Prediction of Protein-Protein Interactions from Amino Acid Sequences Based on Continuous and Discrete Wavelet Transform Features

Abstract
:1. Introduction
2. Results
2.1. Parameter Selection
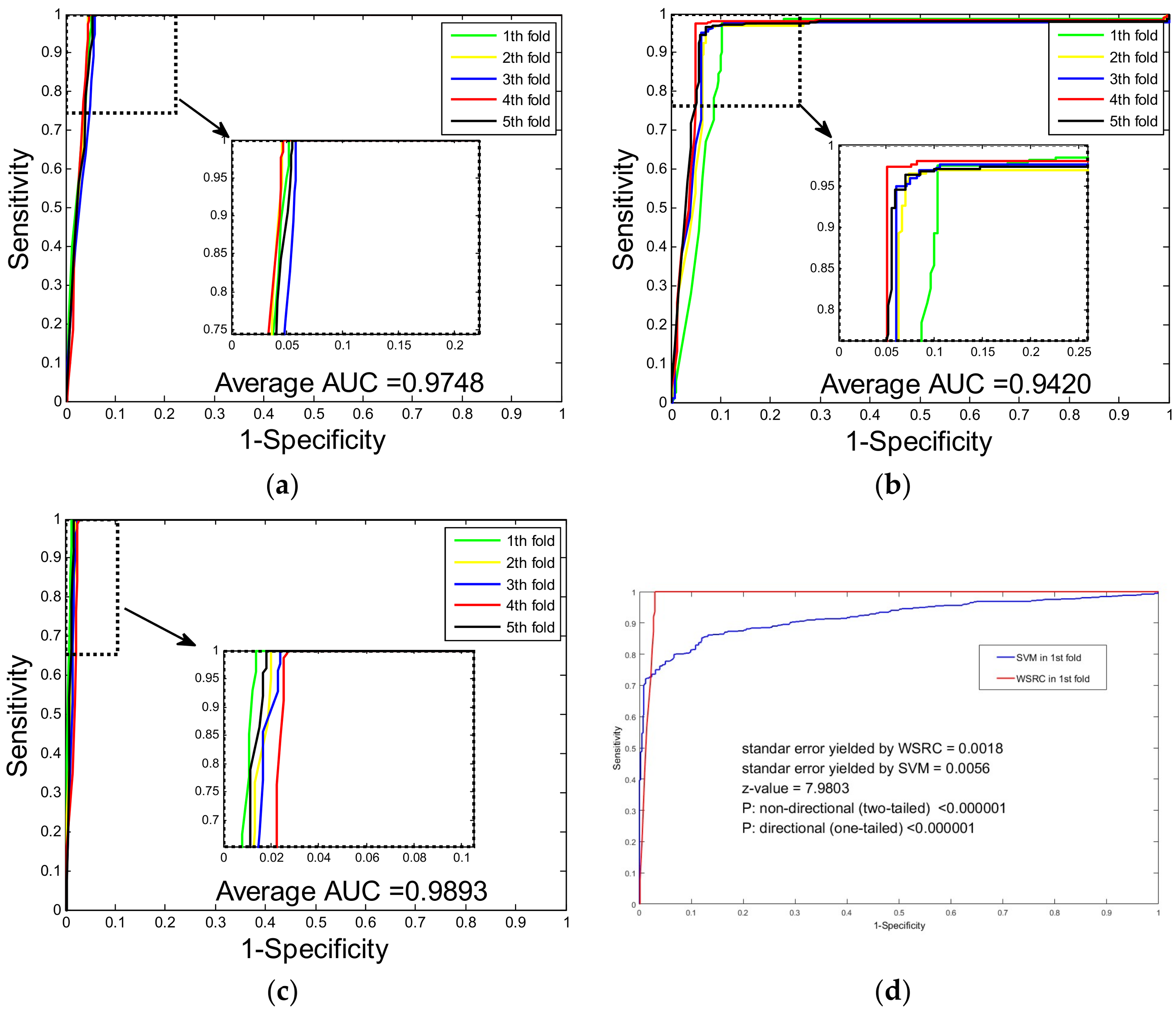
2.2. Assessment of Prediction Ability
2.3. Comparison with SVM and Single Wavelet Feature Descriptor
2.4. Comparison with Other Methods
2.5. Performance on Independent Dataset
3. Discussion
4. Materials and Methods
4.1. Datasets
4.2. Continuous Wavelet Transformation
4.3. Discrete Wavelet Transform
4.4. Weighted Sparse Representation Based Classifier
| Algorithm 1. Weighted Sparse Representation-Based Classifier (WSRC) |
| 1. Input: training samples matrix and any test sample . 2. Normalize the columns of X to have unit l2-norm. 3. Calculate the Gaussian distances between y and each sample in X and make up matrix W. 4. Solve the stable l1-minimization problem defined in Eq.(13) 5. Compute each residual of K classes: (c = 1, 2, …, K) 6. Ouput: assign y to class c by the rule: |
4.5. Evaluation Measures
4.6. Cross-Validation
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Walsh, I.; Di Domenico, T.; Tosatto, S.C. RUBI: Rapid proteomic-scale prediction of lysine ubiquitination and factors influencing predictor performance. Amino Acids 2014, 46, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Chiba, T.; Ozawa, R.; Yoshida, M.; Hattori, M.; Sakaki, Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Nat. Acad. Sci. USA 2001, 98, 4569–4574. [Google Scholar] [CrossRef] [PubMed]
- Pazos, F.; Valencia, A. In silico two-hybrid system for the selection of physically interacting protein pairs. Proteins Struct. Funct. Bioinform. 2002, 47, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Gavin, A.-C.; Bösche, M.; Krause, R.; Grandi, P.; Marzioch, M.; Bauer, A.; Schultz, J.; Rick, J.M.; Michon, A.-M.; Cruciat, C.-M. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002, 415, 141. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.; Gruhler, A.; Heilbut, A.; Bader, G.D.; Moore, L.; Adams, S.-L.; Millar, A.; Taylor, P.; Bennett, K.; Boutilier, K. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 2002, 415, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanzoni, A.; Montecchi-Palazzi, L.; Quondam, M.; Ausiello, G.; Helmer-Citterich, M.; Cesareni, G. MINT: A Molecular INTeraction database. FEBS Lett. 2002, 513, 135–140. [Google Scholar] [CrossRef]
- Xenarios, I.; Rice, D.W.; Salwinski, L.; Baron, M.K.; Marcotte, E.M.; Eisenberg, D. DIP: The database of interacting proteins. Nucleic Acids Res. 2000, 28, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.J.; Helk, B.; Trout, B.L. A computational tool to predict the evolutionarily conserved protein-protein interaction hot-spot residues from the structure of the unbound protein. FEBS Lett. 2014, 588, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Wang, X. Prediction of protein-protein interaction sites using patch-based residue characterization. J. Theor. Biol. 2012, 293, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Tosatto, S.; Toppo, S. Large-scale prediction of protein structure and function from sequence. Curr. Pharm. Des. 2006, 12, 2067–2086. [Google Scholar] [CrossRef] [PubMed]
- Giollo, M.; Minervini, G.; Scalzotto, M.; Leonardi, E.; Ferrari, C.; Tosatto, S.C. BOOGIE: Predicting blood groups from high throughput sequencing data. PLoS ONE 2015, 10, e0124579. [Google Scholar] [CrossRef] [PubMed]
- Zahiri, J.; Yaghoubi, O.; Mohammad-Noori, M.; Ebrahimpour, R.; Masoudi-Nejad, A. PPIevo: Protein-protein interaction prediction from PSSM based evolutionary information. Genomics 2013, 102, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Chanphai, P.; Bekale, L.; Tajmir-Riahi, H. Effect of hydrophobicity on protein-protein interactions. Eur. Polym. J. 2015, 67, 224–231. [Google Scholar] [CrossRef]
- Koike, A.; Takagi, T. Prediction of protein-protein interaction sites using support vector machines. Protein Eng. Des. Sel. 2004, 17, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Wang, X.; Lin, L.; Guan, Y. Exploiting residue-level and profile-level interface propensities for usage in binding sites prediction of proteins. BMC Bioinform. 2007, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Pei, Z.; Qin, S.; Zhao, X. Prediction of protein-protein interactions in saccharomyces cerevisiae based on protein secondary structure. In Proceedings of the 2012 IEEE International Conference on Biomedical Engineering and Biotechnology (iCBEB), Macao, China, 28–30 May 2012; pp. 413–416. [Google Scholar]
- Chen, H.; Zhou, H.X. Prediction of interface residues in protein-protein complexes by a consensus neural network method: Test against NMR data. Proteins Struct. Funct. Bioinform. 2005, 61, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yu, L.; Wen, Z.; Li, M. Using support vector machine combined with auto covariance to predict protein-protein interactions from protein sequences. Nucleic Acids Res. 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Z.; Gao, Y.; Zheng, Y.Y. Prediction of Protein-Protein Interactions Using Local Description of Amino Acid Sequence. In Advances in Computer Science and Education Applications; Springer: Berlin, Germany, 2011; pp. 254–262. [Google Scholar]
- Yang, L.; Xia, J.-F.; Gui, J. Prediction of protein-protein interactions from protein sequence using local descriptors. Protein Pept. Lett. 2010, 17, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-A.; You, Z.-H.; Chen, X.; Yan, G.-Y. Improved protein-protein interactions prediction via weighted sparse representation model combining continuous wavelet descriptor and PseAA composition. BMC Syst. Biol. 2016, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Lumini, A.; Brahnam, S. An empirical study of different approaches for protein classification. Sci. World J. 2014, 2014, 236717. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Brahnam, S.; Lumini, A. Wavelet images and Chou’s pseudo amino acid composition for protein classification. Amino Acids 2012, 43, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Bock, J.R.; Gough, D.A. Whole-proteome interaction mining. Bioinformatics 2003, 19, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L. Hyperplanes for predicting protein-protein interactions. Neurocomputing 2005, 69, 257–263. [Google Scholar] [CrossRef]
- Martin, S.; Roe, D.; Faulon, J.-L. Predicting protein-protein interactions using signature products. Bioinformatics 2005, 21, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Lumini, A. An ensemble of K-local hyperplanes for predicting protein-protein interactions. Bioinformatics 2006, 22, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.-G.; Xia, J.-F.; Li, X.-L.; Huang, D.-S. Predicting protein-protein interactions from sequence using correlation coefficient and high-quality interaction dataset. Amino Acids 2010, 38, 891–899. [Google Scholar] [CrossRef] [PubMed]
- You, Z.-H.; Yu, J.-Z.; Zhu, L.; Li, S.; Wen, Z.-K. A MapReduce based parallel SVM for large-scale predicting protein-protein interactions. Neurocomputing 2014, 145, 37–43. [Google Scholar] [CrossRef]
- Li, F.M.; Li, Q.Z. Predicting Protein Subcellular Location Using Chous Pseudo Amino Acid Composition and Improved Hybrid Approach. Protein Pept. Lett. 2008, 15, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.; Ganesh, A.; Zhou, Z.; Wagner, A.; Ma, Y. Demo: Robust Face Recognition Via Sparse Representation. In Proceedings of the 8th IEEE International Conference on Automatic Face & Gesture Recognition, Amsterdam, The Netherlands, 17–19 September 2008; pp. 1–2. [Google Scholar]
- Candes, E.; Tao, T. Near Optimal Signal Recovery From Random Projections: Universal Encoding Strategies? IEEE Trans. Inf. Theory 2004, 52, 5406–5425. [Google Scholar] [CrossRef]
- Chen, S.S.; Donoho, D.L.; Saunders, M.A. Atomic decomposition by basis pursuit. SIAM Rev. 2001, 43, 129–159. [Google Scholar] [CrossRef]
- Roweis, S.T.; Saul, L.K. Nonlinear Dimensionality Reduction by Locally Linear Embedding. Science 2000, 290, 2323–2326. [Google Scholar] [CrossRef]
- Wang, J.; Yang, J.; Yu, K.; Lv, F.; Huang, T.; Gong, Y. Locality-constrained Linear Coding for image classification. In Proceedings of the 2010 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), San Francisco, CA, USA, 13–18 June 2010; pp. 3360–3367. [Google Scholar]
- Feng, P.-M.; Chen, W.; Lin, H.; Chou, K.-C. iHSP-PseRAAAC: Identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 2013, 442, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.M.; van Berkel, V.H.; Frieboes, H.B. Application of unsupervised analysis techniques to lung cancer patient data. PLoS ONE 2017, 12, e0184370. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Shin, T.H.; Lee, G. DHSpred: Support-vector-machine-based human DNase I hypersensitive sites prediction using the optimal features selected by random forest. Oncotarget 2018, 9, 1944. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Shin, T.H.; Choi, S.; Kim, M.O.; Lee, G. MLACP: Machine-learning-based prediction of anticancer peptides. Oncotarget 2017, 8, 77121. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Lee, J. SVMQA: Support–vector-machine-based protein single-model quality assessment. Bioinformatics 2017, 33, 2496–2503. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Lee, J.; Lee, J. Random forest-based protein model quality assessment (RFMQA) using structural features and potential energy terms. PLoS ONE 2014, 9, e106542. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chen, W.; Chou, K.-C. iDNA6mA-PseKNC: Identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yang, H.; Feng, P.; Ding, H.; Lin, H. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 2017, 33, 3518–3523. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |


{kind=link}
| Test Set | Accuracy (%) | Precision (%) | Sensitivity (%) | MCC (%) | AUC (%) |
|---|---|---|---|---|---|
| 1 | 97.32 | 100.00 | 94.70 | 94.77 | 97.69 |
| 2 | 97.63 | 100.00 | 95.24 | 95.37 | 97.67 |
| 3 | 97.05 | 100.00 | 94.08 | 94.26 | 97.05 |
| 4 | 97.76 | 100.00 | 95.63 | 95.63 | 97.55 |
| 5 | 97.14 | 100.00 | 94.13 | 94.43 | 97.41 |
| Average | 97.38 ± 0.31 | 100.00 ± 0.00 | 94.76 ± 0.68 | 94.89 ± 0.59 | 97.48 ± 0.26 |
| Test Set | Accuracy (%) | Precision (%) | Sensitivity (%) | MCC (%) | AUC (%) |
|---|---|---|---|---|---|
| 1 | 92.28 | 96.72 | 88.04 | 85.72 | 92.72 |
| 2 | 93.83 | 95.94 | 91.23 | 88.38 | 93.74 |
| 3 | 94.68 | 95.99 | 92.93 | 89.91 | 94.20 |
| 4 | 95.20 | 97.70 | 93.40 | 90.81 | 95.36 |
| 5 | 93.66 | 95.70 | 90.41 | 88.01 | 94.99 |
| Average | 93.93 ± 1.11 | 96.41 ± 0.81 | 91.20 ± 2.15 | 88.57 ± 1.95 | 94.20 ± 1.05 |
| Classification Model | Testing Set | Accuracy (%) | Precision (%) | Sensitivity (%) | MCC (%) | AUC (%) |
|---|---|---|---|---|---|---|
| Proposed Method | 1 | 99.19 | 99.86 | 98.38 | 98.39 | 99.41 |
| 2 | 98.88 | 100.00 | 97.60 | 97.78 | 99.03 | |
| 3 | 98.70 | 99.87 | 97.47 | 97.43 | 98.78 | |
| 4 | 98.64 | 100.00 | 97.07 | 97.29 | 98.41 | |
| 5 | 99.20 | 100.00 | 98.33 | 98.40 | 99.03 | |
| Average | 98.92 ± 0.27 | 99.95 ± 0.07 | 97.77 ± 0.57 | 97.86 ± 0.52 | 98.93 ± 0.37 | |
| Combined Wavelet Feature with SVM | 1 | 91.39 | 96.15 | 84.57 | 83.91 | 94.71 |
| 2 | 90.77 | 97.17 | 82.53 | 82.83 | 94.77 | |
| 3 | 88.85 | 96.08 | 80.53 | 79.87 | 93.65 | |
| 4 | 88.79 | 94.53 | 80.56 | 79.62 | 91.50 | |
| 5 | 90.85 | 96.75 | 83.85 | 83.14 | 95.33 | |
| Average | 90.13 ± 1.22 | 96.14 ± 1.00 | 82.41 ± 1.85 | 81.87 ± 1.99 | 93.99 ± 1.52 |
| Method | Approach | Accuracy (%) | Precision (%) | Sensitivity (%) | MCC (%) |
|---|---|---|---|---|---|
| Guos’ work [18] | ACC | 89.33 ± 2.67 | 88.87 ± 6.16 | 89.93 ± 3.68 | N/A |
| AC (Auto Covariance) | 87.36 ± 1.38 | 87.82 ± 4.33 | 87.30 ± 4.68 | N/A | |
| Zhous’ work [19] | SVM + LD | 88.56 ± 0.33 | 89.50 ± 0.60 | 87.37 ± 0.22 | 77.15 ± 0.68 |
| Yangs’ work [20] | Cod1 | 75.08 ± 1.13 | 74.75 ± 1.23 | 75.81 ± 1.20 | N/A |
| Cod2 | 80.04 ± 1.06 | 82.17 ± 1.35 | 76.77 ± 0.69 | N/A | |
| Cod3 | 80.41 ± 0.47 | 81.86 ± 0.99 | 78.14 ± 0.90 | N/A | |
| Cod4 | 86.15 ± 1.17 | 90.24 ± 1.34 | 81.03 ± 1.74 | N/A | |
| Huangs’ work [21] | CW + PseAAC | 92.05 ± 0.59 | 95.87 ± 0.89 | 88.82 ± 0.98 | 86.09 ± 1.02 |
| Our work | WSRC + AM [22] | 96.03 ± 0.55 | 100.00 ± 0.00 | 92.07 ± 1.03 | 92.36 ± 1.01 |
| WSRC + BGR [22] | 96.14 ± 0.43 | 100.00 ± 0.00 | 92.29 ± 0.77 | 92.55 ± 0.80 | |
| WSRC + LBP − HF [23] | 96.60 ± 0.31 | 100.00 ± 0.00 | 93.20 ± 0.69 | 93.42 ± 0.58 | |
| WSRC+ LPQ [23] | 96.25 ± 0.17 | 100.00 ± 0.00 | 92.51 ± 0.45 | 92.77 ± 0.33 | |
| WSRC + CW&DW | 97.38 ± 0.31 | 100.00 ± 0.00 | 94.76 ± 0.68 | 94.89 ± 0.59 |
| Method | Accuracy (%) | Precision (%) | Sensitivity (%) | MCC (%) |
|---|---|---|---|---|
| Phylogenetic Booststrap [24] | 75.80 | 80.20 | 69.80 | N/A |
| HKNN [25] | 84.00 | 84.00 | 86.00 | N/A |
| Signature Products [26] | 83.40 | 85.70 | 79.90 | N/A |
| Ensemble of HKNN [27] | 86.60 | 85.00 | 86.70 | N/A |
| Boosting [28] | 79.52 | 81.69 | 80.37 | 70.64 |
| Proposed Method | 93.93 | 96.41 | 91.20 | 88.57 |
| Species | Test Pairs | Accuracy |
|---|---|---|
| D. mela | 21774 | 97.36% |
| E. coli | 6897 | 86.56% |
| C. elegans | 4013 | 96.64% |
| H. sapien | 1406 | 94.24% |
| H. pylori | 1420 | 95.07% |
| M. musculus | 312 | 94.23% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Li, L.; Huang, Y.-A.; Zhang, H.; Ma, Y.; Zhou, X. Prediction of Protein-Protein Interactions from Amino Acid Sequences Based on Continuous and Discrete Wavelet Transform Features. Molecules 2018, 23, 823. https://doi.org/10.3390/molecules23040823
Wang T, Li L, Huang Y-A, Zhang H, Ma Y, Zhou X. Prediction of Protein-Protein Interactions from Amino Acid Sequences Based on Continuous and Discrete Wavelet Transform Features. Molecules. 2018; 23(4):823. https://doi.org/10.3390/molecules23040823
Chicago/Turabian StyleWang, Tao, Liping Li, Yu-An Huang, Hui Zhang, Yahong Ma, and Xing Zhou. 2018. "Prediction of Protein-Protein Interactions from Amino Acid Sequences Based on Continuous and Discrete Wavelet Transform Features" Molecules 23, no. 4: 823. https://doi.org/10.3390/molecules23040823