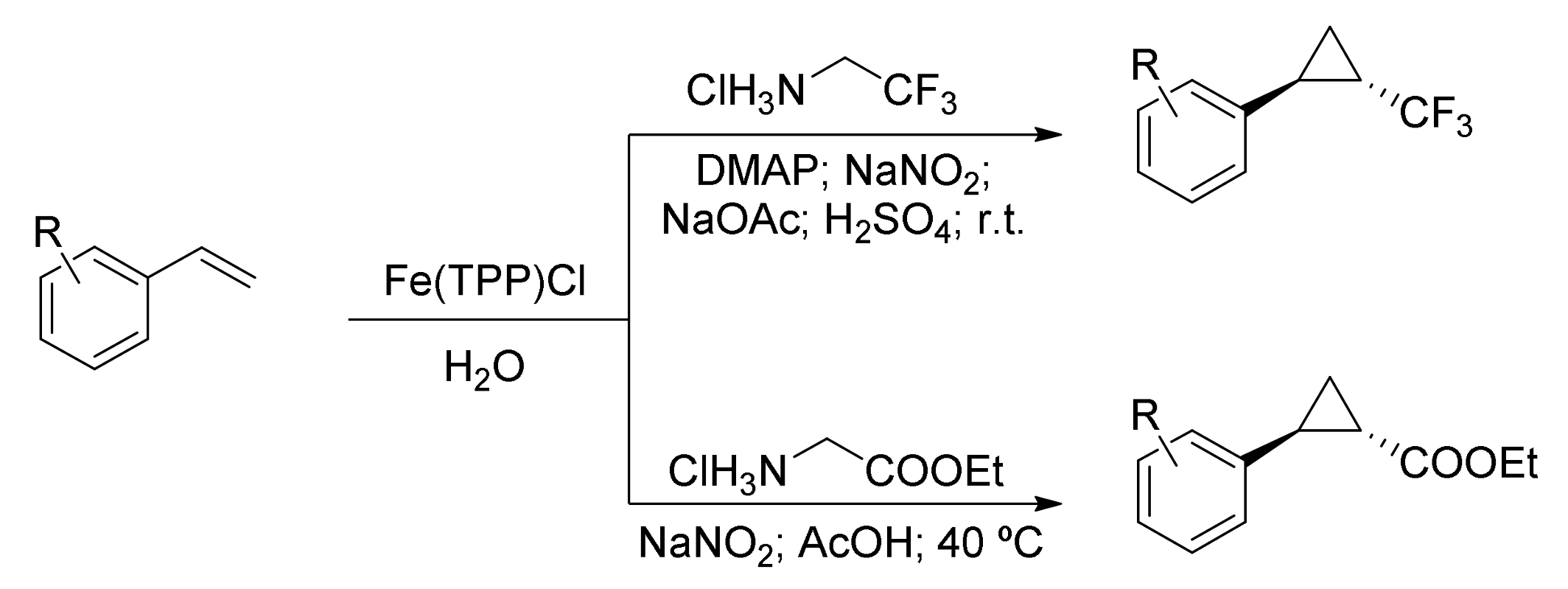
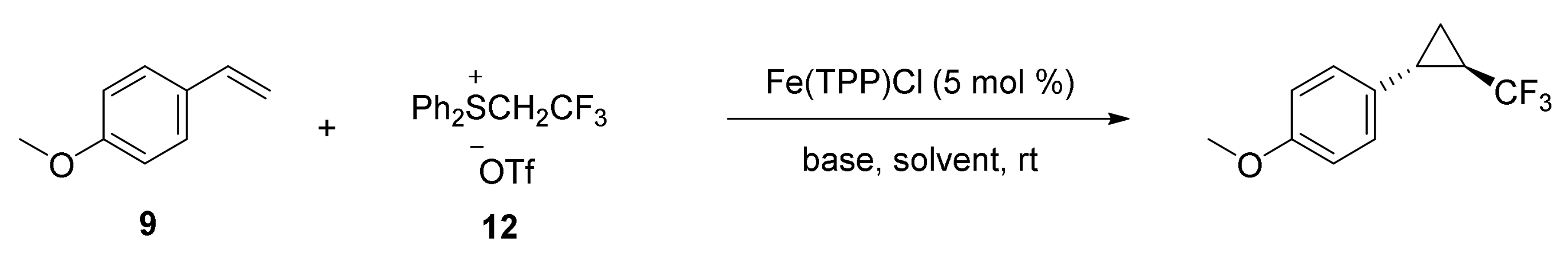
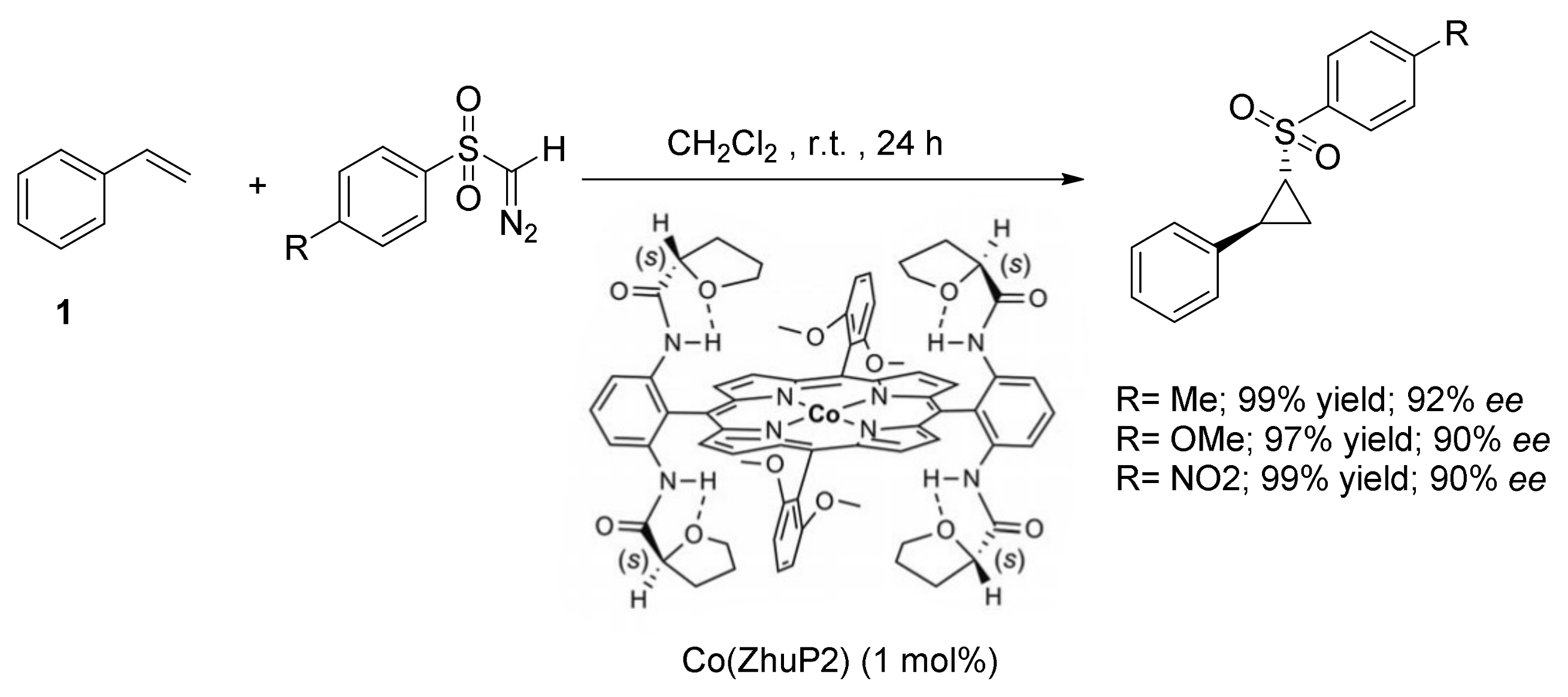
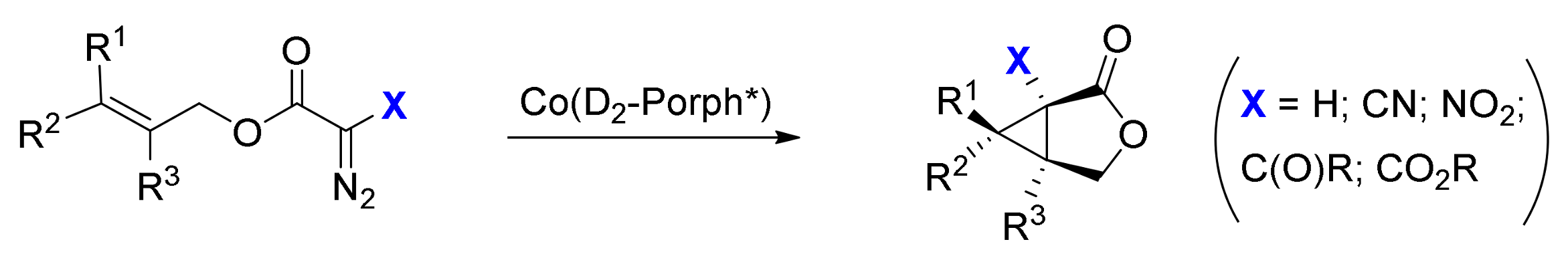
2. Cyclopropanation Reactions
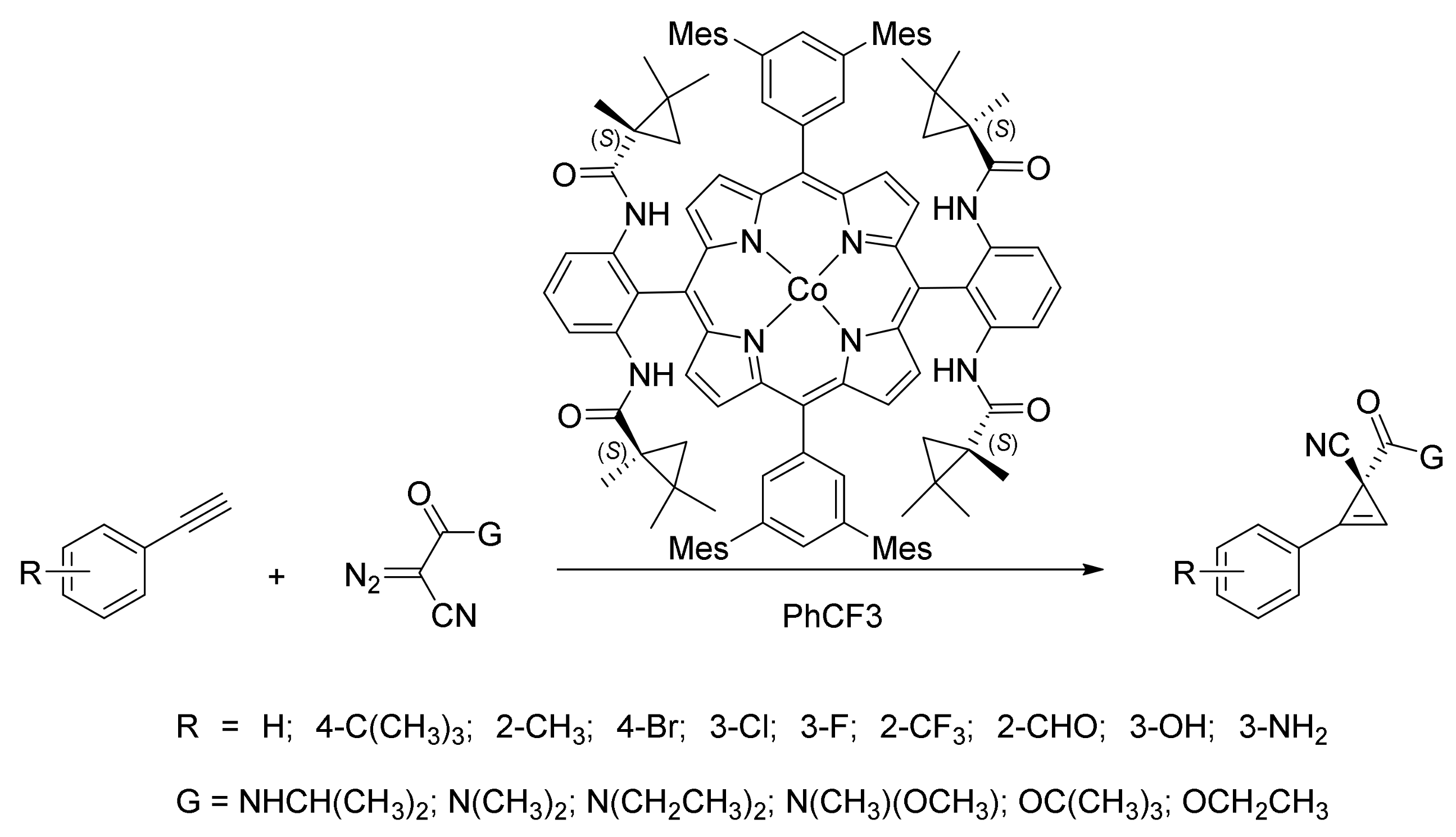
3. Cyclopropenation Reactions
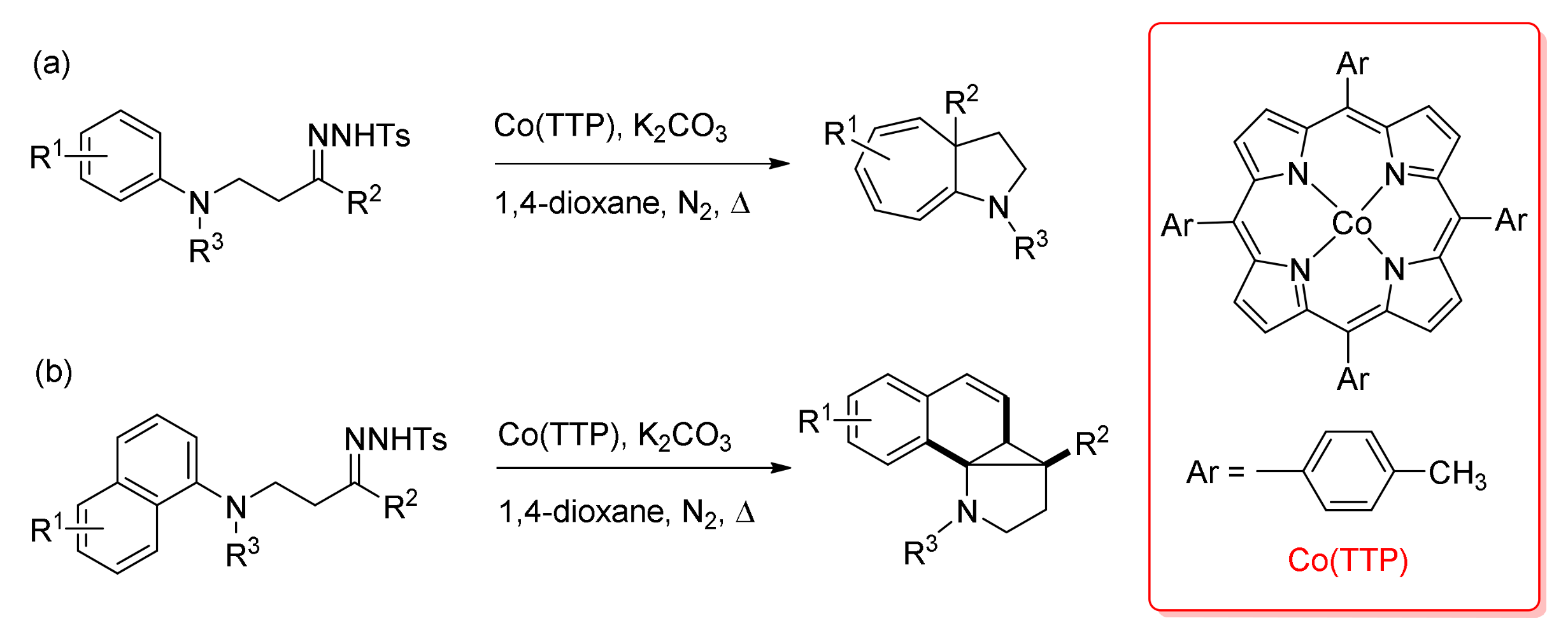
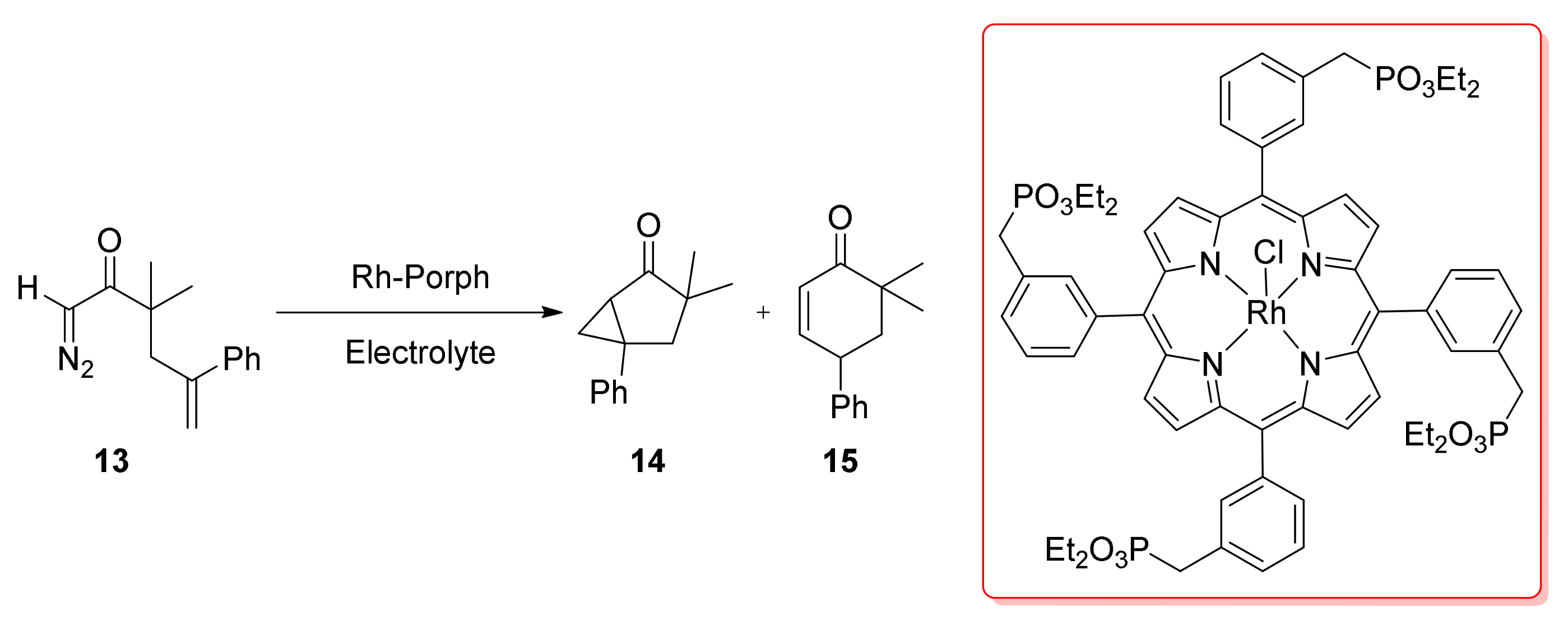
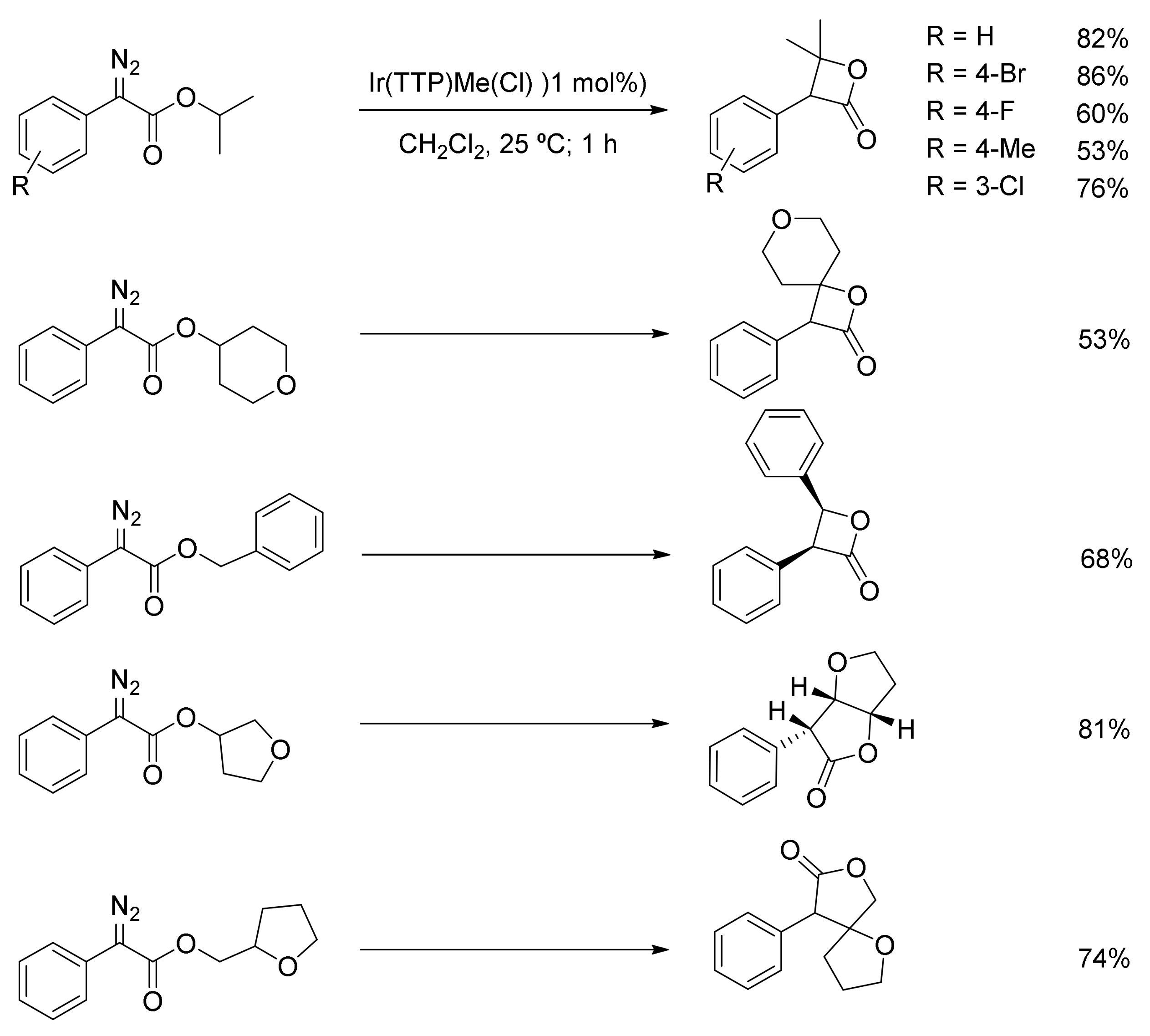
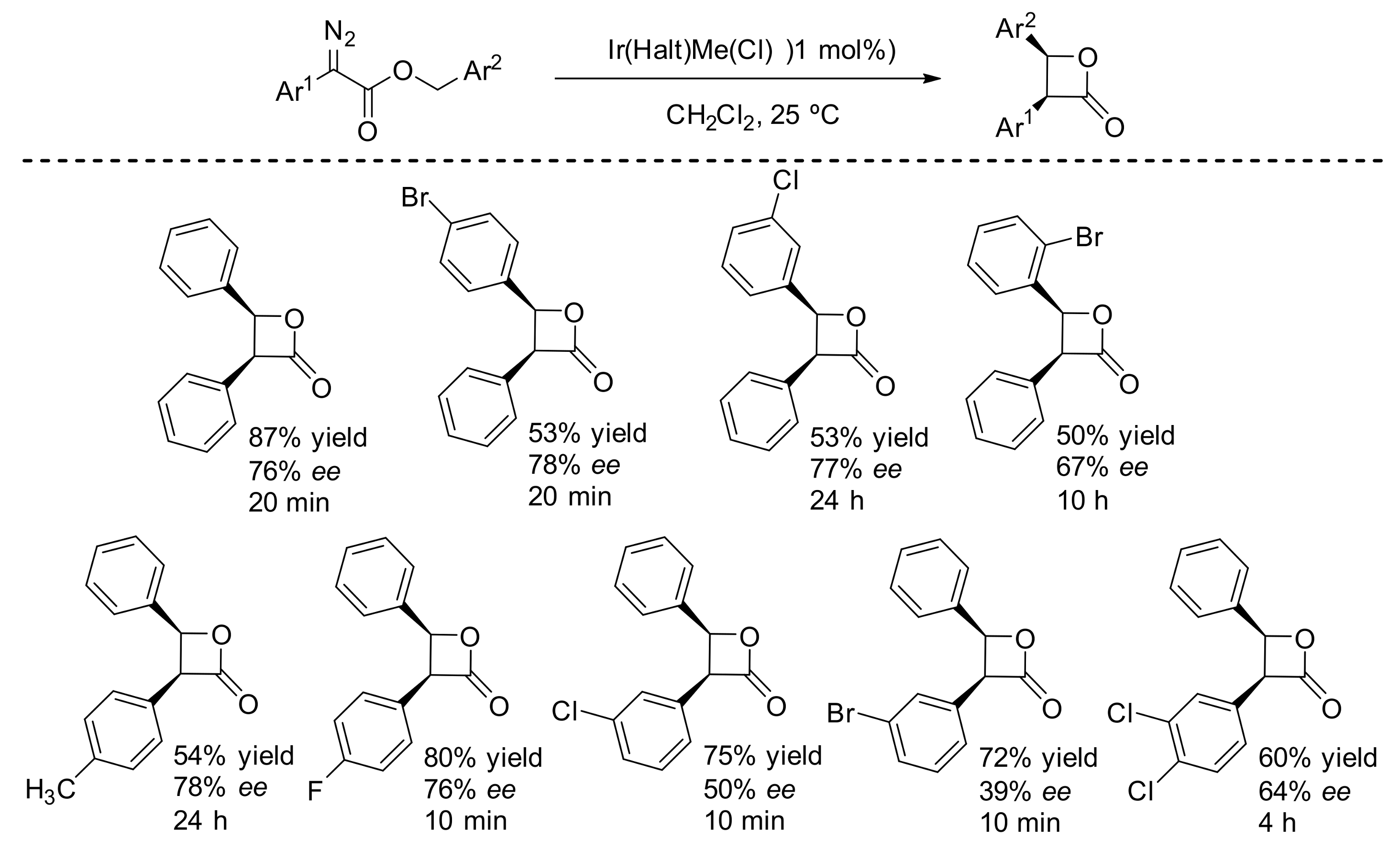
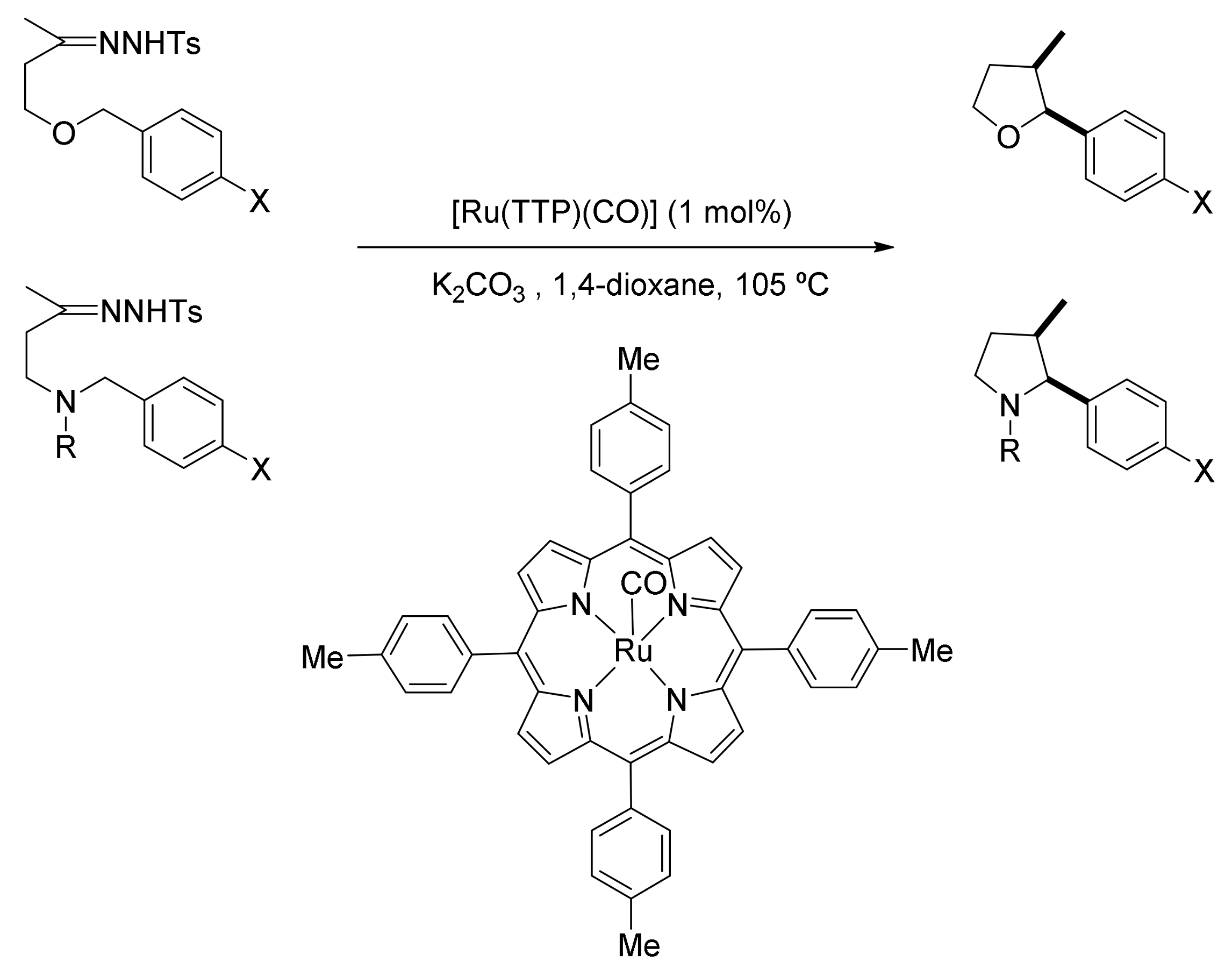
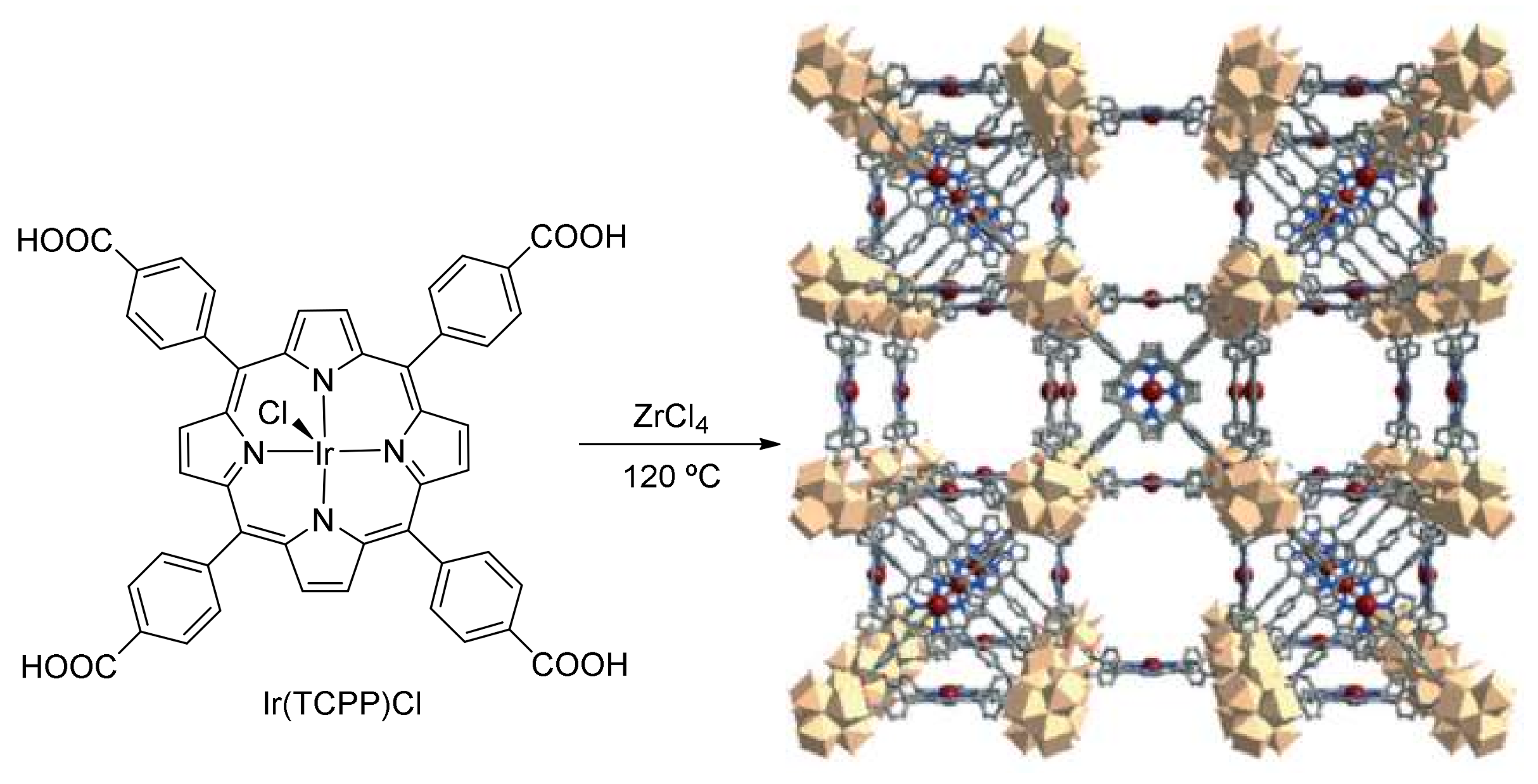
4. Carbene Insertion into C(sp3)-H
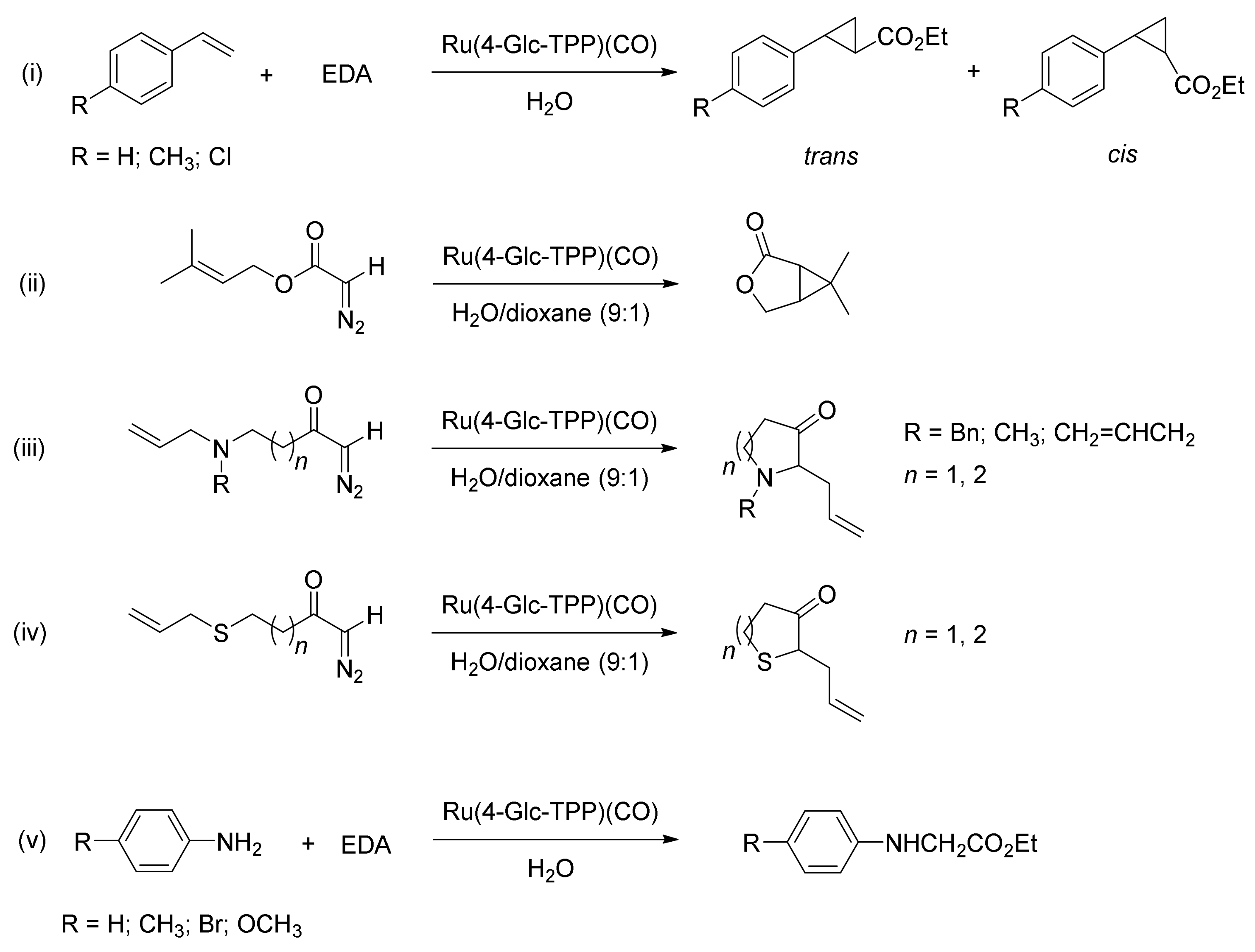
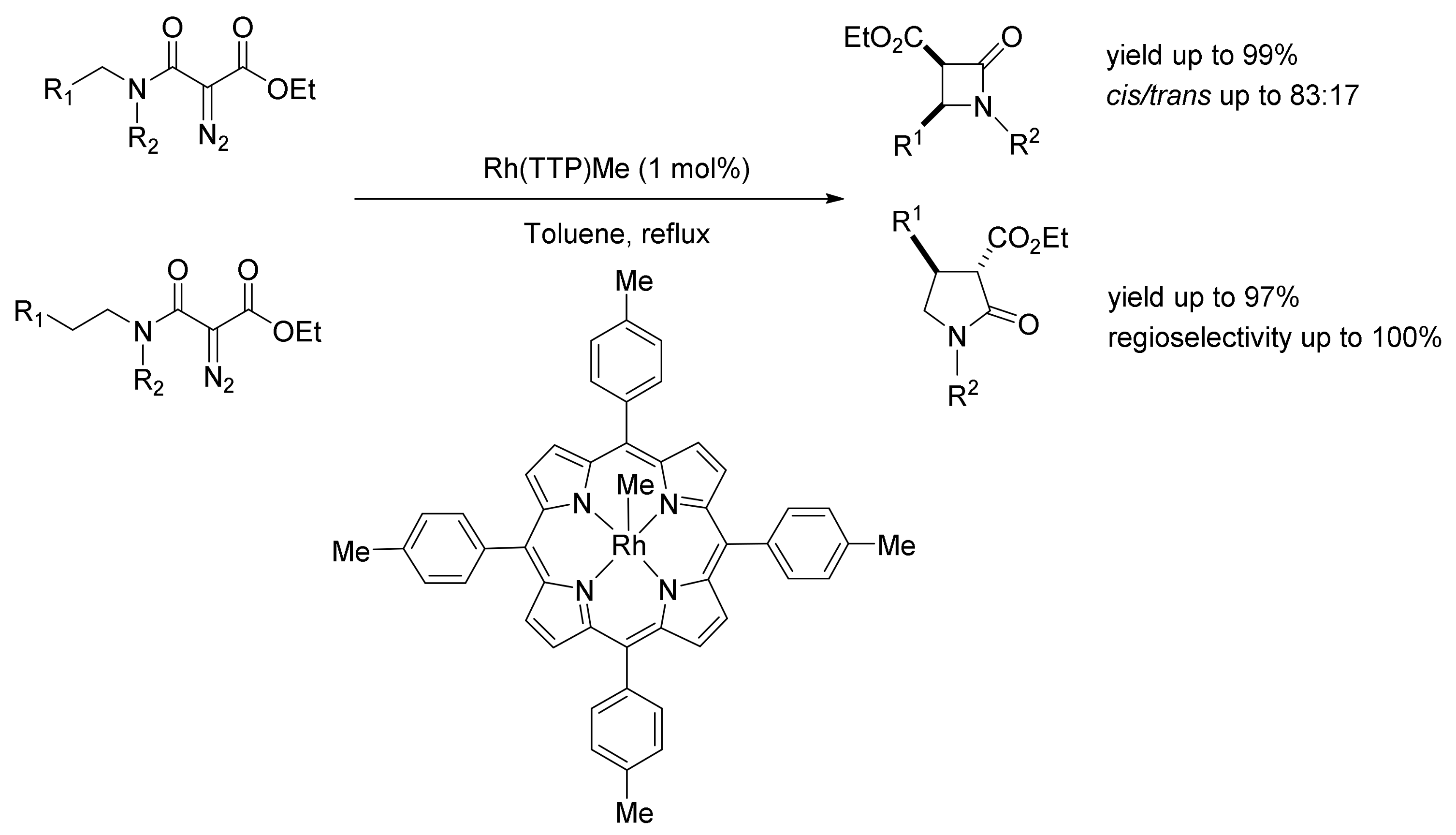
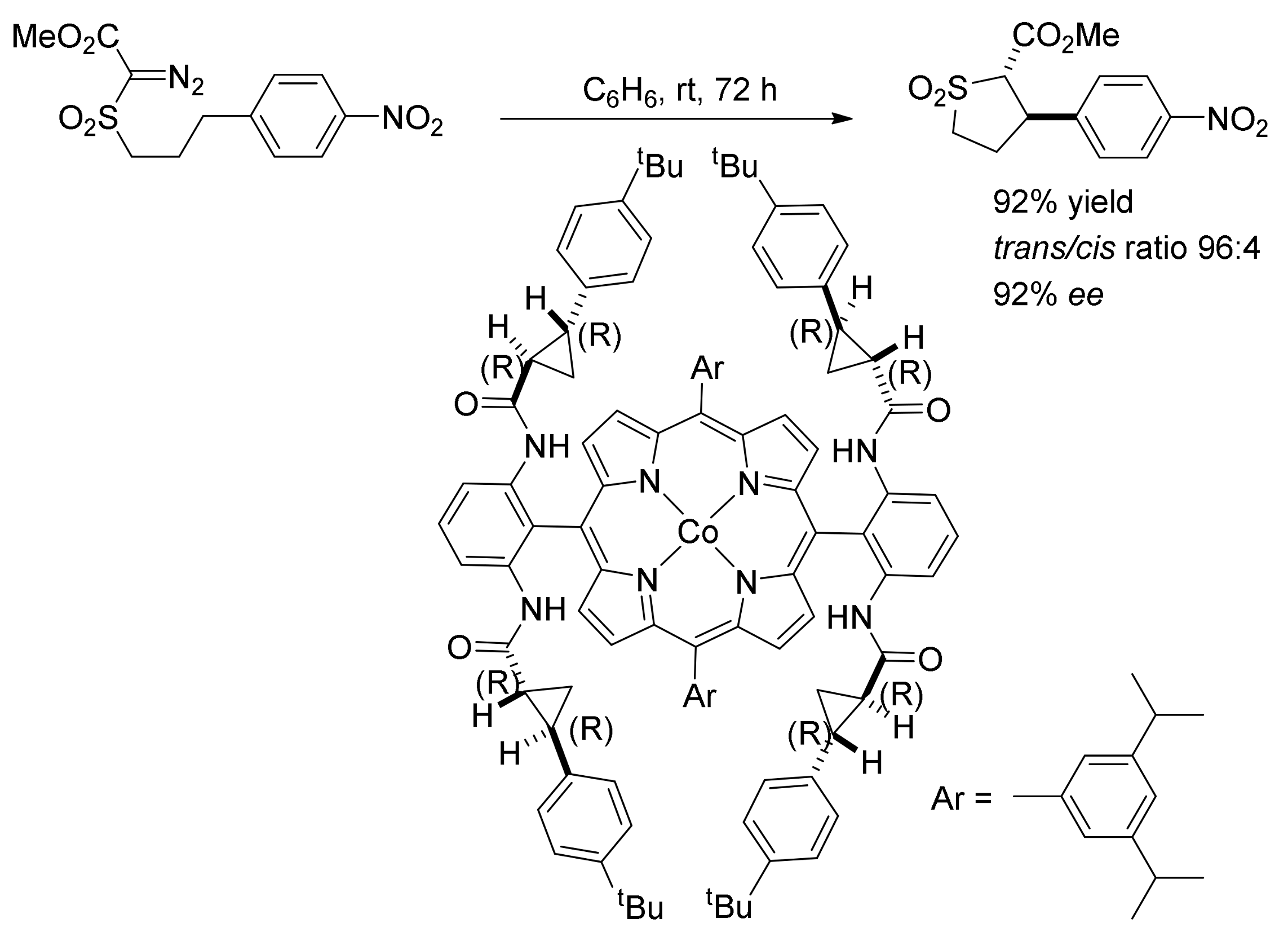
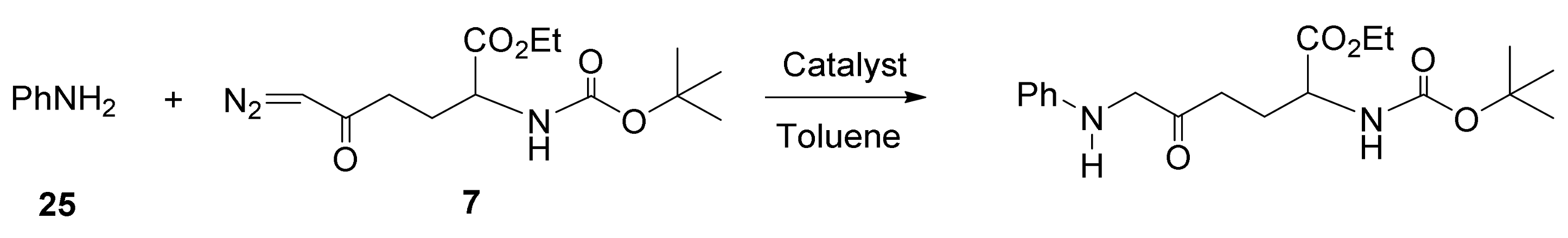
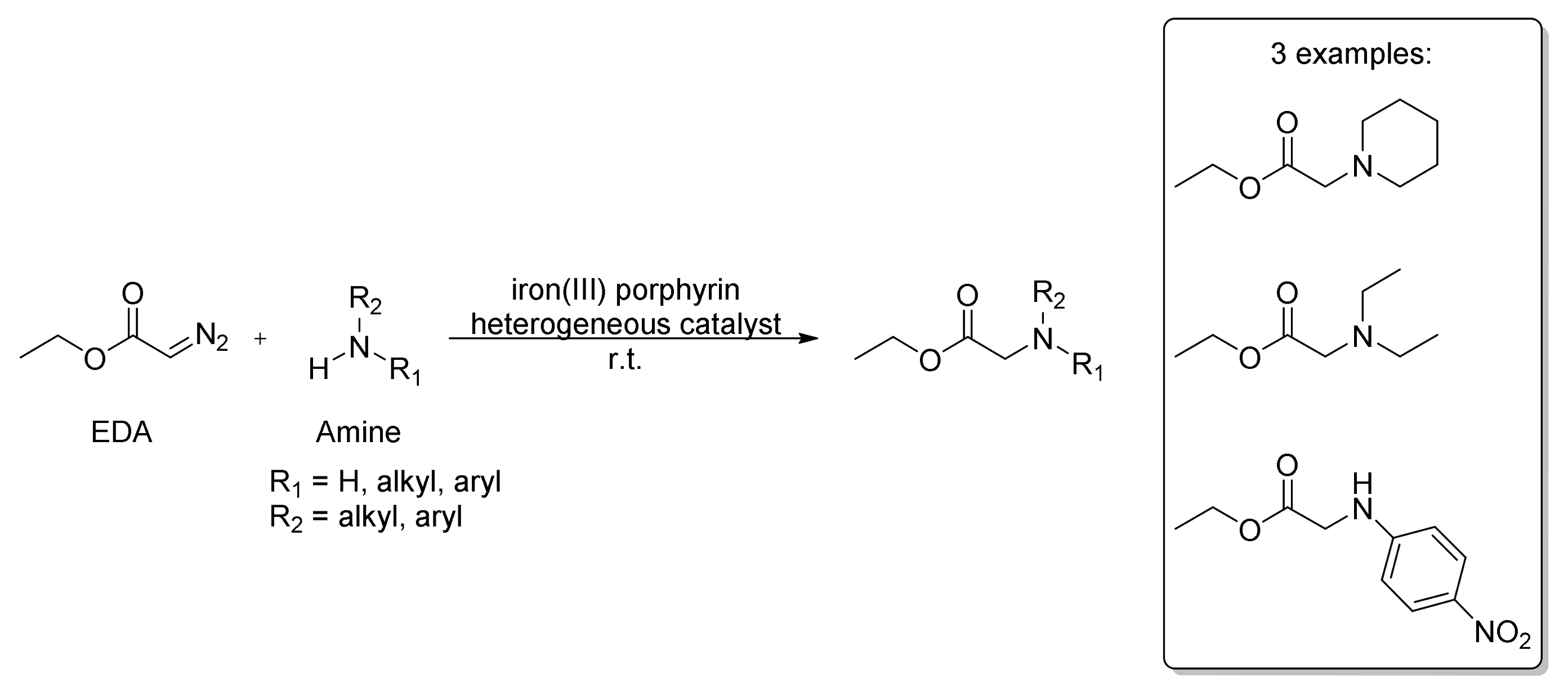
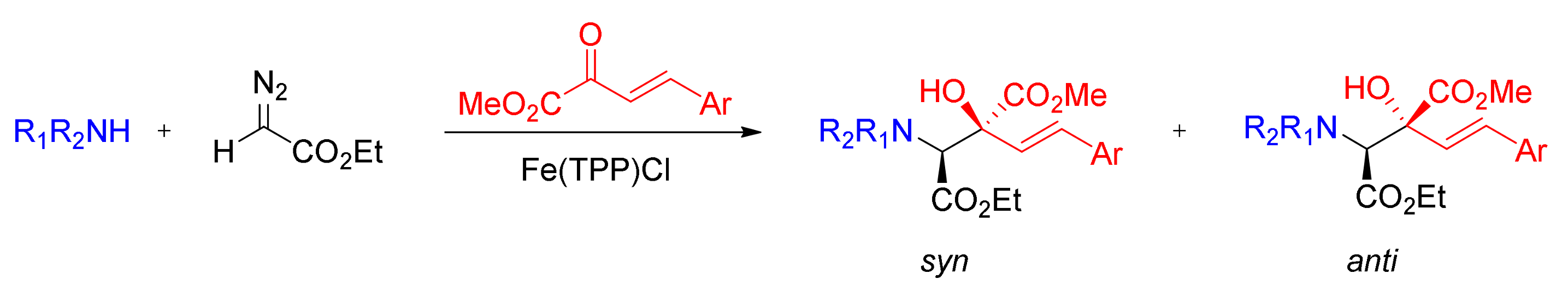
5. Carbene Insertion into N-H
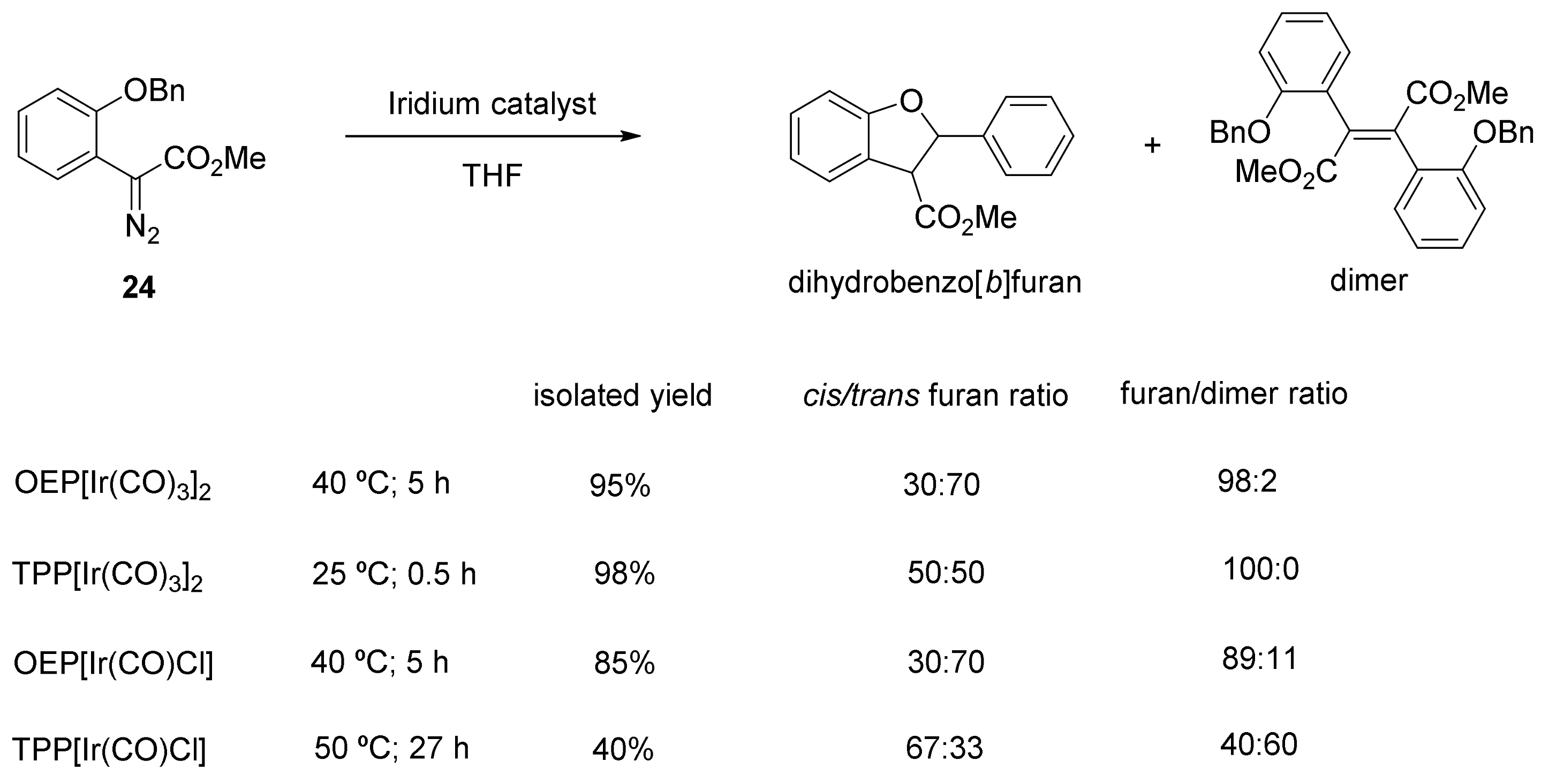
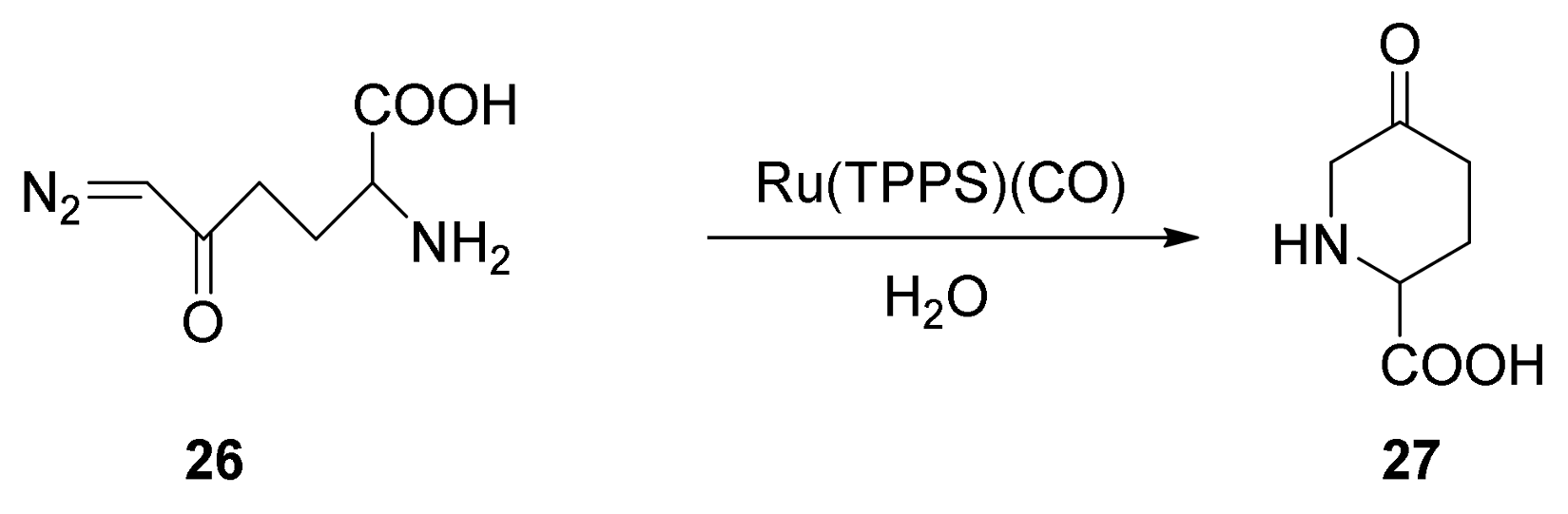
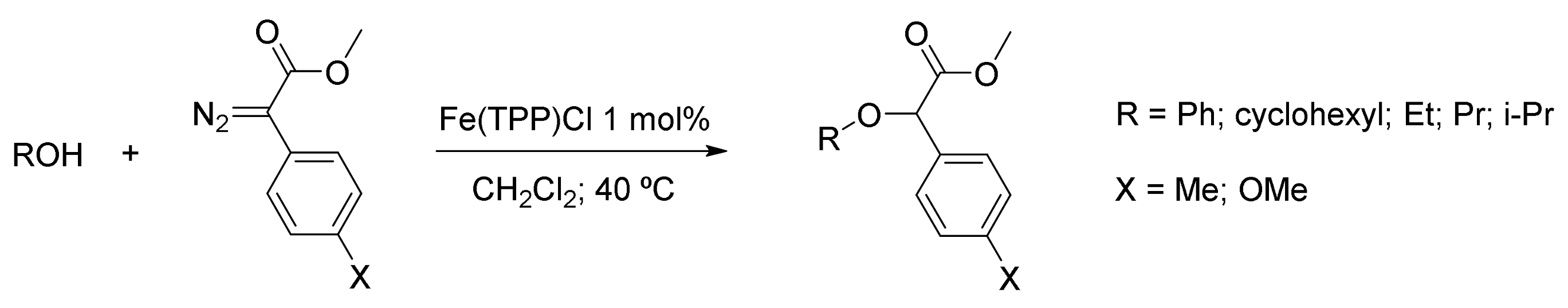
6. Carbene Insertion into O-H
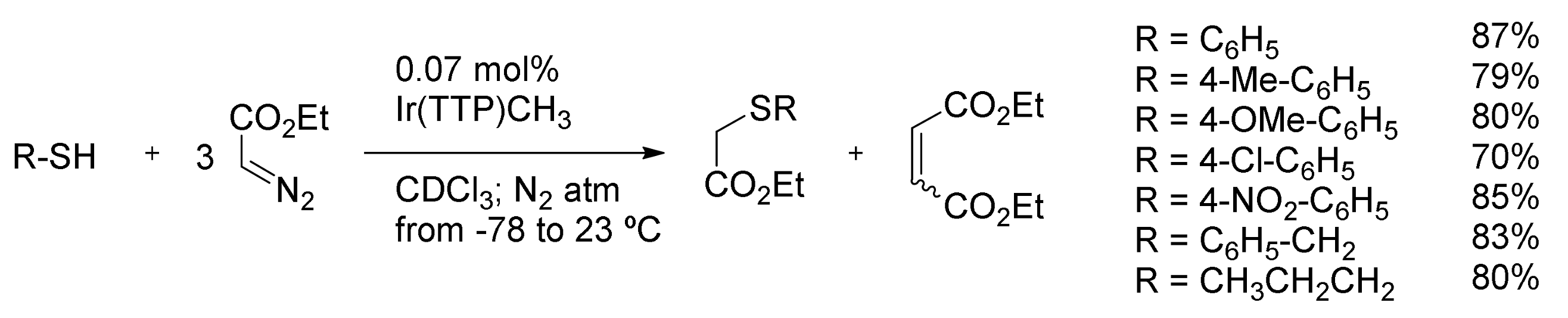
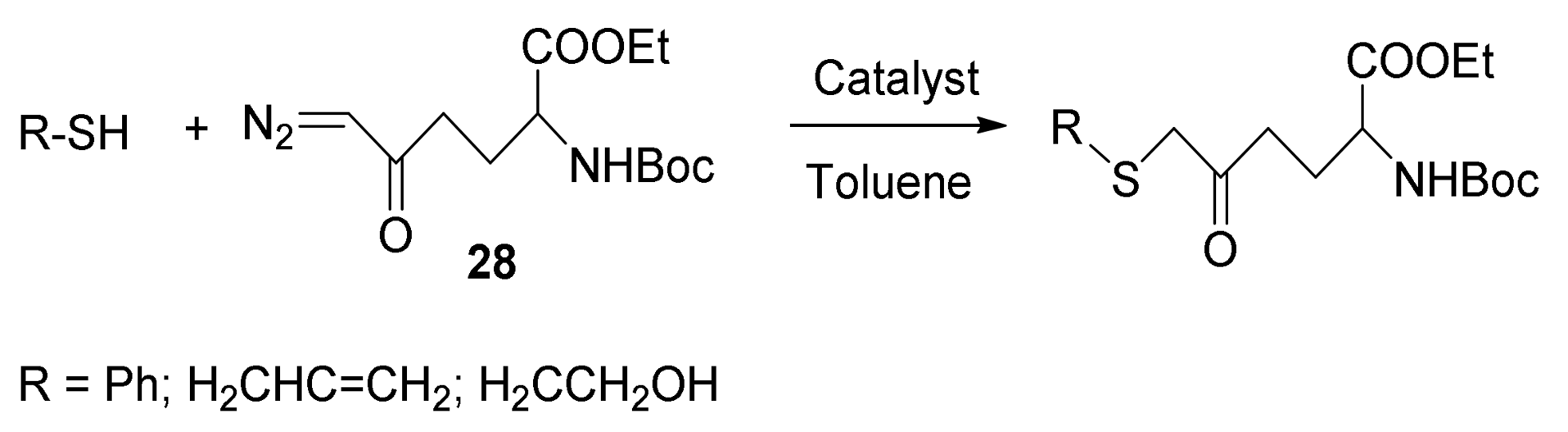
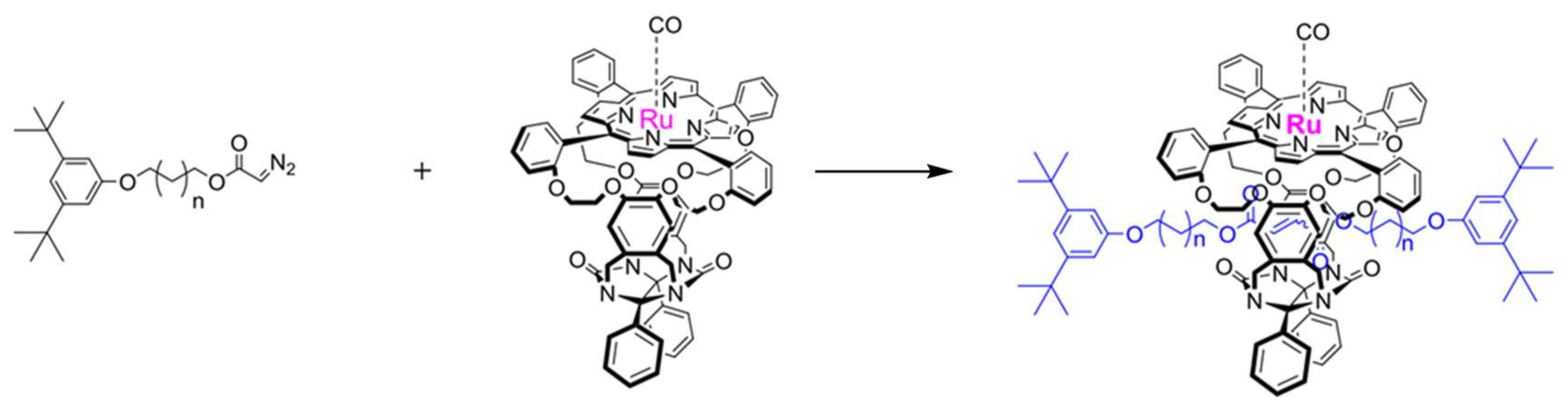
7. Carbene Insertion into S-H
8. Carbene Insertion into Si-H
9. Alkene Formation Reactions
9.1. Coupling of Diazo Compounds
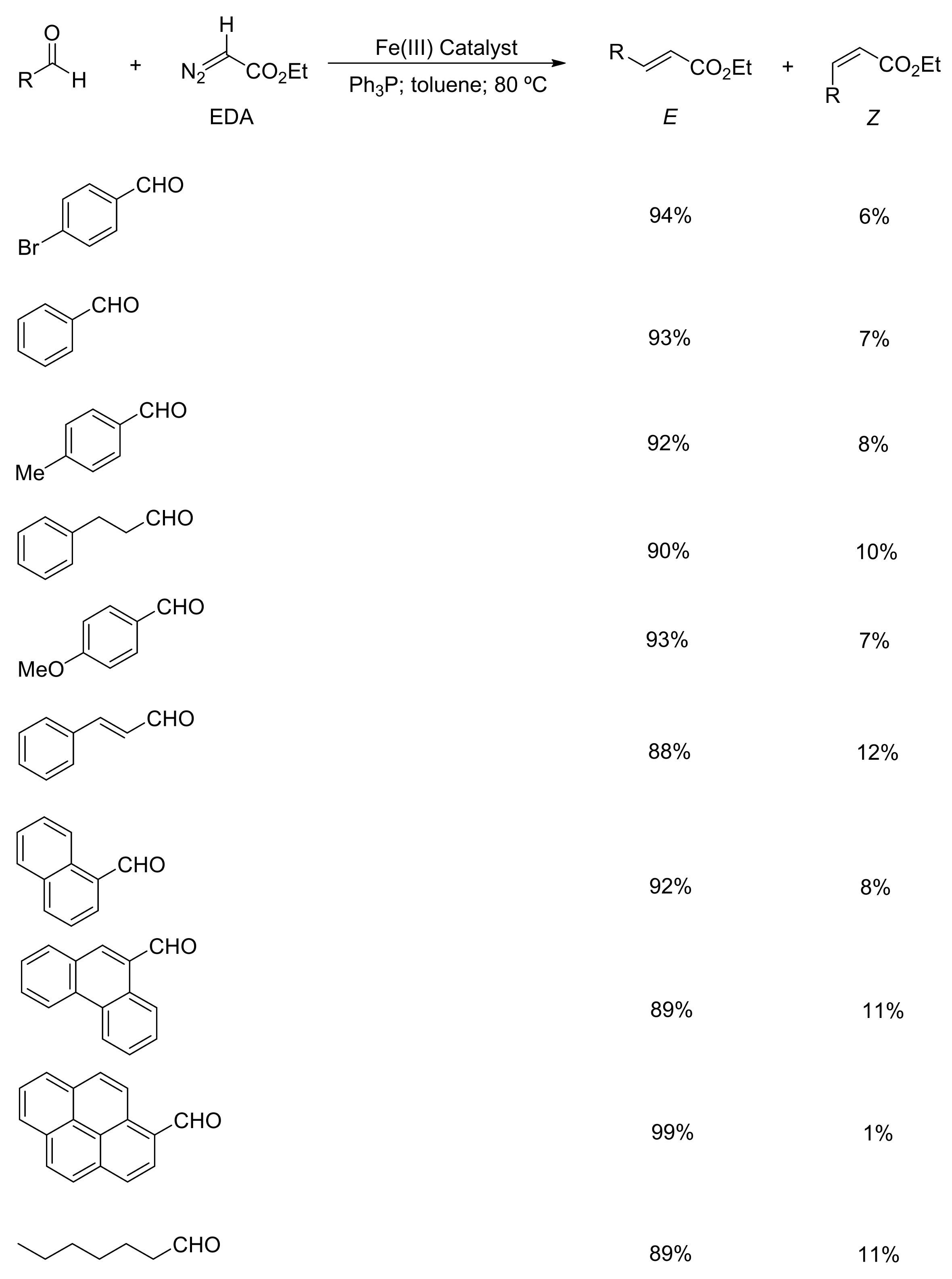
9.2. Olefination of Carbonyl Compounds
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; The “Gold Book”; Blackwell Scientific Publications: Oxford, UK, 2014; Available online: https://goldbook.iupac.org/html/C/C00806.html (accessed on 29 March 2018).
- Chakraborty, J.; Nath, I.; Verpoort, F. Snapshots of encapsulated porphyrins and heme enzymes in metal-organic materials: A prevailing paradigm of heme mimicry. Coord. Chem. Rev. 2016, 326, 135–163. [Google Scholar] [CrossRef]
- Key, H.M.; Dydio, P.; Liu, Z.; Rha, J.Y.-E.; Nazarenko, A.; Seyedkazemi, V.; Clark, D.S.; Hartwig, J.F. Beyond Iron: Iridium-Containing P450 Enzymes for Selective Cyclopropanations of Structurally Diverse Alkenes. ACS Cent. Sci. 2017, 3, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Dydio, P.; Key, H.M.; Nazarenko, A.; Rha, J.Y.-E.; Seyedkazemi, V.; Clark, D.S.; Hartwig, J.F. An artificial metalloenzyme with the kinetics of native enzymes. Science 2016, 354, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Gober, J.G.; Rydeen, A.E.; Gibson-O’Grady, E.J.; Leuthaeuser, J.B.; Fetrow, J.S.; Brustad, E.M. Mutating a Highly Conserved Residue in Diverse Cytochrome P450s Facilitates Diastereoselective Olefin Cyclopropanation. ChemBioChem 2016, 17, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Renata, H.; Lewis, R.D.; Sweredoski, M.J.; Moradian, A.; Hess, S.; Wang, Z.J.; Arnold, F.H. Identification of Mechanism-Based Inactivation in P450-Catalyzed Cyclopropanation Facilitates Engineering of Improved Enzymes. J. Am. Chem. Soc. 2016, 138, 12527–12533. [Google Scholar] [CrossRef] [PubMed]
- Hyster, T.K.; Arnold, F.H. P450BM3-axial mutations: A gateway to non-natural reactivity. Isr. J. Chem. 2015, 55, 14–20. [Google Scholar] [CrossRef]
- Wang, Z.J.; Peck, N.E.; Renata, H.; Arnold, F.H. Cytochrome P450-catalyzed insertion of carbenoids into N-H bonds. Chem. Sci. 2014, 5, 598–601. [Google Scholar] [CrossRef] [PubMed]
- Coelho, P.S.; Brustad, E.M.; Kannan, A.; Arnold, F.H. Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science 2013, 339, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Roiban, G.-D.; Reetz, M.T. Enzyme Promiscuity: Using a P450 Enzyme as a Carbene Transfer Catalyst. Angew. Chem. Int. Ed. 2013, 52, 5439–5440. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.; Davey, S.; Johnson, R.; Pichon, A. Promiscuous by nature. Nat. Chem. 2013, 5, 151. [Google Scholar]
- Narayan, A.R.H.; Sherman, D.H. Re-Engineering Nature’s Catalysts. Science 2013, 339, 283–284. [Google Scholar] [CrossRef] [PubMed]
- Coelho, P.S.; Wang, Z.J.; Ener, M.E.; Baril, S.A.; Kannan, A.; Arnold, F.H.; Brustad, E.M. A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo. Nat. Chem. Biol. 2013, 9, 485–487. [Google Scholar] [CrossRef] [PubMed]
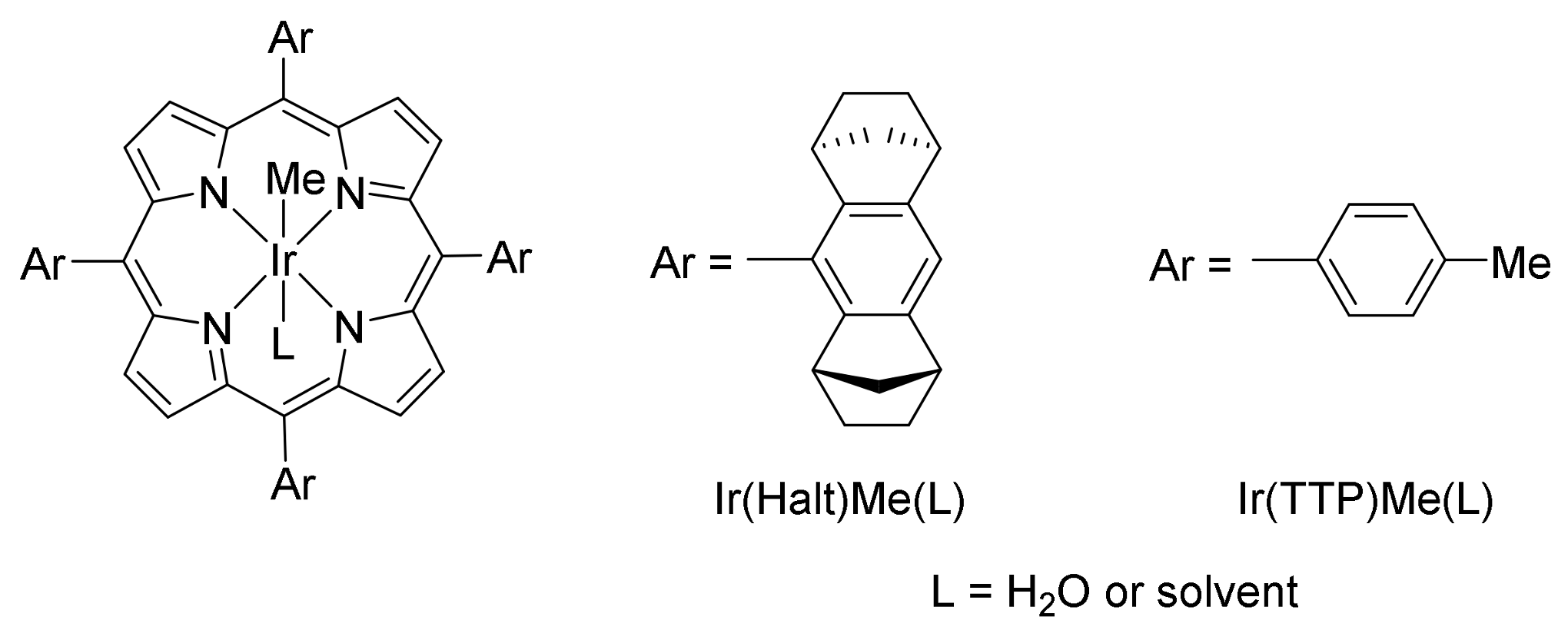
- Key, H.M.; Dydio, P.; Clark, D.S.; Hartwig, J.F. Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Sreenilayam, G.; Fasan, R. Myoglobin-catalyzed intermolecular carbene N-H insertion with arylamine substrates. Chem. Commun. 2015, 51, 1532–1534. [Google Scholar] [CrossRef] [PubMed]
- Bordeaux, M.; Tyagi, V.; Fasan, R. Highly Diastereoselective and Enantioselective Olefin Cyclopropanation Using Engineered Myoglobin-Based Catalysts. Angew. Chem. Int. Ed. 2015, 54, 1744–1748. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, V.; Bonn, R.B.; Fasan, R. Intermolecular carbene S-H insertion catalysed by engineered myoglobin-based catalysts. Chem. Sci. 2015, 6, 2488–2494. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, P.; Sreenilayam, G.; Tyagi, V.; Fasan, R. Gram-Scale Synthesis of Chiral Cyclopropane-Containing Drugs and Drug Precursors with Engineered Myoglobin Catalysts Featuring Complementary Stereoselectivity. Angew. Chem. Int. Ed. 2016, 55, 16110–16114. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, V.; Fasan, R. Myoglobin-Catalyzed Olefination of Aldehydes. Angew. Chem. Int. Ed. 2016, 55, 2512–2516. [Google Scholar] [CrossRef] [PubMed]
- Sreenilayam, G.; Moore, E.J.; Steck, V.; Fasan, R. Metal Substitution Modulates the Reactivity and Extends the Reaction Scope of Myoglobin Carbene Transfer Catalysts. Adv. Synth. Catal. 2017, 359, 2076–2089. [Google Scholar] [CrossRef]
- Tinoco, A.; Steck, V.; Tyagi, V.; Fasan, R. Highly Diastereo- and Enantioselective Synthesis of TrifluoromethylSubstituted Cyclopropanes via Myoglobin-Catalyzed Transfer of Trifluoromethylcarbene. J. Am. Chem. Soc. 2017, 139, 5293–5296. [Google Scholar] [CrossRef] [PubMed]
- Sreenilayam, G.; Moore, E.J.; Steck, V.; Fasan, R. Stereoselective Olefin Cyclopropanation under Aerobic Conditions with an Artificial Enzyme Incorporating an Iron-Chlorin e6 Cofactor. ACS Catal. 2017, 7, 7629–7633. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, C.; Tao, Z.; Pan, Y. Hemin-Catalyzed, Cyclodextrin-Assisted Insertion of Carbenoids into N-H Bonds. Adv. Synth. Catal. 2015, 357, 3341–3345. [Google Scholar] [CrossRef]
- Xu, X.; Li, C.; Xiong, M.; Tao, Z.; Pan, Y. Hemin-catalyzed sulfonium ylide formation and subsequently reactant-controlled chemoselective rearrangements. Chem. Commun. 2017, 53, 6219–6222. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.W.; Vargas, D.A.; Lehnert, N. Engineering of RuMb: Toward a Green Catalyst for Carbene Insertion Reactions. Inorg. Chem. 2017, 56, 5623–5635. [Google Scholar] [CrossRef] [PubMed]
- Oohora, K.; Meichin, H.; Zhao, L.; Wolf, M.W.; Nakayama, A.; Hasegawa, J.; Lehnert, N.; Hayashi, T. Catalytic Cyclopropanation by Myoglobin Reconstituted with Iron Porphycene: Acceleration of Catalysis due to Rapid Formation of the Carbene Species. J. Am. Chem. Soc. 2017, 139, 17265–17268. [Google Scholar] [CrossRef] [PubMed]
- Weissenborn, M.J.; Löw, S.A.; Borlinghaus, N.; Kuhn, M.; Kummer, S.; Rami, F.; Plietker, B.; Hauer, B. Enzyme-Catalyzed Carbonyl Olefination by the E. coli Protein YfeX in the Absence of Phosphines. ChemCatChem 2016, 8, 1636–1640. [Google Scholar] [CrossRef]
- Rybicka-Jasinska, K.; Ciszewskia, L.W.; Gryko, D.T.; Gryko, D. C-C bond forming reactions catalyzed by chiral metalloporphyrins. J. Porphyr. Phthalocyanines 2016, 20, 76–95. [Google Scholar] [CrossRef]
- Intrieri, D.; Carminati, D.M.; Gallo, E. The Ligand Influence in Stereoselective Carbene Transfer Reactions Promoted by Chiral Metal Porphyrin Catalysts. Dalton Trans. 2016, 45, 15746–15761. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.; Miel, H.; Ring, A.; Slattery, C.N.; Maguire, A.R.; McKervey, M.A. Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev. 2015, 115, 9981–10080. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhou, Q. Iron-catalyzed transformations of diazo compounds. Natl. Sci. Rev. 2014, 1, 580–603. [Google Scholar] [CrossRef]
- Intrieri, D.; Caselli, A.; Gallo, E. Cyclopropanation Reactions Mediated by Group 9 Metal Porphyrin Complexes. Eur. J. Inorg. Chem. 2011, 5071–5081. [Google Scholar] [CrossRef]
- Che, C.-M.; Lo, V.K.-Y.; Zhou, C.-Y.; Huang, J.-S. Selective functionalisation of saturated C-H bonds with metalloporphyrin catalysts. Chem. Soc. Rev. 2011, 40, 1950–1975. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhang, X.P. Catalytic C-H functionalization by metalloporphyrins: Recent developments and future directions. Chem. Soc. Rev. 2011, 40, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Che, C.-M.; Zhou, C.; Wong, E.L. Catalysis by Fe = X Complexes (X = NR, CR2). Top. Organomet. Chem. 2011, 33, 111–138. [Google Scholar]
- Zhou, C.-Y.; Huang, J.-S.; Che, C.-M. Ruthenium-Porphyrin-Catalyzed Carbenoid Transfer Reactions. Synlett 2010, 18, 2681–2700. [Google Scholar]
- Simonneaux, G. Optically active ruthenium porphyrins: Chiral recognition and asymmetric catalysis. Coord. Chem. Rev. 2002, 228, 43–60. [Google Scholar] [CrossRef]
- Che, C.-M.; Huang, J.-S. Ruthenium and osmium porphyrin carbene complexes: Synthesis, structure, and connection to the metal-mediated cyclopropanation of alkenes. Coord. Chem. Rev. 2002, 231, 151–164. [Google Scholar] [CrossRef]
- Reissig, H.-U.; Zimmer, R. Donor-Acceptor-Substituted Cyclopropane Derivatives and Their Application in Organic Synthesis. Chem. Rev. 2003, 103, 1151–1196. [Google Scholar] [CrossRef] [PubMed]
- Gnad, F.; Reiser, O. Synthesis and Applications of β-Aminocarboxylic Acids Containing a Cyclopropane Ring. Chem. Rev. 2003, 103, 1603–1624. [Google Scholar] [CrossRef] [PubMed]
- Lebel, H.; Marcoux, J.-F.; Molinaro, C.; Charette, A.B. Stereoselective Cyclopropanation Reactions. Chem. Rev. 2003, 103, 977–1050. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, H. Recent developments in asymmetric cyclopropanation. Tetrahedron 2008, 64, 7041–7095. [Google Scholar] [CrossRef]
- Nicolas, E.; Le Maux, P.; Simonneaux, G. Asymmetric catalytic cyclopropanation reactions in water. Coord. Chem. Rev. 2008, 252, 727–735. [Google Scholar] [CrossRef]
- Caballero, A.; Prieto, A.; Díaz-Requejo, M.M.; Pérez, P.J. Metal-Catalyzed Olefin Cyclopropanation with Ethyl Diazoacetate: Control of the Diastereoselectivity. Eur. J. Inorg. Chem. 2009, 2009, 1137–1144. [Google Scholar] [CrossRef]
- Ebner, C.; Carreira, E.M. Cyclopropanation Strategies in Recent Total Syntheses. Chem. Rev. 2017, 117, 11651–11679. [Google Scholar] [CrossRef] [PubMed]
- Maas, G. Ruthenium-catalyzed carbenoid cyclopropanation reactions with diazo compounds. Chem. Soc. Rev. 2004, 33, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Raoul, N.; Gallo, E.; Rose, E. Synthesis of chiral ruthenium and cobalt (meso-2-amidophenyl)porphyrins and their catalytic activity in cyclopropanation reactions. J. Porphyr. Phthalocyanines 2011, 15, 602–611. [Google Scholar] [CrossRef]
- Intrieri, D.; Le Gac, S.; Caselli, A.; Rose, E.; Boitrel, B.; Gallo, E. Highly diastereoselective cyclopropanation of α-methylstyrene catalyzed by a C2-symmetrical chiral iron porphyrin complex. Chem. Commun. 2014, 50, 1811–1813. [Google Scholar] [CrossRef] [PubMed]
- Carminati, D.M.; Intrieri, D.; Caselli, A.; Le Gac, S.; Boitrel, B.; Toma, L.; Legnani, L.; Gallo, E. Designing ‘Totem’ C2-Symmetrical Iron Porphyrin Catalysts for Stereoselective Cyclopropanations. Chem. Eur. J. 2016, 22, 13599–13612. [Google Scholar] [CrossRef] [PubMed]
- Carminati, D.M.; Intrieri, D.; Le Gac, S.; Roisnel, T.; Boitrel, B.; Toma, L.; Legnani, L.; Gallo, E. Synthesis, characterisation and catalytic use of iron porphyrin amino ester conjugates. New J. Chem. 2017, 41, 5950–5959. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, X.; Cui, X.; Wojtas, L.; Zhang, X.P. Asymmetric Radical Cyclopropanation of Alkenes with In Situ Generated Donor-Substituted Diazo Reagents via Co(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc. 2017, 139, 1049–1052. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Xu, X.; Perman, J.A.; Zhang, X.P. A General and Efficient Cobalt(II)-Based Catalytic System for Highly Stereoselective Cyclopropanation of Alkenes with α-Cyanodiazoacetates. J. Am. Chem. Soc. 2010, 132, 12796–12799. [Google Scholar] [CrossRef] [PubMed]
- Dzik, W.I.; Xu, X.; Zhang, X.P.; Reek, J.N.H.; de Bruin, B. ‘Carbene Radicals’ in CoII(por)-Catalyzed Olefin Cyclopropanation. J. Am. Chem. Soc. 2010, 132, 10891–10902. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhu, S.; Cui, X.; Wojtas, L.; Zhang, X.P. Cobalt(II)-Catalyzed Asymmetric Olefin Cyclopropanation with α-Ketodiazoacetates. Angew. Chem. Int. Ed. 2013, 52, 11857–11861. [Google Scholar] [CrossRef] [PubMed]
- Berkessel, A.; Ertürk, E.; Neudörfl, J.M. Asymmetric cyclopropanation of olefins catalyzed by a chiral cobalt(II) porphyrin. Org. Commun. 2017, 10, 79–89. [Google Scholar] [CrossRef]
- Le Maux, P.; Nicolas, I.; Chevance, S.; Simonneaux, G. Chemical reactivity of 6-diazo-5-oxo-l-norleucine (DON) catalyzed by metalloporphyrins (Fe, Ru). Tetrahedron 2010, 66, 4462–4468. [Google Scholar] [CrossRef]
- Otte, M.; Kuijpers, P.F.; Troeppner, O.; Ivanovic-Burmazovic, I.; Reek, J.N.H.; de Bruin, B. Encapsulation of Metalloporphyrins in a Self-Assembled Cubic M8L6 Cage: A New Molecular Flask for Cobalt-Porphyrin-Catalyzed Radical-Type Reactions. Chem. Eur. J. 2013, 19, 10170–10178. [Google Scholar] [CrossRef] [PubMed]
- Otte, M.; Kuijpers, P.F.; Troeppner, O.; Ivanovic-Burmazovic, I.; Reek, J.N.H.; de Bruin, B. Encapsulated Cobalt-Porphyrin as a Catalyst for Size-Selective Radical-type Cyclopropanation Reactions. Chem. Eur. J. 2014, 20, 4880–4884. [Google Scholar] [CrossRef] [PubMed]
- Ciammaichella, A.; Cardoni, V.; Leoni, A.; Tagliatesta, P. Rhodium Porphyrin Bound to a Merrifield Resin as Heterogeneous Catalyst for the Cyclopropanation Reaction of Olefins. Molecules 2016, 21, 278. [Google Scholar] [CrossRef] [PubMed]
- Oelerich, J.; Roelfes, G. DNA-based asymmetric organometallic catalysis in water. Chem. Sci. 2013, 4, 2013–2017. [Google Scholar] [CrossRef]
- Rioz-Martínez, A.; Oelerich, J.; Ségaud, N.; Roelfes, G. DNA-Accelerated Catalysis of Carbene-Transfer Reactions by a DNA/Cationic Iron Porphyrin Hybrid. Angew. Chem. Int. Ed. 2016, 55, 14136–14140. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.; Balskus, E.P. Interfacing Microbial Styrene Production with a Biocompatible Cyclopropanation Reaction. Angew. Chem. Int. Ed. 2015, 54, 7106–7109. [Google Scholar] [CrossRef] [PubMed]
- Gharaati, S.; Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpoor-Baltork, I.; Barati, B.; Sadegh, F. High-valent tin(IV) porphyrins: Efficient and selective catalysts for cyclopropanation of styrene derivatives with EDA under mild conditions. J. Organomet. Chem. 2013, 741–742, 78–82. [Google Scholar] [CrossRef]
- Anding, B.J.; Ellern, A.; Woo, L.K. Olefin Cyclopropanation Catalyzed by Iridium(III) Porphyrin Complexes. Organometallics 2012, 31, 3628–3635. [Google Scholar] [CrossRef]
- Vins, P.; de Cózar, A.; Rivilla, I.; Nováková, K.; Zangi, R.; Cvacka, J.; Arrastia, I.; Arrieta, A.; Drasar, P.; Miranda, J.I.; Cossío, F.P. Cyclopropanation reactions catalyzed by dendrimers possessing one metalloporphyrin active site at the core: Linear and sigmoidal kinetic behaviour for different dendrimer generations. Tetrahedron 2016, 72, 1120–1131. [Google Scholar] [CrossRef]
- Morandi, B.; Carreira, E.M. Iron-Catalyzed Cyclopropanation in 6 M KOH with in Situ Generation of Diazomethane. Science 2012, 335, 1471–1474. [Google Scholar] [CrossRef] [PubMed]
- Morandi, B.; Carreira, E.M. Iron-Catalyzed Cyclopropanation with Trifluoroethylamine Hydrochloride and Olefins in Aqueous Media: In Situ Generation of Trifluoromethyl Diazomethane. Angew. Chem. Int. Ed. 2010, 49, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Morandi, B.; Dolva, A.; Carreira, E.M. Iron-Catalyzed Cyclopropanation with Glycine Ethyl Ester Hydrochloride in Water. Org. Lett. 2012, 14, 2162–2163. [Google Scholar] [CrossRef] [PubMed]
- Kaschel, J.; Schneider, T.F.; Werz, D.B. One Pot, Two Phases: Iron-Catalyzed Cyclopropanation with In Situ Generated Diazomethane. Angew. Chem. Int. Ed. 2012, 51, 7085–7086. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Lin, J.-H.; Xiao, J.-C.; Gu, Y.-C. A Trifluoromethylcarbene Source. Org. Lett. 2016, 18, 2471–2474. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Cui, X.; Zhang, X.P. Ligand Effect on Cobalt(II)-Catalyzed Asymmetric Cyclopropanation with Diazosulfones Approaching High Stereoselectivity through Modular Design of D2-Symmetric Chiral Porphyrins. Eur. J. Inorg. Chem. 2012, 2012, 430–434. [Google Scholar] [CrossRef]
- Xu, X.; Lu, H.; Ruppel, J.V.; Cui, X.; Lopez de Mesa, S.; Wojtas, L.; Zhang, X.P. Highly Asymmetric Intramolecular Cyclopropanation of Acceptor-Substituted Diazoacetates by Co(II)-Based Metalloradical Catalysis: Iterative Approach for Development of New-Generation Catalysts. J. Am. Chem. Soc. 2011, 133, 15292–15295. [Google Scholar] [CrossRef] [PubMed]
- Ruppel, J.V.; Cui, X.; Xua, X.; Zhang, X.P. Stereoselective intramolecular cyclopropanation of α-diazoacetates via Co(II)-based metalloradical catalysis. Org. Chem. Front. 2014, 1, 515–520. [Google Scholar] [CrossRef] [PubMed]
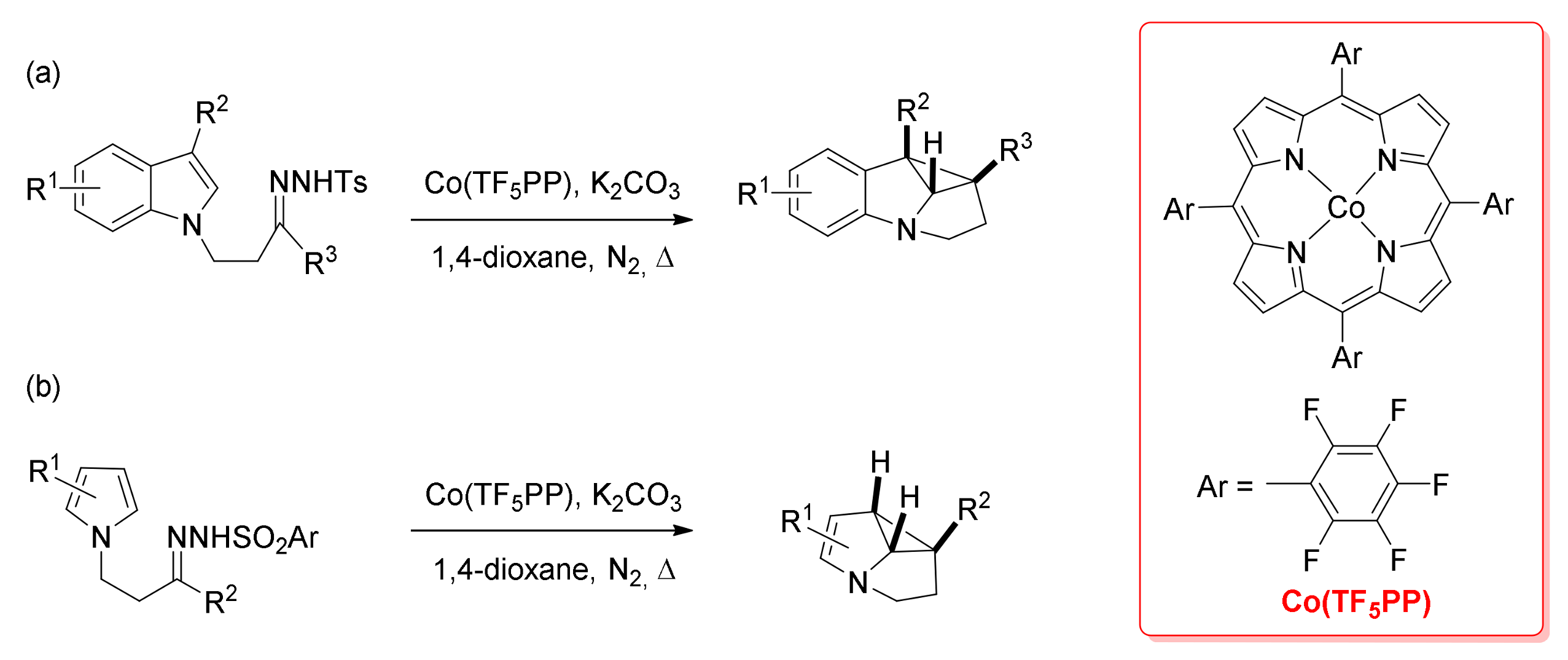
- Reddy, A.R.; Hao, F.; Wu, K.; Zhou, C.-Y.; Che, C.-M. Cobalt(II) Porphyrin-Catalyzed Intramolecular Cyclopropanation of N-Alkyl Indoles/Pyrroles with Alkylcarbene: Efficient Synthesis of Polycyclic N-Heterocycles. Angew. Chem. Int. Ed. 2016, 55, 1810–1815. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhou, C.-Y.; Che, C.-M. Cobalt-Porphyrin-Catalyzed Intramolecular Buchner Reaction and Arene Cyclopropanation of In Situ Generated Alkyl Diazomethanes. Adv. Synth. Catal. 2017, 359, 2253–2258. [Google Scholar] [CrossRef]
- Gorin, C.F.; Beh, E.S.; Bui, Q.M.; Dick, G.R.; Kanan, M.W. Interfacial Electric Field Effects on a Carbene Reaction Catalyzed by Rh Porphyrins. J. Am. Chem. Soc. 2013, 135, 11257–11265. [Google Scholar] [CrossRef] [PubMed]
- Beh, E.S.; Basun, S.A.; Feng, X.; Idehenre, I.U.; Evans, D.R.; Kanan, M.W. Molecular catalysis at polarized interfaces created by ferroelectric BaTiO3. Chem. Sci. 2017, 8, 2790–2794. [Google Scholar] [CrossRef] [PubMed]
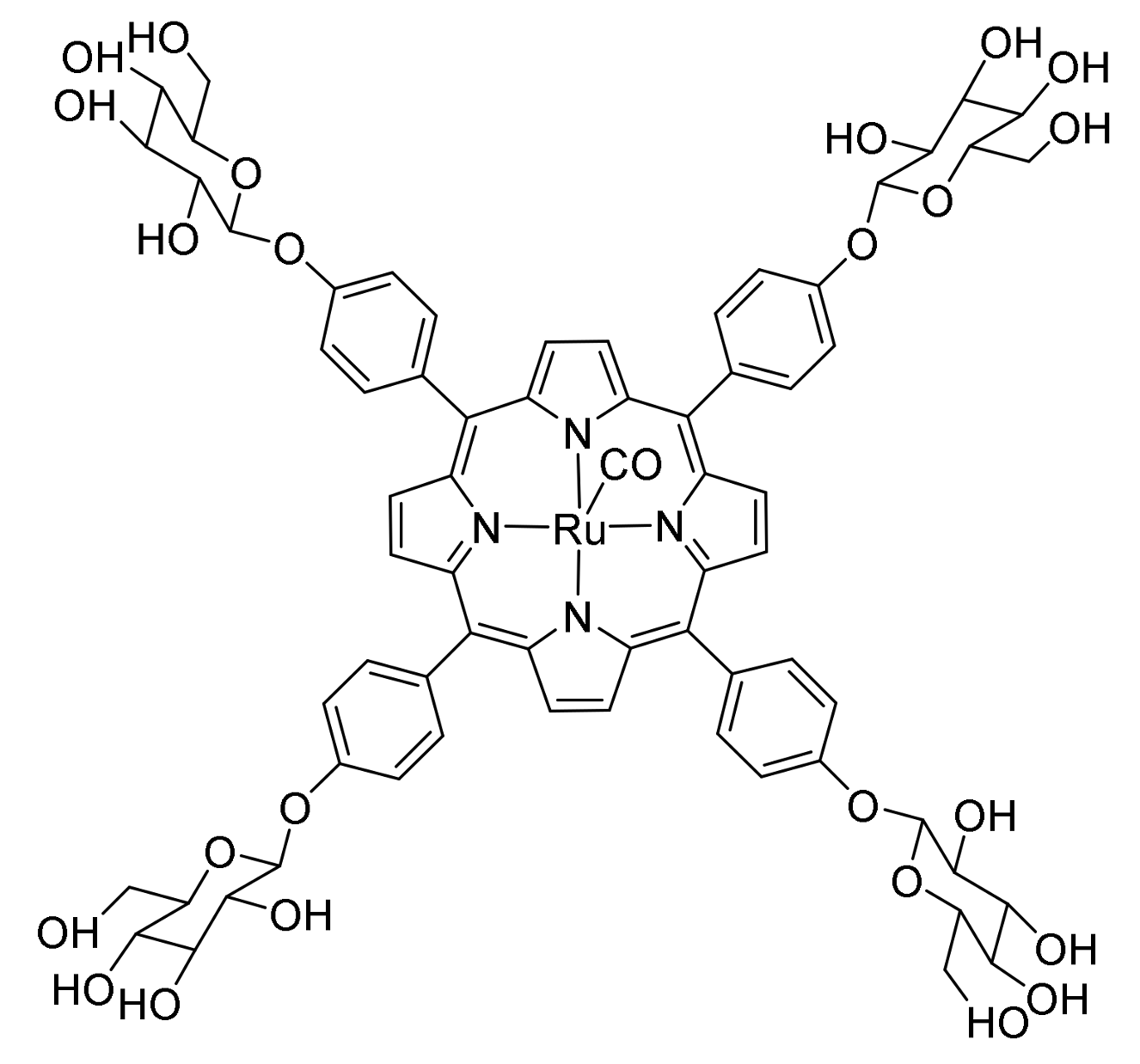
- Ho, C.-M.; Zhang, J.-L.; Zhou, C.-Y.; Chan, O.-Y.; Yan, J.J.; Zhang, F.-Y.; Huang, J.-S.; Che, C.-M. A Water-Soluble Ruthenium Glycosylated Porphyrin Catalyst for Carbenoid Transfer Reactions in Aqueous Media with Applications in Bioconjugation Reactions. J. Am. Chem. Soc. 2010, 132, 1886–1894. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.G.; Blakey, S.B. Ir-Catalyzed enantioselective group transfer reactions. Chem. Soc. Rev. 2015, 44, 5969–5980. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xu, X.; Lu, H.; Zhu, S.; Wojtas, L.; Zhang, X.P. Enantioselective Cyclopropenation of Alkynes with Acceptor/Acceptor-Substituted Diazo Reagents via Co(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc. 2011, 133, 3304–3307. [Google Scholar] [CrossRef] [PubMed]
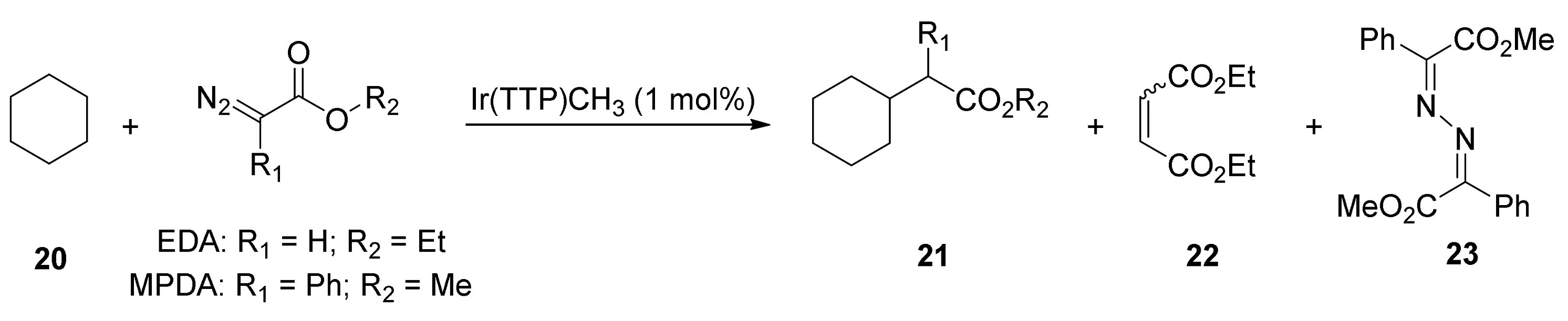
- Wang, B.; Qiu, D.; Zhang, Y.; Wang, J. Recent advances in C(sp3)-H bond functionalization via metal-carbene insertions. Beilstein J. Org. Chem. 2016, 12, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Callot, H.J.; Metz, F. Homologation of n-alkanes using diazoesters and rhodium(III)porphyrins. Enhanced attack on primary C-H bonds. Tetrahedron Lett. 1982, 23, 4321–4324. [Google Scholar] [CrossRef]
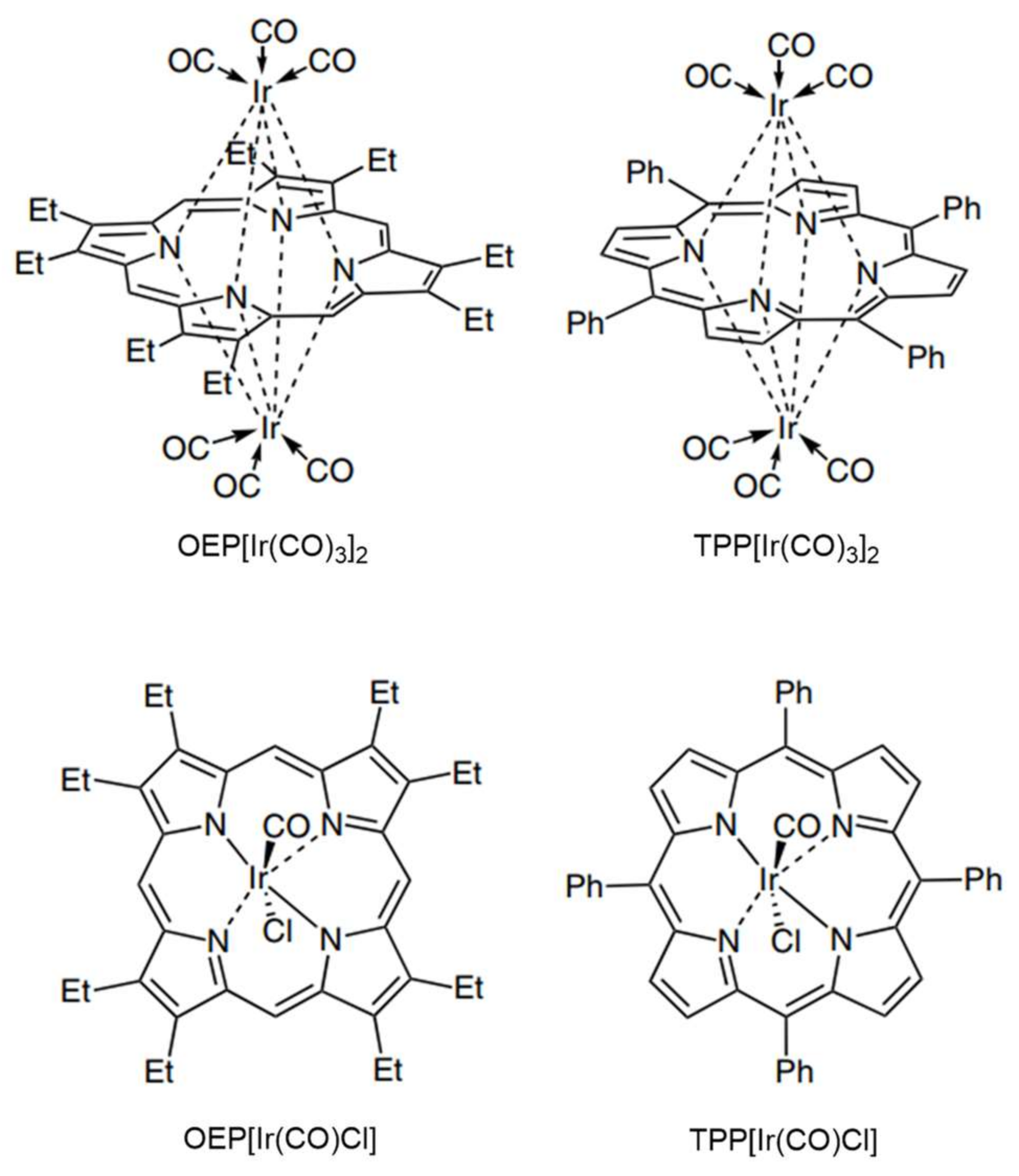
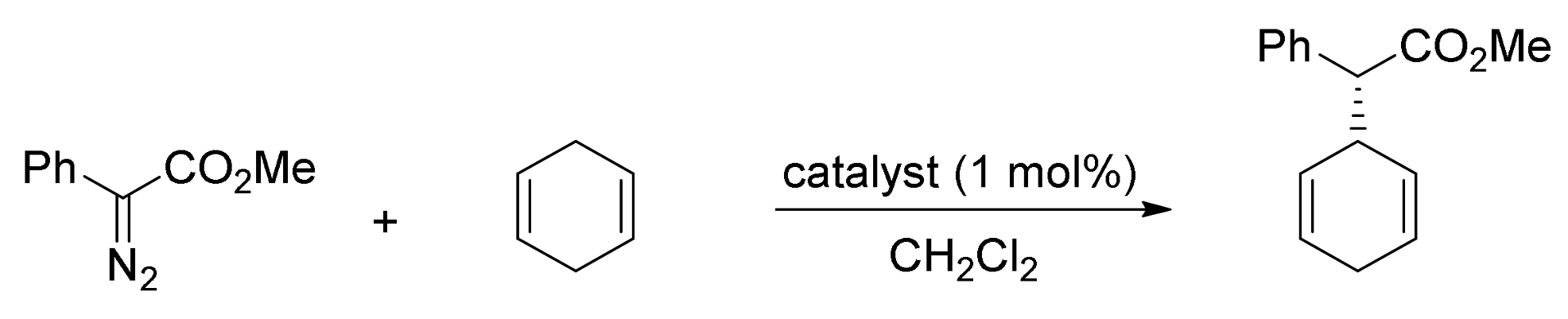
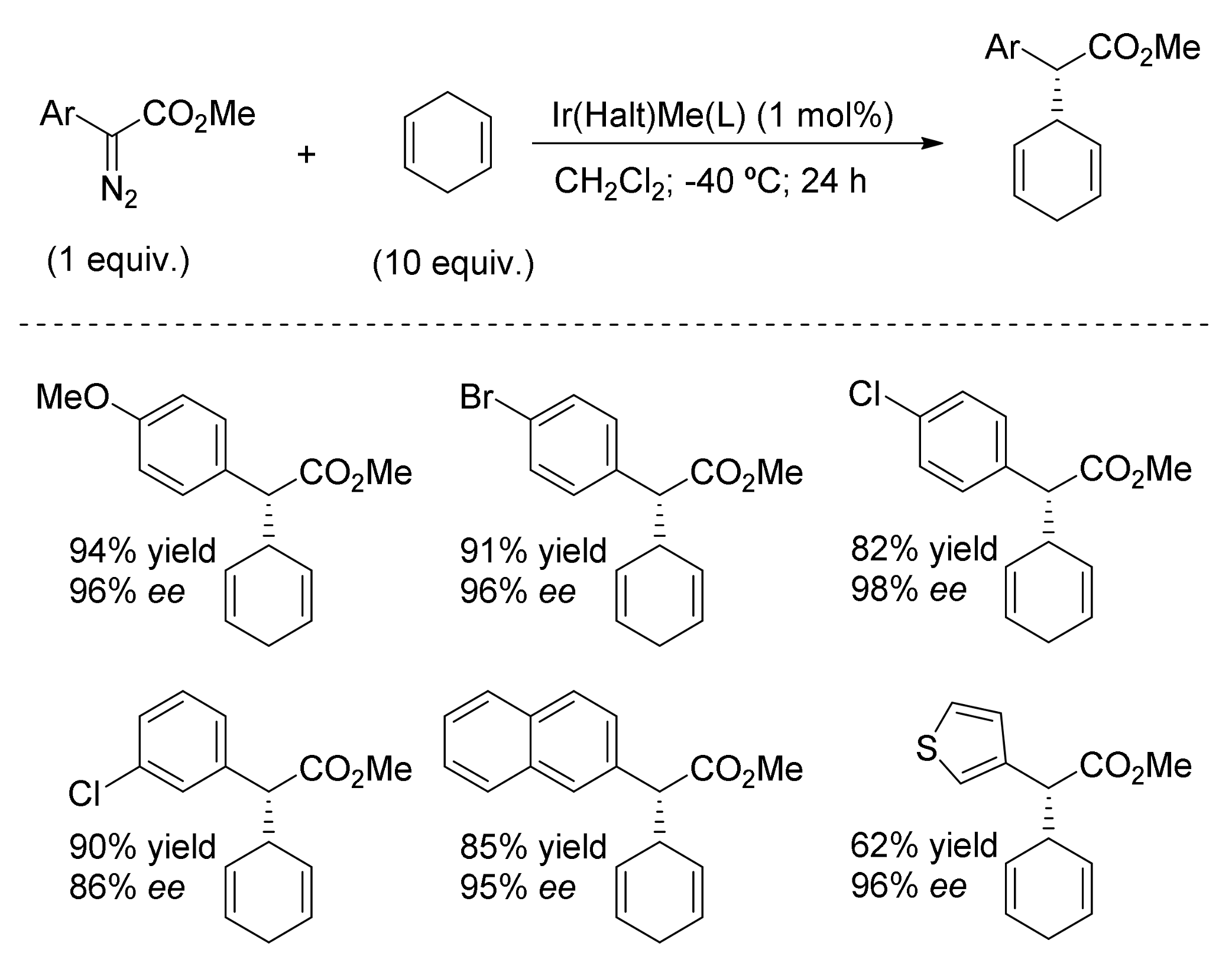
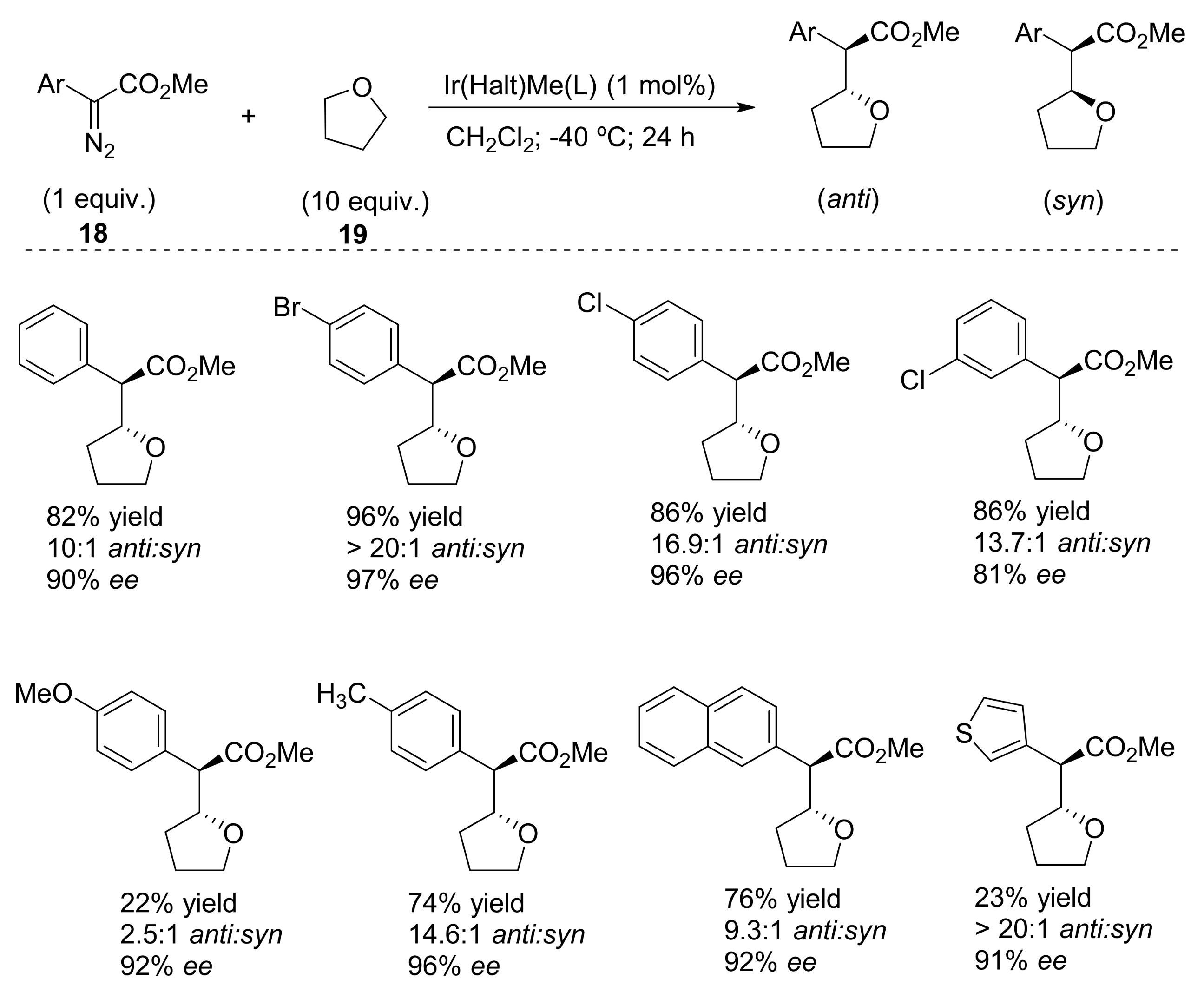
- Wang, J.-C.; Xu, Z.-J.; Guo, Z.; Deng, Q.-H.; Zhou, C.-Y.; Wan, X.-L.; Che, C.-M. Highly enantioselective intermolecular carbene insertion to C-H and Si-H bonds catalyzed by a chiral iridium(III) complex of a D4-symmetric Halterman porphyrin ligand. Chem. Commun. 2012, 48, 4299–4301. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-C.; Zhang, Y.; Xu, Z.-J.; Lo, V.K.-Y.; Che, C.-M. Enantioselective Intramolecular Carbene C-H Insertion Catalyzed by a Chiral Iridium(III) Complex of D4-Symmetric Porphyrin Ligand. ACS Catal. 2013, 3, 1144–1148. [Google Scholar] [CrossRef]
- Anding, B.J.; Brgoch, J.; Miller, G.J.; Woo, L.K. C-H Insertion Catalyzed by Tetratolylporphyrinato Methyliridium via a Metal-Carbene Intermediate. Organometallics 2012, 31, 5586–5590. [Google Scholar] [CrossRef]
- López-Sánchez, C.; Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Efficient Intramolecular C-H Insertion Catalyzed by Iridium Porphyrin Complexes. Synlett 2012, 23, 2469–2472. [Google Scholar]
- Lo, V.K.-Y.; Thu, H.-Y.; Chan, Y.-M.; Lam, T.-L.; Yu, W.-Y.; Che, C.-M. Stereoselective Intramolecular Carbene C-H Insertion Catalyzed by Rhodium(III) Porphyrin Complexes. Synlett 2012, 23, 2753–2757. [Google Scholar] [CrossRef]
- Reddy, A.R.; Zhou, C.; Guo, Z.; Wei, J.; Che, C.-M. Ruthenium-Porphyrin-Catalyzed Diastereoselective Intramolecular Alkyl Carbene Insertion into C-H Bonds of Alkyl Diazomethanes Generated In Situ from N-Tosylhydrazones. Angew. Chem. Int. Ed. 2014, 53, 14175–14180. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xu, X.; Jin, L.; Wojtas, L.; Zhang, X.P. Stereoselective radical C-H alkylation with acceptor/acceptor-substituted diazo reagents via Co(II)-based metalloradical catalysis. Chem. Sci. 2015, 6, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Mbuvi, H.M.; Klobukowski, E.R.; Roberts, G.M.; Woo, L.K. O-H insertion and tandem N-H insertion/cyclization reactions using an iron porphyrin as catalyst with diazo compounds as carbene sources. J. Porphyr. Phthalocyanines 2010, 14, 284–292. [Google Scholar] [CrossRef]
- Srour, H.F.; Le Maux, P.; Chevance, S.; Carrié, D.; Le Yondre, N.; Simonneaux, G. Diazo ester insertion in N-H bonds of amino acid derivatives and insulin catalysed by water-soluble iron and ruthenium porphyrin complexes (FeTSPPCl) as application of carbenoid transfer in aqueous media. J. Mol. Catal. A: Chem. 2015, 407, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.; Ko, J.H.; Choi, J.; Cho, K.; Lee, S.M.; Kim, H.J.; Ko, Y.-J.; Park, K.H.; Son, S.U. Dual role of Cu2O nanocubes as templates and networking catalysts for hollow and microporous Fe-porphyrin networks. Chem. Commun. 2017, 53, 2598–2601. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Park, N.; Park, J.H.; Park, J.H.; Kang, S.; Lee, S.M.; Kim, H.J.; Jo, H.; Park, J.; Son, S.U. Magnetically Separable Microporous Fe−Porphyrin Networks for Catalytic Carbene Insertion into N−H Bonds. ACS Catal. 2015, 5, 350–355. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, H.; Xiong, L.; He, Z.; Wang, T.; Xu, Y.; Huang, K. Fe-Porphyrin functionalized microporous organic nanotube networks and their application for the catalytic olefination of aldehydes and carbene insertion into N-H bonds. Polym. Chem. 2017, 8, 3721–3730. [Google Scholar] [CrossRef]
- Nakazawa, J.; Smith, B.J.; Stack, T.D.P. Discrete Complexes Immobilized onto Click-SBA-15 Silica: Controllable Loadings and the Impact of Surface Coverage on Catalysis. J. Am. Chem. Soc. 2012, 134, 2750–2759. [Google Scholar] [CrossRef] [PubMed]
- Anding, B.J.; Woo, L.K. Iridium Porphyrin Catalyzed N-H Insertion Reactions: Scope and Mechanism. Organometallics 2013, 32, 2599–2607. [Google Scholar] [CrossRef]
- Anding, B.J.; Dairo, T.O.; Woo, L.K. Reactivity Comparison of Primary Aromatic Amines and Thiols in E-H Insertion Reactions with Diazoacetates Catalyzed by Iridium(III) Tetratolylporphyrin. Organometallics 2017, 36, 1842–1847. [Google Scholar] [CrossRef]
- Sharon, D.A.; Mallick, D.; Wang, B.; Shaik, S. Computation Sheds Insight into Iron Porphyrin Carbenes’ Electronic Structure, Formation, and N-H Insertion Reactivity. J. Am. Chem. Soc. 2016, 138, 9597–9610. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Xing, D.; Zhai, C.; Che, J.; Liu, S.; Wang, J.; Hu, W. Iron Porphyrin-Catalyzed Three-Component Reaction of Ethyl Diazoacetate with Aliphatic Amines and β,γ-Unsaturated α-Keto Esters. Org. Lett. 2013, 15, 6140–6143. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Wang, Y.; Wang, Y.; Fan, Y.-Z.; Zhang, L.; Su, C.-Y. A stable and porous iridium(III)-porphyrin metal-organic framework: Synthesis, structure and catalysis. CrystEngComm 2016, 18, 2203–2209. [Google Scholar] [CrossRef]
- Dairo, T.O.; Woo, L.K. Scope and Mechanism of Iridium Porphyrin-Catalyzed S-H Insertion Reactions between Thiols and Diazo Esters. Organometallics 2017, 36, 927–934. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, H.; Wei, Z.-W.; Wang, H.-P.; Zhang, L.; Su, C.-Y. Engineering catalytic coordination space in a chemically stable Ir-porphyrin MOF with a confinement effect inverting conventional Si-H insertion chemoselectivity. Chem. Sci. 2017, 8, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Van den Boomen, O.I.; Coumans, R.G.E.; Akeroyd, N.; Peters, T.P.J.; Schlebos, P.P.J.; Smits, J.; de Gelder, R.; Elemans, J.A.A.W.; Nolte, R.J.M.; Rowan, A.E. Carbenoid transfer reactions catalyzed by a ruthenium porphyrin macrocycle. Tetrahedron 2017, 73, 5029–5037. [Google Scholar] [CrossRef]



















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1,2-Disubstituted Substrates | EDA (equiv.) | Product (s) |
|---|---|---|
 | 1 |  |
 | 1 |  |
 | 1 |  |
 | 2 |  |
 | 1 |  |
 | 2 |  |
 | 60 ° C12 h (without EDA) |  |
 | 2 |  |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simões, M.M.Q.; Gonzaga, D.T.G.; Cardoso, M.F.C.; Forezi, L.D.S.M.; Gomes, A.T.P.C.; Da Silva, F.D.C.; Ferreira, V.F.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Carbene Transfer Reactions Catalysed by Dyes of the Metalloporphyrin Group. Molecules 2018, 23, 792. https://doi.org/10.3390/molecules23040792
Simões MMQ, Gonzaga DTG, Cardoso MFC, Forezi LDSM, Gomes ATPC, Da Silva FDC, Ferreira VF, Neves MGPMS, Cavaleiro JAS. Carbene Transfer Reactions Catalysed by Dyes of the Metalloporphyrin Group. Molecules. 2018; 23(4):792. https://doi.org/10.3390/molecules23040792
Chicago/Turabian StyleSimões, Mário M. Q., Daniel T. G. Gonzaga, Mariana F. C. Cardoso, Luana Da S. M. Forezi, Ana T. P. C. Gomes, Fernando De C. Da Silva, Vítor F. Ferreira, Maria G. P. M. S. Neves, and José A. S. Cavaleiro. 2018. "Carbene Transfer Reactions Catalysed by Dyes of the Metalloporphyrin Group" Molecules 23, no. 4: 792. https://doi.org/10.3390/molecules23040792