
Design, Synthesis, and Evaluation of Novel Immunomodulatory Small Molecules Targeting the CD40–CD154 Costimulatory Protein-Protein Interaction



Abstract
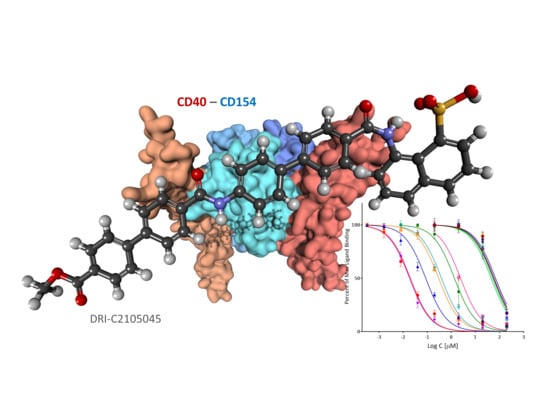
:
1. Introduction
2. Results
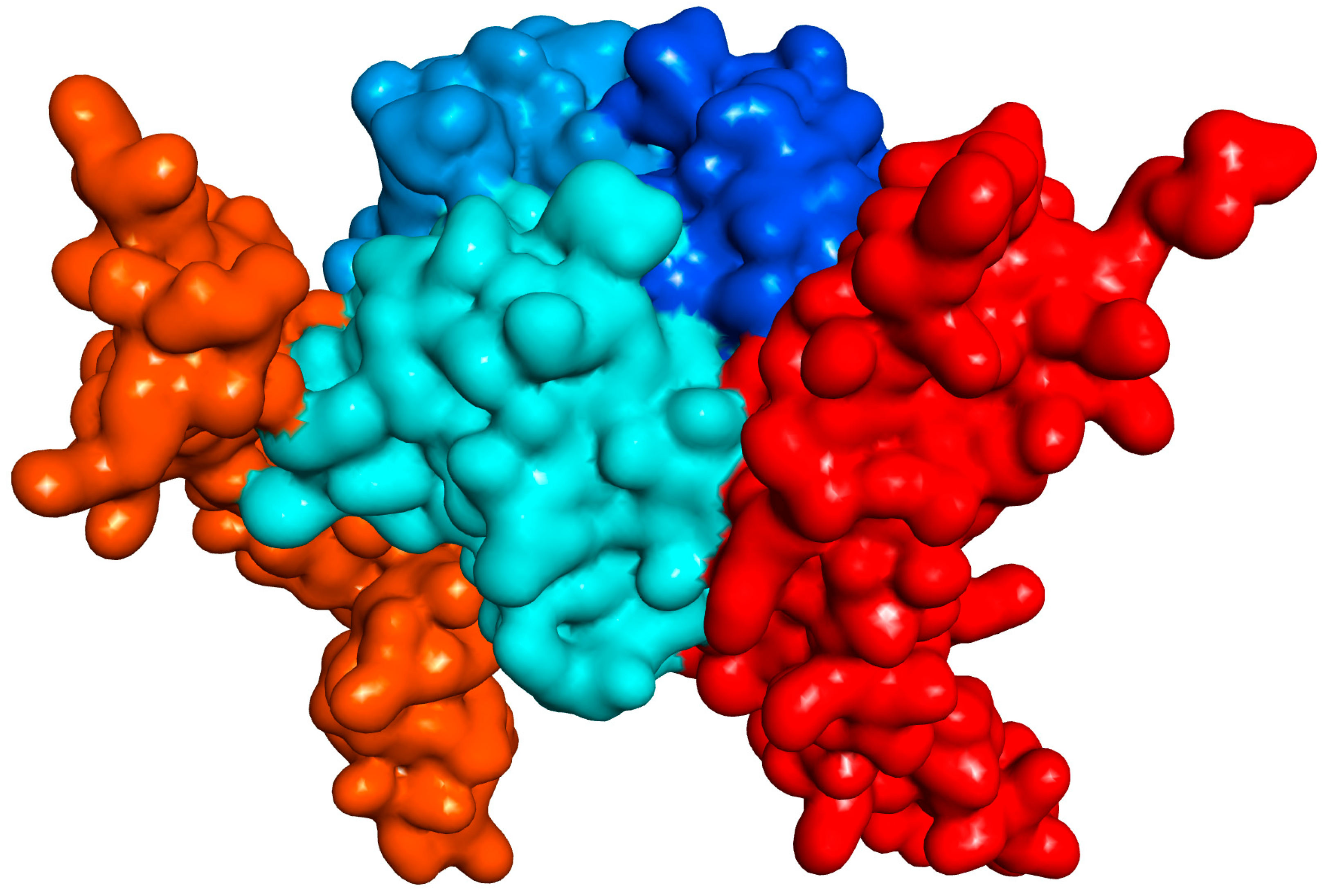
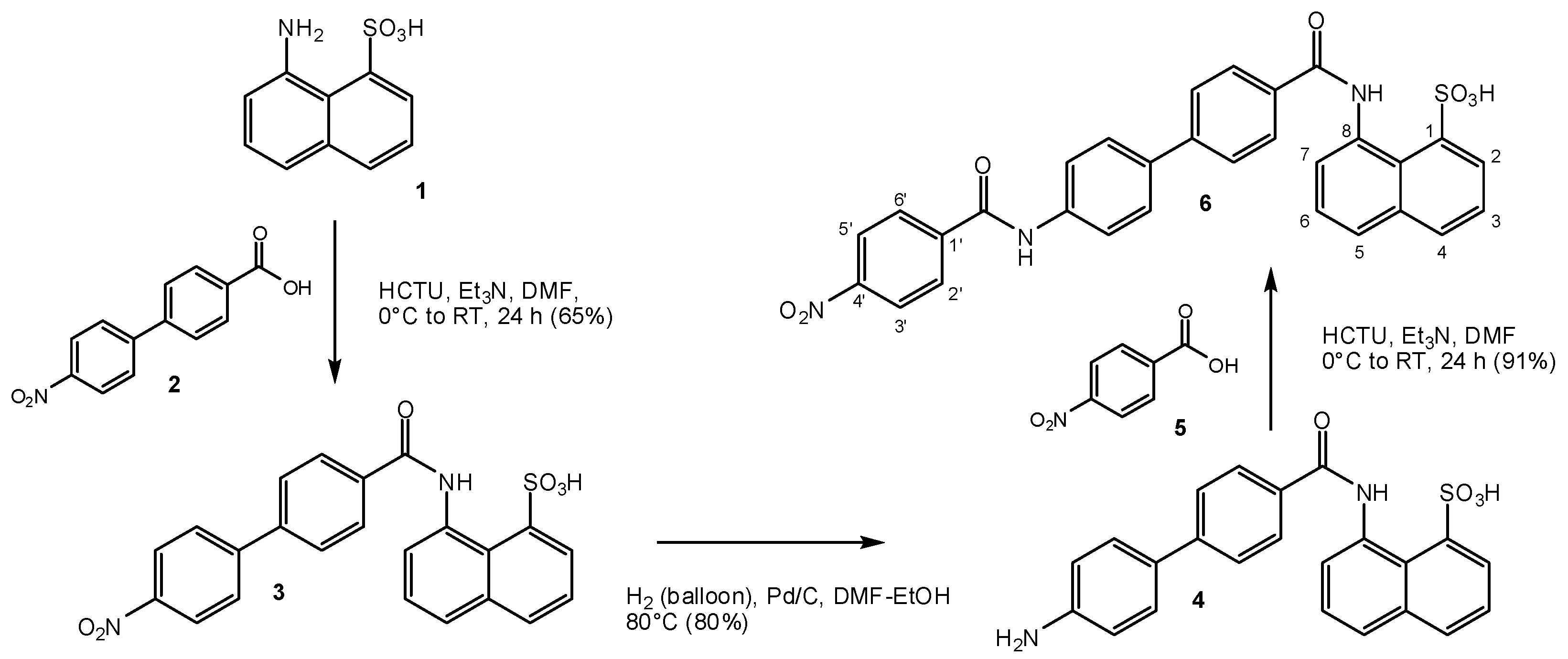
2.1. Design and Synthesis
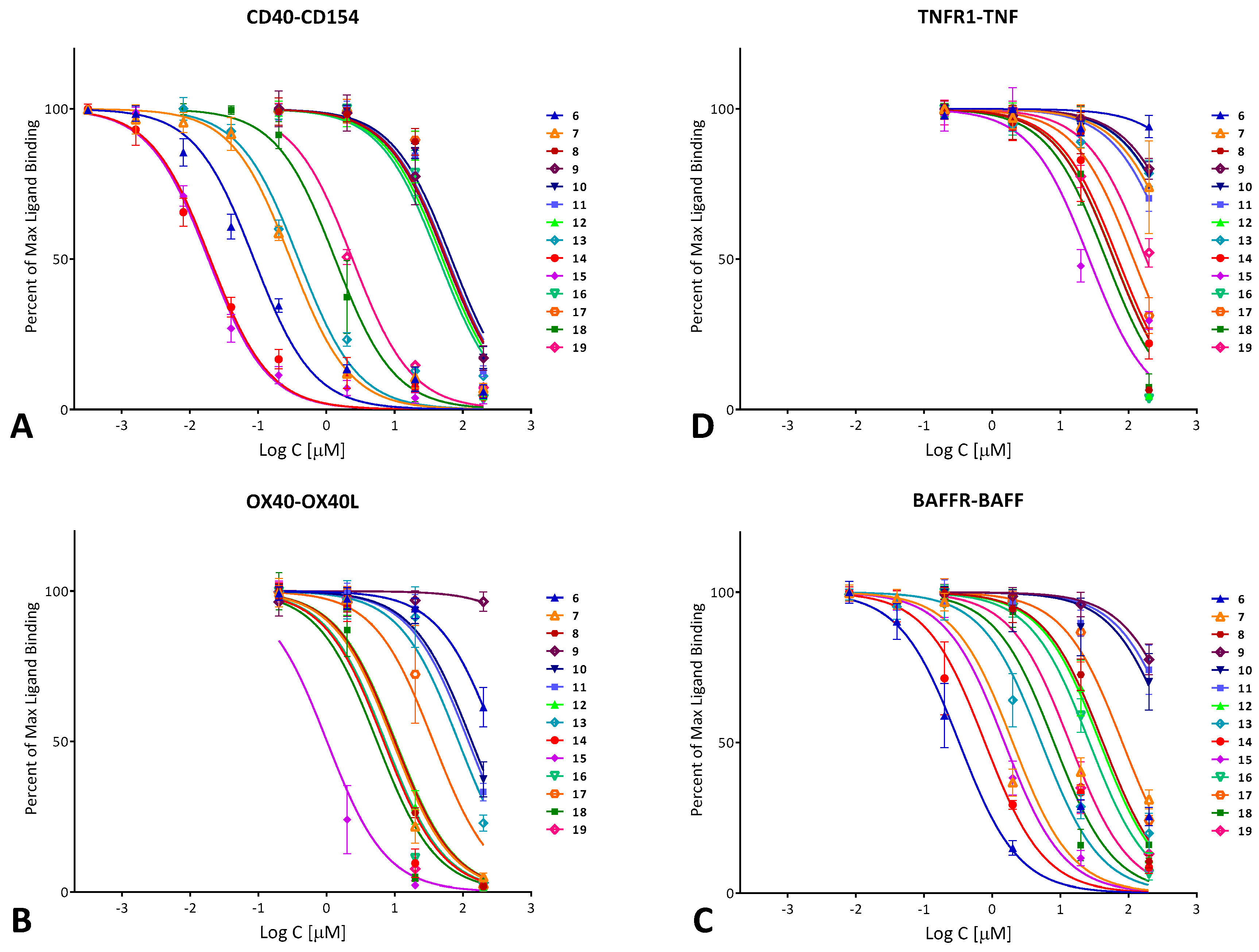
2.2. Binding Inhibition Assays
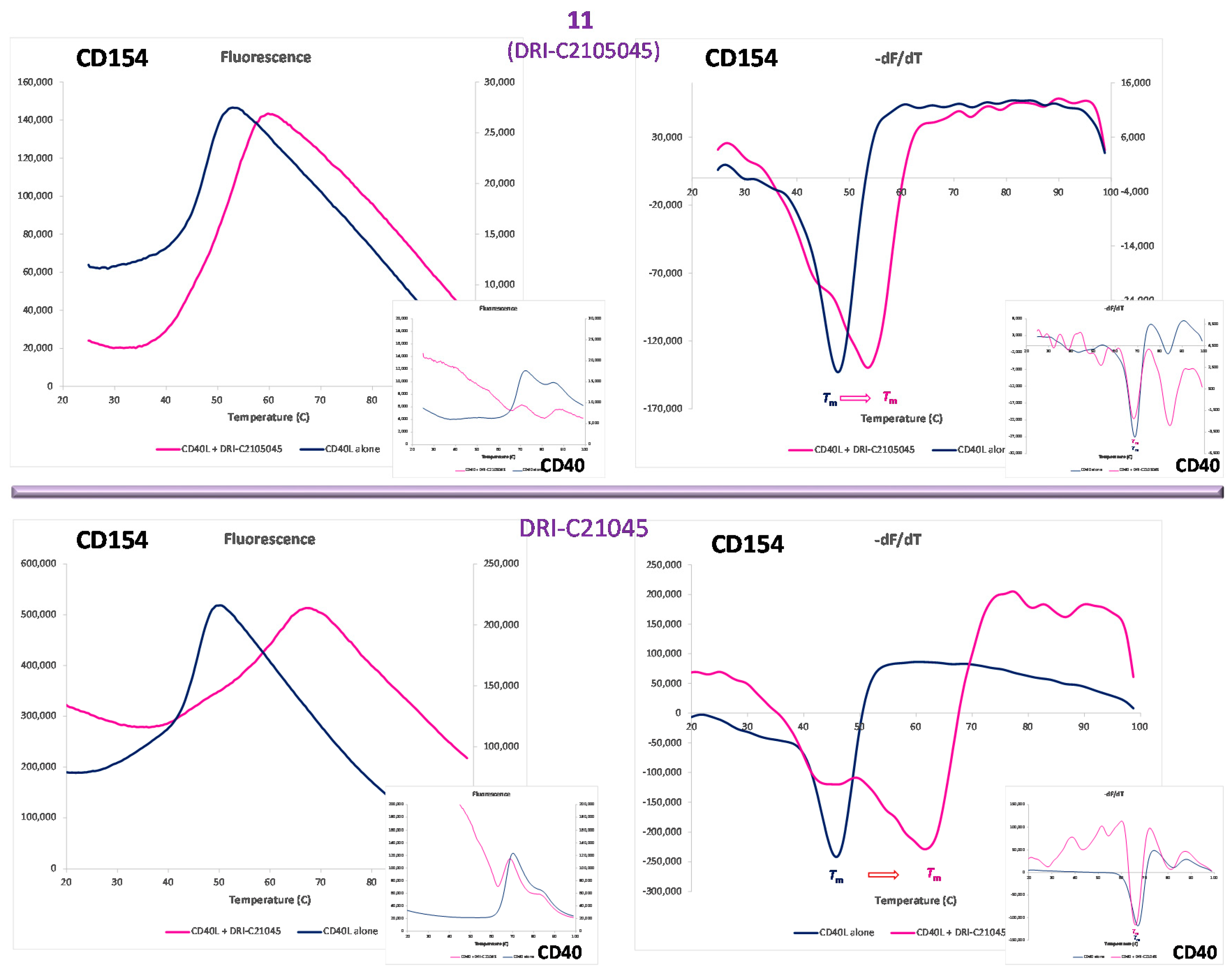
2.3. Binding Partner (Protein Thermal Shift)
2.4. In Vitro Activity
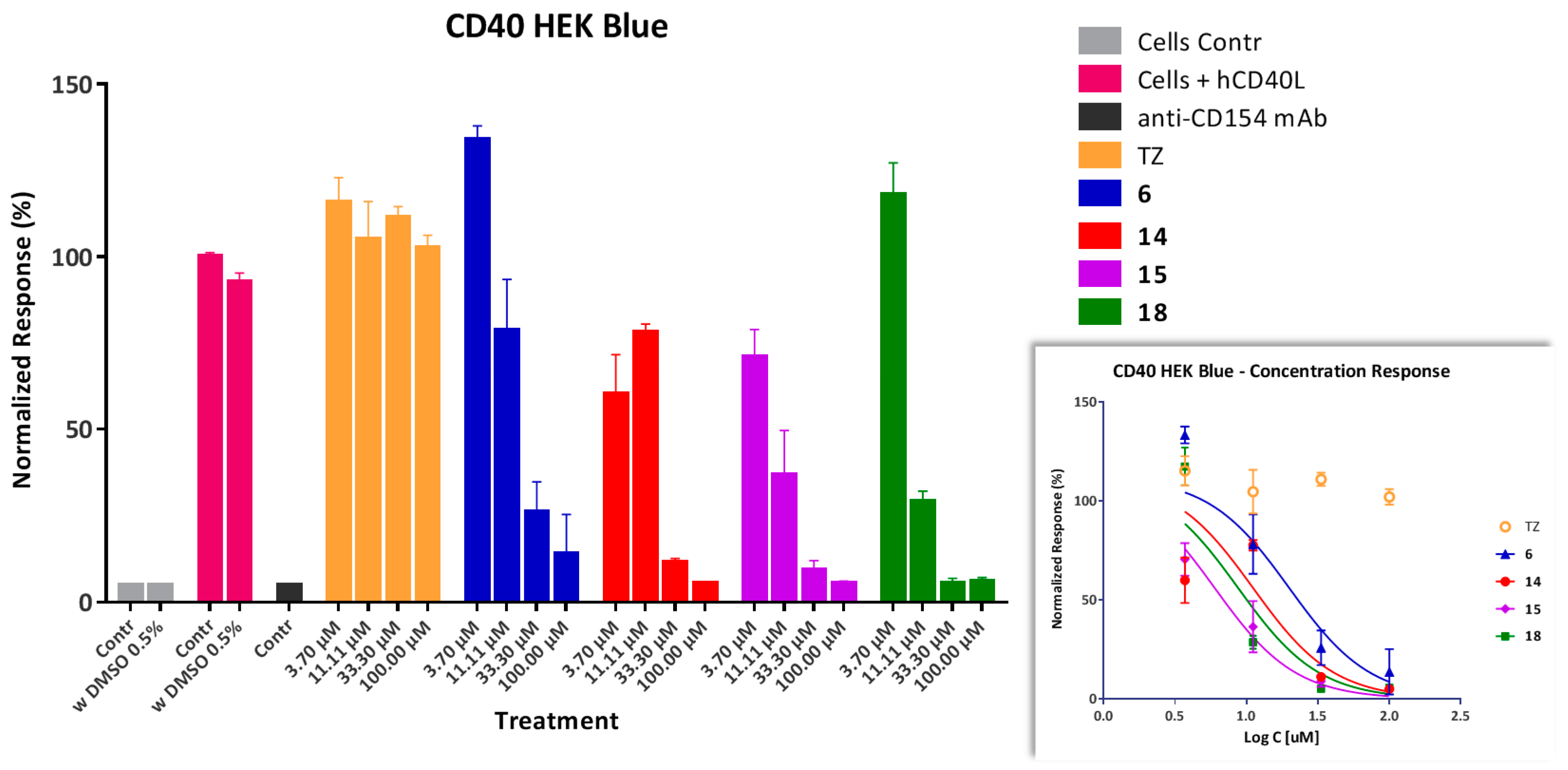
Inhibition of CD154-Induced NF-κB Activation in Sensor Cells
2.5. In Vivo Activity
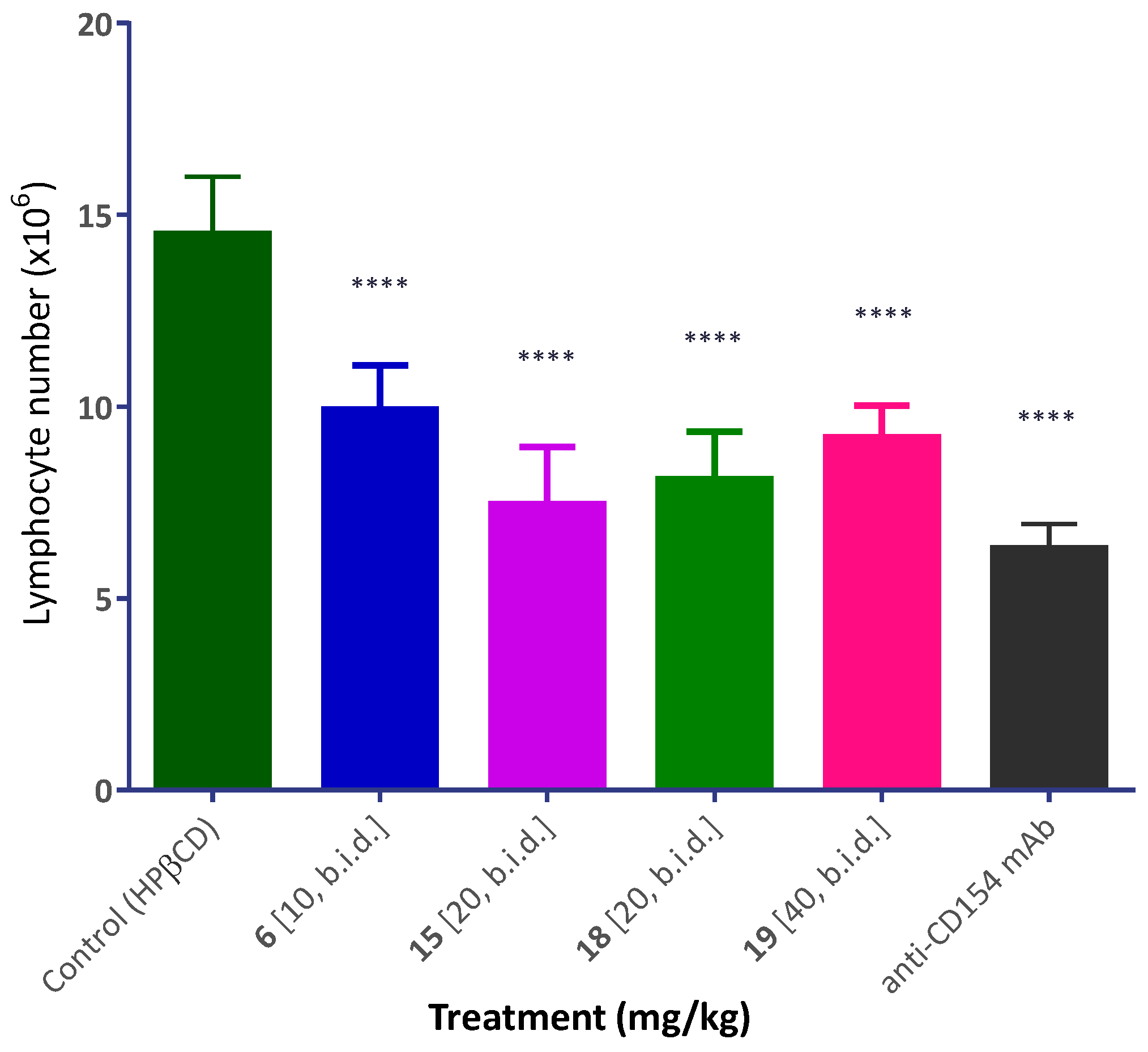
Inhibition of Alloantigen-Induced Immune Response
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Chemistry
4.3. Binding Assays
4.4. Protein Thermal Shift (Differential Scanning Fluorimetry)
4.5. CD40 Sensor Cell Assay
4.6. Cytotoxicity Assay
4.7. Animal Care and Treatment
4.8. Draining Lymph Node
4.9. Statistics and Data Fitting
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| CI | confidence interval |
| PPI | protein-protein interaction |
| T1D | type 1 diabetes |
| TNF | tumor necrosis factor |
| TNFSF | TNF superfamily |
References
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Vincenti, F.; Luggen, M. T cell costimulation: A rational target in the therapeutic armamentarium for autoimmune diseases and transplantation. Annu. Rev. Med. 2007, 58, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Rothstein, D.M.; Sayegh, M.H. Costimulatory pathways in transplantation: challenges and new developments. Immunol. Rev. 2009, 229, 271–293. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.L.; Stunz, L.L.; Bishop, G.A. CD40 and autoimmunity: The dark side of a great activator. Semin. Immunol. 2009, 21, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Benedict, C.A.; Ware, C.F. Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug Discov. 2013, 12, 147–168. [Google Scholar] [CrossRef] [PubMed]
- Giuroiu, I.; Weber, J. Novel checkpoints and cosignaling molecules in cancer immunotherapy. Cancer J. 2017, 23, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Szymkowski, D.E. The TNF superfamily in 2009: New pathways, new indications, and new drugs. Drug Discov. Today 2009, 14, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Hoos, A. Development of immuno-oncology drugs—From CTLA4 to PD1 to the next generations. Nat. Rev. Drug Discov. 2016, 15, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidou, M.; Zarganes-Tzitzikas, T.; Magiera, K.; Holak, T.A.; Dömling, A. Immune checkpoint PD-1/PD-L1: Is there life beyond antibodies? Angew. Chem. Int. Ed. Engl. 2018, 57. [Google Scholar] [CrossRef] [PubMed]
- Schönbeck, U.; Libby, P. The CD40/CD154 receptor/ligand dyad. Cell Mol. Life Sci. 2001, 58, 4–43. [Google Scholar] [PubMed]
- An, H.J.; Kim, Y.J.; Song, D.H.; Park, B.S.; Kim, H.M.; Lee, J.D.; Paik, S.G.; Lee, J.O.; Lee, H. Crystallographic and mutational analysis of the CD40-CD154 complex and its implications for receptor activation. J. Biol. Chem. 2011, 286, 11226–11235. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, N.S.; Chatzipetrou, M.; Masetti, M.; Ranuncoli, A.; Oliveira, M.; Wagner, J.L.; Kirk, A.D.; Harlan, D.M.; Burkly, L.C.; Ricordi, C. Long-term survival and function of intrahepatic islet allografts in rhesus monkeys treated with humanized anti-CD154. Proc. Natl. Acad. Sci. USA 1999, 96, 8132–8137. [Google Scholar] [CrossRef] [PubMed]
- Cardona, K.; Korbutt, G.S.; Milas, Z.; Lyon, J.; Cano, J.; Jiang, W.; Bello-Laborn, H.; Hacquoil, B.; Strobert, E.; Gangappa, S.; et al. Long-term survival of neonatal porcine islets in nonhuman primates by targeting costimulation pathways. Nat. Med. 2006, 12, 304–306. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Jarvinen, L.Z.; Lind, E.F.; Noelle, R.J. CD40/CD154 interactions at the interface of tolerance and immunity. Annu. Rev. Immunol. 2004, 22, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.H., Jr.; Vaitaitis, G.; Sanderson, R.; Poulin, M.; Dobbs, C.; Haskins, K. Expression of CD40 identifies a unique pathogenic T cell population in type 1 diabetes. Proc. Natl. Acad. Sci. USA 2002, 99, 3782–3787. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.L.; Mallevaey, T.; Gapin, L.; Haskins, K. T cells interact with T cells via CD40-CD154 to promote autoimmunity in type 1 diabetes. Eur. J. Immunol. 2012, 42, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Vaitaitis, G.M.; Olmstead, M.H.; Waid, D.M.; Carter, J.R.; Wagner, D.H., Jr. A CD40-targeted peptide controls and reverses type 1 diabetes in NOD mice. Diabetologia 2014, 57, 2366–2373. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.L.; Wagner, D.H., Jr.; Haskins, K. CD40 on NOD CD4 T cells contributes to their activation and pathogenicity. J. Autoimmun. 2008, 31, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.H., Jr. Overlooked mechanisms in type 1 diabetes etiology: How unique costimulatory molecules contribute to diabetogenesis. Front. Endocrinol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Vaitaitis, G.M.; Waid, D.M.; Yussman, M.G.; Wagner, D.H., Jr. CD40-mediated signalling influences trafficking, T-cell receptor expression, and T-cell pathogenesis, in the NOD model of type 1 diabetes. Immunology 2017, 152, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Andrews, D.; Colvin, R.B.; Sachs, D.H.; Cosimi, A.B. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat. Med. 2000, 6. [Google Scholar] [CrossRef] [PubMed]
- Koyama, I.; Kawai, T.; Andrews, D.; Boskovic, S.; Nadazdin, O.; Wee, S.L.; Sogawa, H.; Wu, D.L.; Smith, R.N.; Colvin, R.B.; et al. Thrombophilia associated with anti-CD154 monoclonal antibody treatment and its prophylaxis in nonhuman primates. Transplantation 2004, 77, 460–462. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Zuckermann, A.; Klepetko, W.; Wolner, E.; Ankersmit, H.J.; Moser, B.; Volf, I. Thrombophilia associated with anti-CD154 monoclonal antibody treatment and its prophylaxis in nonhuman primates. Transplantation 2004, 78, 1238–1239. [Google Scholar] [CrossRef] [PubMed]
- Mirabet, M.; Barrabes, J.A.; Quiroga, A.; Garcia-Dorado, D. Platelet pro-aggregatory effects of CD40L monoclonal antibody. Mol. Immunol. 2008, 45, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Pinelli, D.F.; Ford, M.L. Novel insights into anti-CD40/CD154 immunotherapy in transplant tolerance. Immunotherapy 2015, 7, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.H.; Yamniuk, A.P.; Borowski, V.; Kuhn, R.; Susulic, V.; Rex-Rabe, S.; Yang, X.; Zhou, X.; Zhang, Y.; Gillooly, K.; et al. Engineering of a novel anti-CD40L domain antibody for treatment of autoimmune diseases. J. Immunol. 2014, 192, 4083–4092. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Wakwe, W.; Higginbotham, L.B.; Mathews, D.V.; Breeden, C.P.; Stephenson, A.C.; Jenkins, J.; Strobert, E.; Price, K.; Price, L.; et al. Fc-Silent anti-CD154 domain antibody effectively prevents nonhuman primate renal allograft rejection. Am. J. Transplant. 2017, 17, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Downing, N.S.; Shah, N.D.; Aminawung, J.A.; Pease, A.M.; Zeitoun, J.D.; Krumholz, H.M.; Ross, J.S. Postmarket safety events among novel therapeutics approved by the US Food and Drug Administration between 2001 and 2010. JAMA 2017, 317, 1854–1863. [Google Scholar] [CrossRef] [PubMed]
- Sathish, J.G.; Sethu, S.; Bielsky, M.C.; de Haan, L.; French, N.S.; Govindappa, K.; Green, J.; Griffiths, C.E.; Holgate, S.; Jones, D.; et al. Challenges and approaches for the development of safer immunomodulatory biologics. Nat. Rev. Drug Discov. 2013, 12, 306–324. [Google Scholar] [CrossRef] [PubMed]
- Deambrosis, I.; Lamorte, S.; Giaretta, F.; Tei, L.; Biancone, L.; Bussolati, B.; Camussi, G. Inhibition of CD40-CD154 costimulatory pathway by a cyclic peptide targeting CD154. J. Mol. Med. 2009, 87, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef] [PubMed]
- Giannoukakis, N.; Phillips, B.; Trucco, M. Toward a cure for type 1 diabetes mellitus: diabetes-suppressive dendritic cells and beyond. Pediatr. Diabetes 2008, 9, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, G.M.; Horne, R.; Chanez, P. Compliance in asthma. Respir. Med. 1999, 93, 763–769. [Google Scholar] [CrossRef]
- Moia, M.; Mantovani, L.G.; Carpenedo, M.; Scalone, L.; Monzini, M.S.; Cesana, G.; Mannucci, P.M. Patient preferences and willingness to pay for different options of anticoagulant therapy. Intern. Emerg. Med. 2013, 8, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Hess, C.; Palmer, E. What is the role of autoimmunity in type 1 diabetes? A clinical perspective. Diabetologia 2014, 57, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, L.M.; Haller, M.J.; Schatz, D.A. Understanding pre-type 1 diabetes: The key to prevention. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Skyler, J.S.; Ricordi, C. Stopping type 1 diabetes: Attempts to prevent or cure type 1 diabetes in man. Diabetes 2011, 60, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lernmark, Å.; Larsson, H.E. Immune therapy in type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2013, 9, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Skyler, J.S. Prevention and reversal of type 1 diabetes—Past challenges and future opportunities. Diabetes Care 2015, 38, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, P. Small-molecule protein-protein interaction inhibitors: Therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB Life 2010, 62, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Milroy, L.G.; Grossmann, T.N.; Hennig, S.; Brunsveld, L.; Ottmann, C. Modulators of protein-protein interactions. Chem. Rev. 2014, 114, 4695–4748. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Buchwald, P. TNF superfamily protein-protein interactions: Feasibility of small-molecule modulation. Curr. Drug. Targets 2015, 16, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Huck, B.R.; Kötzner, L.; Urbahns, K. Small molecules drive big improvements in immuno-oncology therapies. Angew. Chem. Int. Ed. Engl. 2018, 57, 4412–4428. [Google Scholar] [CrossRef] [PubMed]
- Bojadzic, D.; Buchwald, P. Toward small-molecule inhibition of protein-protein interactions: General aspects and recent progress in targeting costimulatory and coinhibitory (immune checkpoint) interactions. Curr. Top. Med. Chem. 2018, in press. [Google Scholar]
- Mullard, A. 2015 FDA drug approvals. Nat. Rev. Drug Discov. 2016, 15, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Gadek, T.R.; Burdick, D.J.; McDowell, R.S.; Stanley, M.S.; Marsters, J.C., Jr.; Paris, K.J.; Oare, D.A.; Reynolds, M.E.; Ladner, C.; Zioncheck, K.A.; et al. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science 2002, 295, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Margolles-Clark, E.; Umland, O.; Kenyon, N.S.; Ricordi, C.; Buchwald, P. Small molecule costimulatory blockade: organic dye inhibitors of the CD40–CD154 interaction. J. Mol. Med. 2009, 87, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Margolles-Clark, E.; Kenyon, N.S.; Ricordi, C.; Buchwald, P. Effective and specific inhibition of the CD40–CD154 costimulatory interaction by a naphthalenesulphonic acid derivative. Chem. Biol. Drug Des. 2010, 76, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Song, Y.; Bojadzic, D.; Tamayo-Garcia, A.; Landin, A.M.; Blomberg, B.B.; Buchwald, P. Small-molecule inhibitors of the CD40-CD40L costimulatory protein-protein interaction. J. Med. Chem. 2017, 60, 8906–8922. [Google Scholar] [CrossRef] [PubMed]
- Venkatraj, M.; Messagie, J.; Joossens, J.; Lambeir, A.M.; Haemers, A.; Van der Veken, P.; Augustyns, K. Synthesis and evaluation of non-basic inhibitors of urokinase-type plasminogen activator (uPA). Bioorg. Med. Chem. 2012, 20, 1557–1568. [Google Scholar] [CrossRef] [PubMed]
- Kassack, M.U.; Braun, K.; Ganso, M.; Ullmann, H.; Nickel, P.; Boing, B.; Muller, G.; Lambrecht, G. Structure-activity relationships of analogues of NF449 confirm NF449 as the most potent and selective known P2X1 receptor antagonist. Eur. J. Med. Chem. 2004, 39, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Margolles-Clark, E.; Jacques-Silva, M.C.; Ganesan, L.; Umland, O.; Kenyon, N.S.; Ricordi, C.; Berggren, P.-O.; Buchwald, P. Suramin inhibits the CD40–CD154 costimulatory interaction: A possible mechanism for immunosuppressive effects. Biochem. Pharmacol. 2009, 77, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, C.; Bertozzi, C.; Georg, G.I.; Kiessling, L.; Lindsley, C.; Liotta, D.; Merz, K.M., Jr.; Schepartz, A.; Wang, S. The ecstasy and agony of assay interference compounds. J. Med. Chem. 2017, 60, 2165–2168. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.Y.; Shoichet, B.K. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, L.; Margolles-Clark, E.; Song, Y.; Buchwald, P. The food colorant erythrosine is a promiscuous protein-protein interaction inhibitor. Biochem. Pharmacol. 2011, 81, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; Partch, C.L. Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci. 2015, 79. [Google Scholar] [CrossRef]
- Grasberger, B.L.; Lu, T.; Schubert, C.; Parks, D.J.; Carver, T.E.; Koblish, H.K.; Cummings, M.D.; LaFrance, L.V.; Milkiewicz, K.L.; Calvo, R.R.; et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 2005, 48, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Bodor, N.; Buchwald, P. Retrometabolic Drug Design and Targeting, 1st ed.; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Burrell, B.E.; Lu, G.; Li, X.C.; Bishop, D.K. OX40 costimulation prevents allograft acceptance induced by CD40–CD40L blockade. J. Immunol. 2009, 182, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef]
- Karpusas, M.; Hsu, Y.M.; Wang, J.H.; Thompson, J.; Lederman, S.; Chess, L.; Thomas, D. 2 Å Crystal structure of an extracellular fragment of human CD40 ligand. Structure 1995, 3, 1031–1039. [Google Scholar] [CrossRef]
- Ganesan, L.; Vidović, D.; Schürer, S.C.; Buchwald, P. Exploratory computational assessment of possible binding modes for small molecule inhibitors of the CD40–CD154 costimulatory interaction. Pharmazie 2012, 67, 374–379. [Google Scholar] [PubMed]
- Silvian, L.F.; Friedman, J.E.; Strauch, K.; Cachero, T.G.; Day, E.S.; Qian, F.; Cunningham, B.; Fung, A.; Sun, L.; Shipps, G.W.; et al. Small molecule inhibition of the TNF family cytokine CD40 ligand through a subunit fracture mechanism. ACS Chem. Biol. 2011, 6, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development setting. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- DeGoey, D.A.; Chen, H.J.; Cox, P.B.; Wendt, M.D. Beyond the rule of 5: Lessons learned from AbbVie’s drugs and compound collection. J. Med. Chem. 2017, 61, 2636–2651. [Google Scholar] [CrossRef] [PubMed]
- Still, W.C.; Kahn, M.; Mitra, A. Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 1978, 43, 2923–2925. [Google Scholar] [CrossRef]
- Adachi, Y.; Nakagawa, H.; Matsuo, K.; Suzuki, T.; Miyata, N. Photoactivatable HNO-releasing compounds using the retro-Diels-Alder reaction. Chem. Commun. 2008. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Margolles-Clark, E.; Bayer, A.; Buchwald, P. Small-molecule modulators of the OX40–OX40L costimulatory protein–protein interaction. Br. J. Pharmacol. 2014, 171, 4955–4969. [Google Scholar] [CrossRef] [PubMed]
- Cechin, S.R.; Buchwald, P. Effects of representative glucocorticoids on TNFa- and CD40L-induced NF-kB activation in sensor cells. Steroids 2014, 85, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, P. A three-parameter two-state model of receptor function that incorporates affinity, efficacy, and signal amplification. Pharmacol. Res. Perspect. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Name | Formula | R1 a | R2 a | CD40 IC50 [μM] | OX40 IC50 [μM] | BAFF IC50 [μM] | TNF IC50 [μM] | NF-κB IC50 [μM] |
|---|---|---|---|---|---|---|---|---|---|
| 6 | DRI-C21041 | C30H21N3O7S | 1-SO3H | 4′-NO2 | 0.087 | 317.9 | 0.328 | >1000 | 19.9 |
| 7 | DRI-C24041 | C30H21N3O7S | 4-SO3H | 4′-NO2 | 0.299 | 9.34 | 2.007 | 554.6 | |
| 8 | DRI-21047a | C31H21F3N2O5S | 1-SO3H | 4′-CF3 | 57.85 | 10.07 | 42.18 | 61.07 | |
| 9 | DRI-C24541 | C32H23N3O6 | 4-CO2Me | 4′-NO2 | 55.99 | >1000 | 685.6 | 795.8 | |
| 10 | DRI-C2105041 | C30H21N3O10S2 | 1,5-(SO3H)2 | 4′-NO2 | 68.31 | 135.0 | 439.7 | 688.5 | |
| 11 | DRI-C2105045 | C32H24N2O10S2 | 1,5-(SO3H)2 | 4′-CO2Me | 60.07 | 120.4 | 540.1 | 456.1 | |
| 12 | DRI-C2104121 | C30H20N4O9S | 1-SO3H | 2′,4′-(NO2)2 | 50.50 | 10.31 | 38.14 | 61.68 | |
| 13 | DRI-C2104531 | C32H23N3O9S | 1-SO3H | 3′-NO2-4′-CO2Me | 0.390 | 86.19 | 5.27 | 674.4 | |
| 14 | DRI-C21091 | C36H25N3O7S | 1-SO3H | 4′-(4-NO2)C6H4 | 0.020 | 6.88 | 0.784 | 70.35 | 11.1 |
| 15 | DRI-C21095 | C38H28N2O7S | 1-SO3H | 4′-(4-CO2Me)C6H4 | 0.019 | 1.013 | 1.517 | 26.55 | 6.0 |
| 16 | DRI-C31041 | C32H25N3O7S | 1-SO3H | 4′-NO2 | 44.80 | 7.28 | 26.73 | 61.48 | |
| 17 | DRI-C41041 | C28H19N3O7S | 1-SO3H | 4′-NO2 | 57.45 | 36.63 | 81.38 | 118.5 | |
| 18 | DRI-C61041 | C29H20N4O7S | 1-SO3H | 4′-NO2 | 1.34 | 5.48 | 8.07 | 46.94 | 8.7 |
| 19 | DRI-C61045 | C31H23N3O7S | 1-SO3H | 4′-CO2Me | 2.41 | 6.97 | 13.76 | 182.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bojadzic, D.; Chen, J.; Alcazar, O.; Buchwald, P. Design, Synthesis, and Evaluation of Novel Immunomodulatory Small Molecules Targeting the CD40–CD154 Costimulatory Protein-Protein Interaction. Molecules 2018, 23, 1153. https://doi.org/10.3390/molecules23051153
Bojadzic D, Chen J, Alcazar O, Buchwald P. Design, Synthesis, and Evaluation of Novel Immunomodulatory Small Molecules Targeting the CD40–CD154 Costimulatory Protein-Protein Interaction. Molecules. 2018; 23(5):1153. https://doi.org/10.3390/molecules23051153
Chicago/Turabian StyleBojadzic, Damir, Jinshui Chen, Oscar Alcazar, and Peter Buchwald. 2018. "Design, Synthesis, and Evaluation of Novel Immunomodulatory Small Molecules Targeting the CD40–CD154 Costimulatory Protein-Protein Interaction" Molecules 23, no. 5: 1153. https://doi.org/10.3390/molecules23051153
APA StyleBojadzic, D., Chen, J., Alcazar, O., & Buchwald, P. (2018). Design, Synthesis, and Evaluation of Novel Immunomodulatory Small Molecules Targeting the CD40–CD154 Costimulatory Protein-Protein Interaction. Molecules, 23(5), 1153. https://doi.org/10.3390/molecules23051153