Computational Prediction and Analysis of Associations between Small Molecules and Binding-Associated S-Nitrosylation Sites



Abstract
1. Introduction
2. Experimental Data
3. Method
3.1. Encoding Small Molecules
3.2. Encoding Protein Peptides
3.3. Random Forest
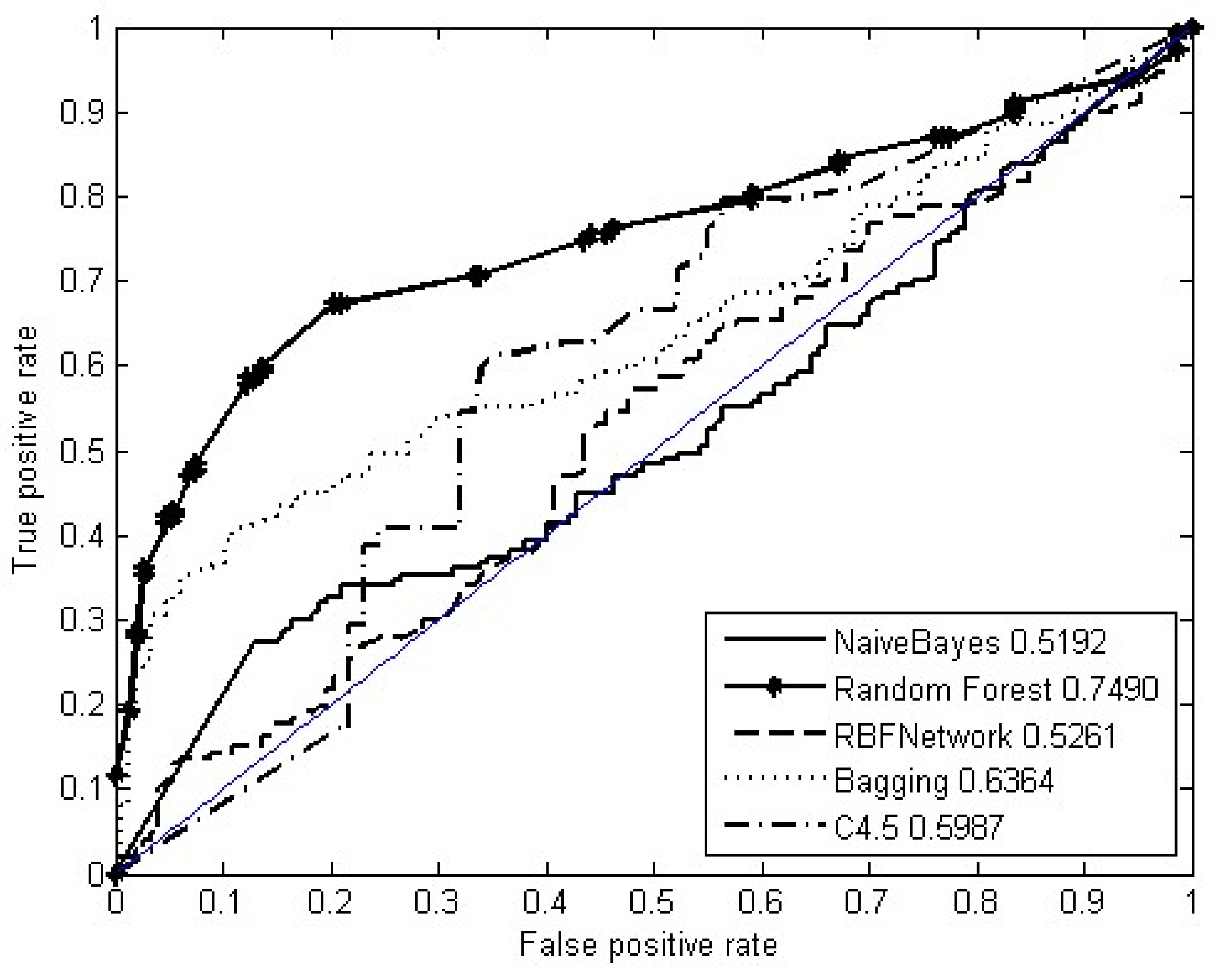
3.3.1. Cross Validation and Metrics
3.3.2. Computational Environment
4. Results and Discussion
4.1. Computational Analysis of Associations between Drugs and SNO-Proteins
4.2. Large-Scale Prediction of Unknown Associations of Drugs and SNO-Proteins
4.3. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Craig, J. Complex Diseases: Research and Applications. Nat. Educ. 2008, 1, 184. [Google Scholar]
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.P.; Brantner, V.V. Estimating the cost of new drug development: Is it really $802 million? Health Aff. 2006, 25, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Kotera, M.; Moriya, Y.; Sawada, R.; Kanehisa, M.; Goto, S. DINIES: Drug-target interaction network inference engine based on supervised analysis. Nucleic Acids Res. 2014, 42, W39–W45. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Kotera, M.; Kanehisa, M.; Goto, S. Drug-target interaction prediction from chemical, genomic and pharmacological data in an integrated framework. Bioinformatics 2010, 26, i246–i254. [Google Scholar] [CrossRef] [PubMed]
- Sawada, R.; Kotera, M.; Yamanishi, Y. Benchmarking a Wide Range of Chemical Descriptors for Drug-Target Interaction Prediction Using a Chemogenomic Approach. Mol. Inform. 2014, 33, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, C.C.; Zhang, X.; Zhang, X.; Dai, F.; Yin, J.; Zhang, Y. Drug-target interaction prediction: Databases, web servers and computational models. Brief. Bioinform. 2016, 17, 696–712. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Liu, C.; Jiang, J.; Lu, W.; Li, W.; Liu, G.; Zhou, W.; Huang, J.; Tang, Y. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput. Biol. 2012, 8, e1002503. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, Z. A semi-supervised method for drug-target interaction prediction with consistency in networks. PLoS ONE 2013, 8, e62975. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, M.X.; Yan, G.Y. Drug-target interaction prediction by random walk on the heterogeneous network. Mol. Biosyst. 2012, 8, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, M.A.; Goh, K.I.; Cusick, M.E.; Barabasi, A.L.; Vidal, M. Drug-target network. Nat. Biotechnol. 2007, 25, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Feng, K.; Li, X.; Peng, Y. Large-Scale Prediction of Drug Targets Based on Local and Global Consistency of Chemical-Chemical Networks. Comb. Chem. High Throughput Screen. 2016, 19, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.F.; Chen, L.; Huang, G.H.; Zhang, T.; Feng, K.Y.; Li, H.P.; Jiang, Y. Prediction of drugs target groups based on ChEBI ontology. BioMed Res. Int. 2013, 2013, 132724. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Zhang, C.H.; Deng, N.Y.; Wang, Y. Kernel-based data fusion improves the drug-protein interaction prediction. Comput. Biol. Chem. 2011, 35, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Wu, L.Y.; Zhou, X.; Wong, S.T. Semi-supervised drug-protein interaction prediction from heterogeneous biological spaces. BMC Syst. Biol. 2010, 4 (Suppl. 2), S6. [Google Scholar] [CrossRef] [PubMed]
- Vina, D.; Uriarte, E.; Orallo, F.; Gonzalez-Diaz, H. Alignment-free prediction of a drug-target complex network based on parameters of drug connectivity and protein sequence of receptors. Mol. Pharm. 2009, 6, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.P.; Kwoh, C.K.; Yang, P.; Li, X.L.; Zheng, J. Drug-target interaction prediction by learning from local information and neighbors. Bioinformatics 2013, 29, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Eichmann, C.; Tzitzilonis, C.; Nakamura, T.; Kwiatkowski, W.; Maslennikov, I.; Choe, S.; Lipton, S.A.; Riek, R. S-Nitrosylation Induces Structural and Dynamical Changes in a Rhodanese Family Protein. J. Mol. Biol. 2016, 428, 3737–3751. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.W.; Hess, D.T.; Stamler, J.S. Protein S-nitrosylation in health and disease: A current perspective. Trends Mol. Med. 2009, 15, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Won, J.S.; Singh, A.K.; Sharma, A.K.; Singh, I. STAT3 regulation by S-nitrosylation: Implication for inflammatory disease. Antioxid. Redox Signal. 2014, 20, 2514–2527. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Tu, S.; Akhtar, M.W.; Sunico, C.R.; Okamoto, S.; Lipton, S.A. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 2013, 78, 596–614. [Google Scholar] [CrossRef] [PubMed]
- Zahid, S.; Khan, R.; Oellerich, M.; Ahmed, N.; Asif, A.R. Differential S-nitrosylation of proteins in Alzheimer's disease. Neuroscience 2014, 256, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Prikhodko, O.A.; Pirie, E.; Nagar, S.; Akhtar, M.W.; Oh, C.K.; McKercher, S.R.; Ambasudhan, R.; Okamoto, S.; Lipton, S.A. Aberrant protein S-nitrosylation contributes to the pathophysiology of neurodegenerative diseases. Neurobiol. Dis. 2015, 84, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.F.; Yu, J.T.; Tan, L. S-Nitrosylation in Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. Protein S-nitrosylation and cancer. Cancer Lett. 2012, 320, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Ben-Lulu, S.; Ziv, T.; Weisman-Shomer, P.; Benhar, M. Nitrosothiol-Trapping-Based Proteomic Analysis of S-Nitrosylation in Human Lung Carcinoma Cells. PLoS ONE 2017, 12, e0169862. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Lipton, S.A. Protein S-Nitrosylation as a Therapeutic Target for Neurodegenerative Diseases. Trends Pharmacol. Sci. 2016, 37, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.Y.; Su, M.G.; Kao, H.J.; Hsieh, Y.C.; Jhong, J.H.; Cheng, K.H.; Huang, H.D.; Lee, T.Y. dbPTM 2016: 10-year anniversary of a resource for post-translational modification of proteins. Nucleic Acids Res. 2016, 44, D435–D446. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.T.; Huang, K.Y.; Su, M.G.; Lee, T.Y.; Bretana, N.A.; Chang, W.C.; Chen, Y.J.; Chen, Y.J.; Huang, H.D. DbPTM 3.0: An informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic Acids Res. 2013, 41, D295–D305. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. UniProt: A hub for protein information. Nucleic Acids Res. 2014, 43, D204–D212. [Google Scholar] [CrossRef] [PubMed]
- Magrane, M.; Consortium, U. UniProt Knowledgebase: A hub of integrated protein data. Database 2011, 2011, bar009. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010, 38, D142–D148. [Google Scholar]
- UniProt, C. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2014, 42, D191–D198. [Google Scholar]
- UniProt, C. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013, 41, D43–D47. [Google Scholar]
- Breiman, L. Random forests. MLear 2001, 45, 5–32. [Google Scholar]
- Christie, B.D.; Leland, B.A.; Nourse, J.G. Structure searching in chemical databases by direct lookup methods. J. Chem. Inf. Comput. Sci. 1993, 33, 545–547. [Google Scholar] [CrossRef]
- Johnson, M.A.; Maggiora, G.M. Concepts and Applications of Molecular Similarity; Wiley: New York, NY, USA, 1990. [Google Scholar]
- McGregor, M.J.; Pallai, P.V. Clustering of large databases of compounds: Using the MDL “keys” as structural descriptors. J. Chem. Inf. Comput. Sci. 1997, 37, 443–448. [Google Scholar] [CrossRef]
- Hall, L.H.; Kier, L.B. Electrotopological state indices for atom types: A novel combination of electronic, topological, and valence state information. J. Chem. Inf. Comput. Sci. 1995, 35, 1039–1045. [Google Scholar] [CrossRef]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. Chapter 12—PubChem: Integrated Platform of Small Molecules and Biological Activities. In Annual Reports in Computational Chemistry; Wheeler, R.A., Spellmeyer, D.C., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 4, pp. 217–241. [Google Scholar]
- Chen, B.; Wild, D.; Guha, R. PubChem as a source of polypharmacology. J. Chem. Inf. Model. 2009, 49, 2044–2055. [Google Scholar] [CrossRef] [PubMed]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Cereto-Massague, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallve, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An open-source Java library for chemo-and bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, C.; Hoppe, C.; Kuhn, S.; Floris, M.; Guha, R.; Willighagen, E.L. Recent developments of the chemistry development kit (CDK)—An open-source java library for chemo- and bioinformatics. Curr. Pharm. Des. 2006, 12, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Cao, D.-S.; Miao, H.-Y.; Liu, S.; Deng, B.-C.; Yun, Y.-H.; Wang, N.-N.; Lu, A.-P.; Zeng, W.-B.; Chen, A.F. ChemDes: An integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminform. 2015, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Deng, M.; Zheng, L.; He, R.L.; Yang, J.; Yau, S.S. DFA7, a new method to distinguish between intron-containing and intronless genes. PLoS ONE 2014, 9, e101363. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Deng, M.; Cheng, S.Y.; Yau, S.C.; He, R.L.; Yau, S.S. Protein space: A natural method for realizing the nature of protein universe. J. Theor. Biol. 2013, 318, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.D.; Ricardo, P.W.; Jen, C.H.; Chou, K.C. Application of SVM to predict membrane protein types. J. Theor. Biol. 2004, 226, 373–376. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Carr, K.; Murray, E.; Armah, E.; He, R.L.; Yau, S.S. A rapid method for characterization of protein relatedness using feature vectors. PLoS ONE 2010, 5, e9550. [Google Scholar] [CrossRef] [PubMed]
- Li, B.Q.; Zhang, Y.C.; Huang, G.H.; Cui, W.R.; Zhang, N.; Cai, Y.D. Prediction of aptamer-target interacting pairs with pseudo-amino acid composition. PLoS ONE 2014, 9, e86729. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2008, 36, D202–D205. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Wan, S.; Yang, Z.; Teschendorff, A.E.; Zou, Q. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 2018, 34, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Li, B.-Q.; Gao, S.; Ruan, J.-S.; Cai, Y.-D. Computational prediction and analysis of protein γ-carboxylation sites based on a random forest method. Mol. Biosyst. 2012, 8, 2946–2955. [Google Scholar] [CrossRef] [PubMed]
- Hamby, S.E.; Hirst, J.D. Prediction of glycosylation sites using random forests. BMC Bioinform. 2008, 9, 500. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, A. SUMOhunt: Combining Spatial Staging between Lysine and SUMO with Random Forests to Predict SUMOylation. ISRN Bioinform. 2013, 2013, 671269. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.; Kusalik, A. Computational phosphorylation site prediction in plants using random forests and organism-specific instance weights. Bioinformatics 2013, 29, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, J.R. C4.5: Programs for Machine Learning; Morgan Kaufmann Publishers: San Mateo, CA, USA, 1993. [Google Scholar]
- Russell, S.; Norvig, P. Artificial Intelligence: A Modern Approach; Prentice-Hall: Egnlewood Cliffs, NJ, USA, 1995; Volume 25. [Google Scholar]
- Schwenker, F.; Kestler, H.A.; Palm, G. Three learning phases for radial-basis-function networks. Neural Netw. 2001, 14, 439–458. [Google Scholar] [CrossRef]
- Polikar, R. Ensemble based systems in decision making. IEEE Circuits Syst. Mag. 2006, 6, 21–45. [Google Scholar] [CrossRef]
- Wu, X.; Kumar, V.; Ross Quinlan, J.; Ghosh, J.; Yang, Q.; Motoda, H.; McLachlan, G.J.; Ng, A.; Liu, B.; Yu, P.S.; et al. Top 10 algorithms in data mining. Knowl. Inf. Syst. 2007, 14, 1–37. [Google Scholar] [CrossRef]
- Oliver, S. Proteomics: Guilt-by-association goes global. Nature 2000, 403, 601–603. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular similarity in medicinal chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef] [PubMed]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef] [PubMed]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2011, 39, D1035–D1041. [Google Scholar] [CrossRef]
- Alaimo, S.; Pulvirenti, A.; Giugno, R.; Ferro, A. Drug-target interaction prediction through domain-tuned network-based inference. Bioinformatics 2013, 29, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Gonen, M. Predicting drug-target interactions from chemical and genomic kernels using Bayesian matrix factorization. Bioinformatics 2012, 28, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhang, J.; Shi, X.H.; Hu, L.L.; Kong, X.; Cai, Y.D.; Chou, K.C. Predicting drug-target interaction networks based on functional groups and biological features. PLoS ONE 2010, 5, e9603. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, J. Predicting drug-target interactions using restricted Boltzmann machines. Bioinformatics 2013, 29, i126–i134. [Google Scholar] [CrossRef] [PubMed]
- Campillos, M.; Kuhn, M.; Gavin, A.C.; Jensen, L.J.; Bork, P. Drug target identification using side-effect similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Takarabe, M.; Kotera, M.; Nishimura, Y.; Goto, S.; Yamanishi, Y. Drug target prediction using adverse event report systems: A pharmacogenomic approach. Bioinformatics 2012, 28, i611–i618. [Google Scholar] [CrossRef] [PubMed]
- Bleakley, K.; Yamanishi, Y. Supervised prediction of drug-target interactions using bipartite local models. Bioinformatics 2009, 25, 2397–2403. [Google Scholar] [CrossRef] [PubMed]
- Dunkel, M.; Gunther, S.; Ahmed, J.; Wittig, B.; Preissner, R. SuperPred: Drug classification and target prediction. Nucleic Acids Res. 2008, 36, W55–W59. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Zuo, Y.; Zou, Q. O-GlcNAcPRED-II: An integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique. Bioinformatics 2018. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Shi, G.; Ji, Z.L.; Zou, Q. Fast prediction of protein methylation sites using a sequence-based feature selection technique. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 1. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not Available. |






{kind=link}
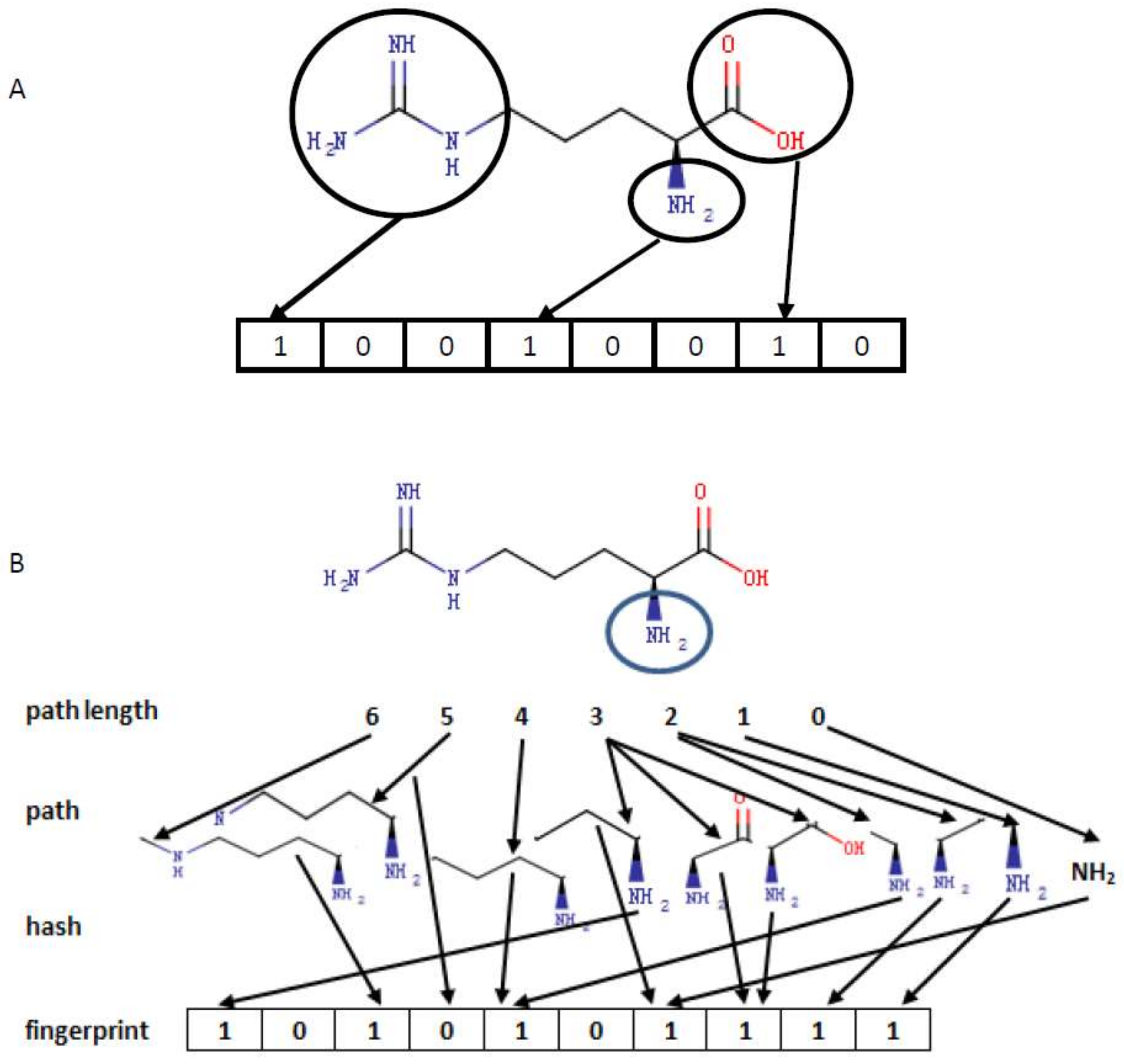{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Number of Bits | Category | Number of Bits |
|---|---|---|---|
| E-state | 79 | Klekota–Roth | 4860 |
| Daylight | 1024 | MACCs | 166 |
| CDK extended | 1024 | CDK substructure | 307 |
| CDK graph | 1024 | PubChem | 881 |
| CDK hybridization | 1024 |
| Predicted Associations | Similar Small Molecules | SNO-Proteins Targeted by Similar Molecules |
|---|---|---|
| DB04427–P18031-215 | DB06887, DB07719, DB08003, DB08549, DB08591, DB08593, DB02827 | P18031-215 |
| DB01960–P15121-299 | DB02338, DB03461 | P15121-299 |
| DB04315–P15121-299 | DB02338, DB03461, DB08772 | P15121-299 |
| DB08213–P18031-215 | DB02827, DB06887, DB07134, DB07719, DB07730, DB08003, DB08549, DB08591, DB08593 | P18031-215 |
| DB04502–P18031-215 | DB01962, DB03483, DB03557, DB06887, DB07719, DB08003, DB08549, DB08591 | P18031-215 |
| DB00114–P18031-215 | DB01962, DB07480 | P18031-215 |
| DB02051–P18031-215 | DB06887, DB07719, DB08549, DB08591 | P18031-215 |
| DB07905–P18031-215 | not existing similar drugs | P18031-215 |
| DB08607–P18031-215 | DB02072, DB02827, DB03102, DB03670, DB07298 | P18031-215 |
| DB02200–P18031-215 | DB03483, DB03557, DB03714, DB06887, DB07651, DB07719, DB08003, DB08549, DB08783 | P18031-215 |
| DB00171–P15121-299 | DB02338, DB03461 | P15121-299 |
| DB00155–P43235-139 | DB04276, DB04523, DB07592 | P43235-139 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, G.; Li, J.; Zhao, C. Computational Prediction and Analysis of Associations between Small Molecules and Binding-Associated S-Nitrosylation Sites. Molecules 2018, 23, 954. https://doi.org/10.3390/molecules23040954
Huang G, Li J, Zhao C. Computational Prediction and Analysis of Associations between Small Molecules and Binding-Associated S-Nitrosylation Sites. Molecules. 2018; 23(4):954. https://doi.org/10.3390/molecules23040954
Chicago/Turabian StyleHuang, Guohua, Jincheng Li, and Chenglin Zhao. 2018. "Computational Prediction and Analysis of Associations between Small Molecules and Binding-Associated S-Nitrosylation Sites" Molecules 23, no. 4: 954. https://doi.org/10.3390/molecules23040954
APA StyleHuang, G., Li, J., & Zhao, C. (2018). Computational Prediction and Analysis of Associations between Small Molecules and Binding-Associated S-Nitrosylation Sites. Molecules, 23(4), 954. https://doi.org/10.3390/molecules23040954