
Tetrel Bonds with π-Electrons Acting as Lewis Bases—Theoretical Results and Experimental Evidences


Abstract
:1. Introduction
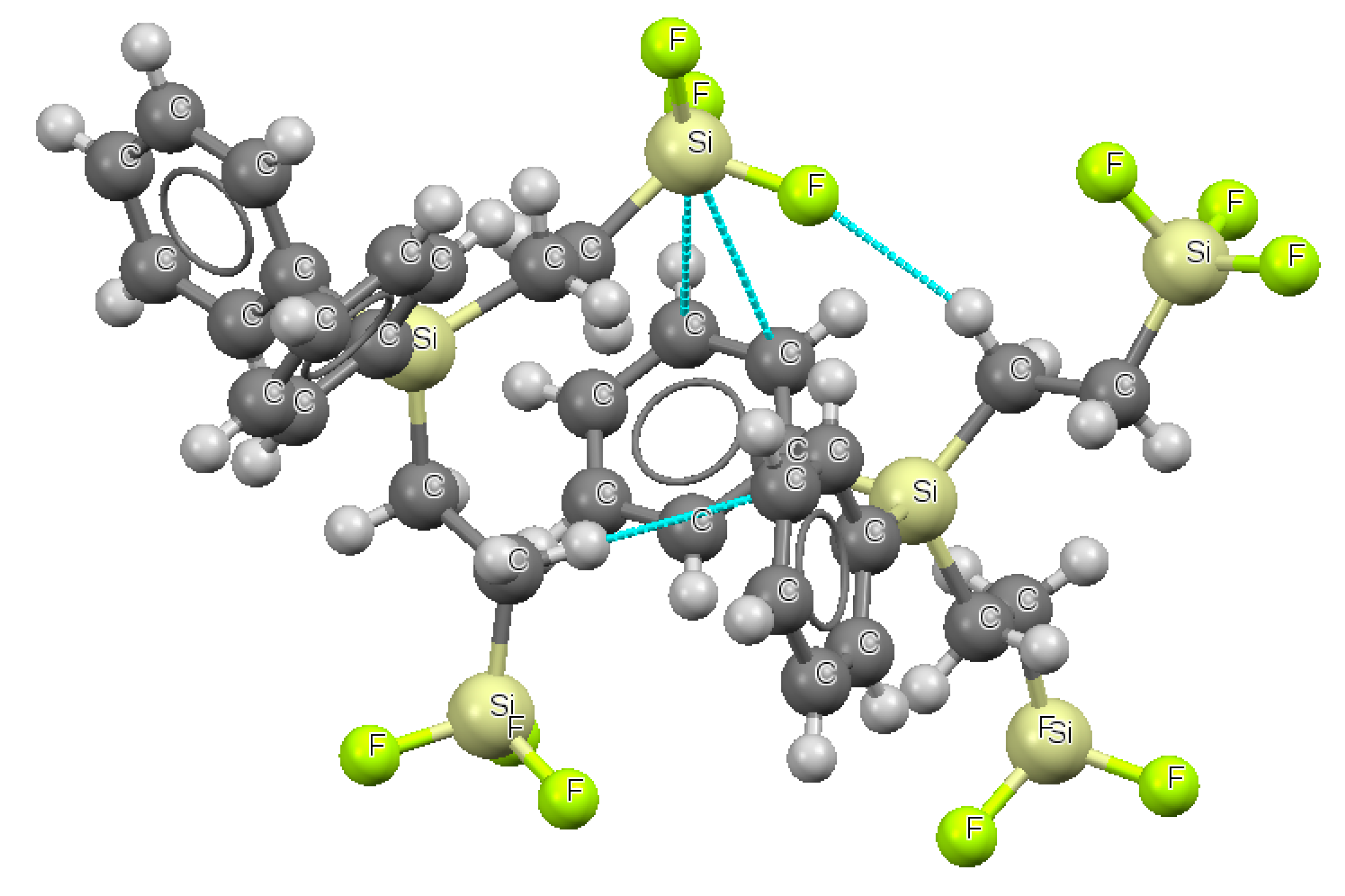
2. Results and Discussion
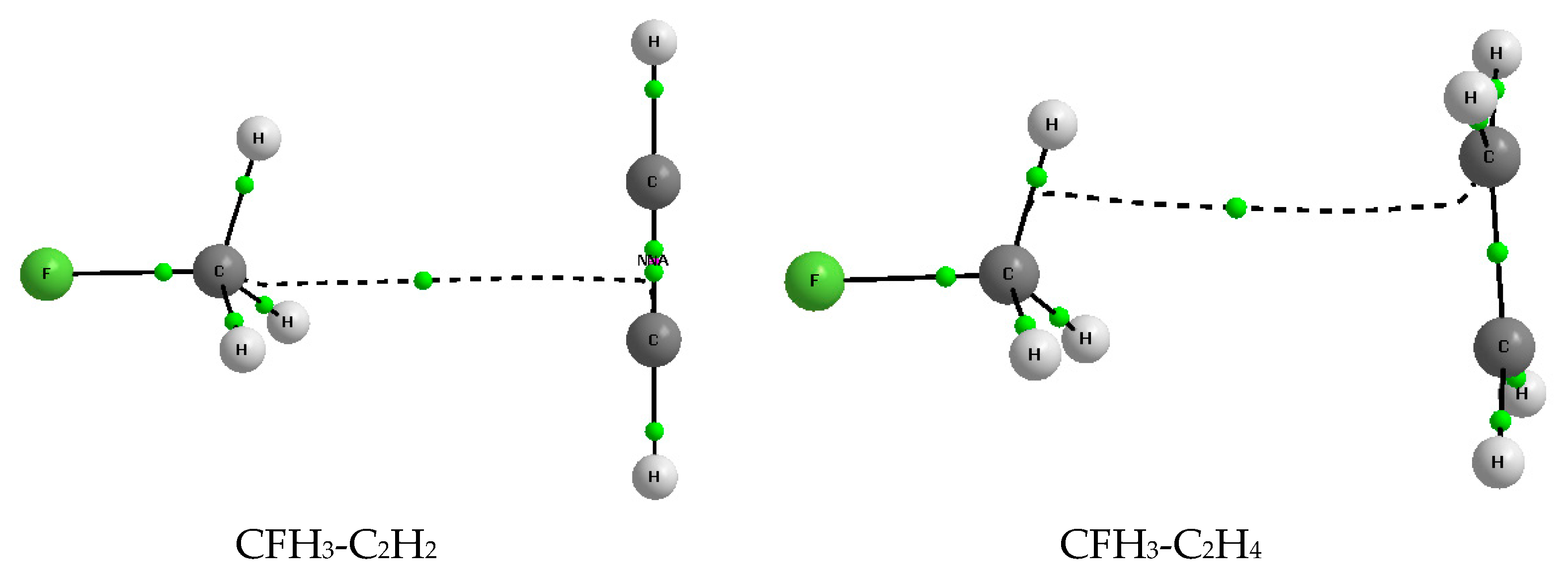
2.1. Energetic and Geometric Parameters
2.2. Nature of Interactions—Decomposition of Interaction Energy
2.3. Quantum Theory of Atoms in Molecules Parameters
3. Computational Details
4. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- García-LLinás, X.; Bauzá, A.; Seth, S.K.; Frontera, A. Importance of R-CF3···O Tetrel Bonding Interactions in Biological Systems. J. Phys. Chem. A 2017, 121, 5371–5376. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Hydrogen bonds, and σ-hole and π-hole bonds–mechanisms protecting doublet and octet electron structures. Phys. Chem. Chem. Phys. 2017, 19, 29742–29759. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Bundhun, A.; Ramasami, P.; Murray, J.S.; Politzer, P. Trends in σ-hole strengths and interactions of F3MX molecules (M = C, Si, Ge and X = F, Cl, Br, I). J. Mol. Model. 2013, 19, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Lane, P.; Concha, M.C. Electrostatically Driven Complexes of SiF4 with Amines. Int. J. Quantum Chem. 2009, 109, 3773–3780. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Chehayber, J.M.; Nagy, S.T.; Lin, C.S. Ab initio studies of complexes between SiF4 and ammonia. Can. J. Chem. 1984, 62, 27–31. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular Complexes between Silicon Derivatives and Electron-Rich Groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Tetrel bond-σ-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Helminiak, H.M.; Knauf, R.R.; Danforth, S.J.; Phillips, J.A. Structural and Energetic Properties of Acetonitrile–Group IV (A & B) Halide Complexes. J. Phys. Chem. A 2014, 118, 4266–4277. [Google Scholar] [PubMed]
- Mani, D.; Arunan, E. The X-C···Y (X = O/F, Y = O/S/F/Cl/Br/N/P) ‘carbon bond’ and hydrophobic interactions. Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-Z.; Zhuo, H.-Y.; Li, H.-B.; Liu, Z.-B.; Li, W.-Z.; Cheng, J.-B. Tetrel-Hydride Interaction between XH3F (X = C, Si, Ge, Sn) and HM (M = Li, Na, BeH, MgH). J. Phys. Chem. A 2015, 119, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Systematic Elucidation of Factors That Influence the Strength of Tetrel Bonds. J. Phys. Chem. A 2017, 121, 5561–5568. [Google Scholar] [CrossRef] [PubMed]
- Kubelka, J.; Bickelhaupt, F.M. Activation Strain Analysis of SN2 Reactions at C, N, O, and F centers. J. Phys. Chem. A 2017, 121, 885–891. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Dronskowski, R. Tetrel Bonds in Infinite Molecular Chains by Electronic Structure Theory and Their Role for Crystal Stabilization. J. Phys. Chem. A 2017, 121, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Marín-Luna, M.; Alkorta, I.; Elguero, J. A theoretical study of the HnF4−nSi:N-base (n = 1–4) tetrel-bonded complexes. Theor. Chem. Acc. 2017, 136, 41. [Google Scholar] [CrossRef]
- Liu, M.; Li, Q.; Scheiner, S. Comparison of tetrel bonds in neutral and protonated complexes of pyridine TF3 and furan TF3 (T = C, Si, and Ge) with NH3. Phys. Chem. Chem. Phys. 2017, 19, 5550–5559. [Google Scholar] [CrossRef] [PubMed]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Implications of monomer deformations for tetrel and pnicogen bonds. Phys. Chem. Chem. Phys. 2018, 20, 8832–8841. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Steric Crowding in tetrel Bonds. J. Phys. Chem. A 2018, 122, 2550–2562. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J.; Sokalski, W.A. Are Various σ-Hole Bonds Steered by the Same Mechanisms? ChemPhysChem 2017, 18, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Drew, M.G.B.; Bauzá, A.; Frontera, A.; Chattopadhyay, S. Non-covalent tetrel bonding interactions in hemidirectional lead (II) complexes with nickel(II)–salen type metalloligands. New J. Chem. 2018, 42, 6062–6076. [Google Scholar]
- Grabowski, S.J. Tetrel bonds, penta- and hexa-coordinated tin and lead centres. Appl. Organomet. Chem. 2017, 31, e3727. [Google Scholar] [CrossRef]
- Grabowski, S.J. Lewis Acid Properties of Tetrel tetrafluorides—The Coincidence of the σ-Hole Concept with the QTAIM Approach. Crystals 2017, 7, 43. [Google Scholar] [CrossRef]
- Nishio, M.; Hirota, M.; Umezawa, Y. The CH/π Interaction, Evidence, Nature, and Consequences; Wiley-VCH: New York, NY, USA, 1998. [Google Scholar]
- Vasilyev, A.V.; Lindeman, S.V.; Kochi, J.K. Noncovalent binding of the halogens to aromatic donors. Discrete structures of labile Br2 complexes with benzene and toluene. Chem. Commun. 2001, 10, 909–910. [Google Scholar] [CrossRef]
- Duarte, D.J.R.; de las Vallejos, M.M.; Peruchena, N.M. Topological analysis of aromatic halogen/hydrogen bonds by electron charge density and electrostatic potentials. J. Mol. Model. 2010, 16, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, H.; Li, Q.; Li, W.; Cheng, J. Is π halogen bonding or lone pair···π interaction formed between borazine and some halogenated compounds. Phys. Chem. Chem. Phys. 2014, 16, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zeng, Y.; Li, X.; Meng, L.; Zhang, X. A comprehensive analysis of P···π pnicogen bonds: Substitution effects and comparison with Br···π halogen bonds. J. Mol. Model. 2015, 21, 143. [Google Scholar] [CrossRef] [PubMed]
- Fau, S.; Frenking, G. Theoretical investigation of the weakly bonded donor-acceptor complexes X3B-H2, X3B-C2H4, and X3B-C2H2 (X = H, F, Cl). Mol. Phys. 1999, 96, 519–527. [Google Scholar]
- Grabowski, S.J. Triel Bonds, π-Hole-π-Electrons Interactions in Complexes of Boron and Aluminium Trihalides and Trihydrides with Acetylene and Ethylene. Molecules 2015, 20, 11297–11316. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. New Type of Halogen Bond: Multivalent Halogen Interacting with π- and σ-Electrons. Molecules 2017, 22, 2150. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules, a Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Weinhold, F.; Landis, C. Valency and Bonding, a Natural Bond Orbital Donor—Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Morokuma, K. Molecular Orbital Studies of Hydrogen Bonds. III. C=O···H-O Hydrogen Bond in H2CO···H2O and H2CO···2H2O. J. Chem. Phys. 1971, 55, 1236–1244. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree–Fock Slater method. Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. Molecular electrostatic potentials and noncovalent interactions. WIREs Comput. Mol. Sci. 2017, 7, e1326. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Sokalski, W.A. Different types of hydrogen bonds: Correlation analysis of interaction energy components. J. Phys. Org. Chem. 2005, 18, 779–784. [Google Scholar] [CrossRef]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Evidence for resonance-assisted hydrogen bonding. 4. Covalent nature of the strong homonuclear hydrogen bond. Study of the O-H···O system by crystal structure correlation methods. J. Am. Chem. Soc. 1994, 116, 909–915. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. Molecular Surfaces, van der Waals Radii and Electrostatic Potentials in Relation to Noncovalent Interactions. Croat. Chem. Acta 2009, 82, 267–275. [Google Scholar]
- Pauling, L. The Nature of the Chemical Bond, 3rd ed.; Cornell University Press: New York, NY, USA, 1960. [Google Scholar]
- Batsanov, S.S. Van der Waals Radii of Elements. Inorg. Mater. 2001, 37, 871–885. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen bonds and other interactions as a response to protect doublet/octet electron structure. J. Mol. Model. 2018, 24, 38. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Solà, M.; Bickelhaupt, F.M. Hydrogen-Hydrogen Bonding in Planar Biphenyl, Predicted by Atoms-In-Molecules Theory, Does Not Exist. Chem. Eur. J. 2006, 12, 2889–2895. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Pauli Repulsions Exist Only in the Eye of the Beholder. Chem. Eur. J. 2006, 12, 2896–2901. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Solà, M.; Bickelhaupt, F.M. A Model of the Chemical Bond Must Be Rooted in Quantum Mechanics, Provide Insight, and Possess Predictive Power. Chem. Eur. J. 2006, 12, 2902–2905. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Ab Initio Calculations on Conventional and Unconventional Hydrogen Bonds–Study of the Hydrogen Bond Strength. J. Phys. Chem. A 2001, 105, 10739–10746. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Ugalde, J.M. Bond Paths Show Preferable Interactions: Ab Initio and QTAIM Studies on the X-H···π Hydrogen Bond. J. Phys. Chem. A 2010, 114, 7223–7229. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03, Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. III. The second row atoms, Al-Ar. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Metz, B.; Stoll, H.; Dolg, M. Small-core multiconfiguration-Dirac-Hartree-Fock-adjusted pseudopotentials for post-d main group elements: Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563–2569. [Google Scholar] [CrossRef]
- Peterson, K.A. Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the post-d group 13-15 elements. J. Chem. Phys. 2003, 119, 11099–11112. [Google Scholar] [CrossRef]
- Piela, L. Ideas of Quantum Chemistry; Elsevier Science Publishers: Amsterdam, The Netherlands, 2007; pp. 684–691. [Google Scholar]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–561. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll (Version 11.08.23); TK Gristmill Software: Overland Park, KS, USA, 2011. Available online: aim.tkgristmill.com.
- Grabowski, S.J. Dihydrogen bond and X-H···σ interaction as sub-classes of hydrogen bond. J. Phys. Org. Chem. 2013, 26, 452–459. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavious. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Velde, G.T.E.; Bickelhaupt, F.M.; Baerends, E.J.; Guerra, C.F.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Wong, R.; Allen, F.H.; Willett, P. The scientific impact of the Cambridge Structural Database: A citation-based study. J. Appl. Crystallogr. 2010, 43, 811–824. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the author. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Distance | Eint | Ebin | Edef | BSSE |
|---|---|---|---|---|---|
| CFH3-C2H2 | 3.428 (+0.53) | −1.3 | −1.2 | 0.0 | 0.3 |
| CFH3-C2H4 | 3.458 (+0.56) | −1.3 | −1.3 | 0.0 | 0.4 |
| CFH3-C6H6 | 3.398 (+0.50) | −2.7 | −2.7 | 0.0 | 1.0 |
| CFH3-C5H5− | 3.241 (+0.34) | −10.8 | −10.5 | 0.3 | 1.2 |
| SiFH3-C2H2 | 3.344 (+0.04) | −2.4 | −2.3 | 0.0 | 0.6 |
| SiFH3-C2H4 | 3.315 (−0.02) | −2.7 | −2.6 | 0.1 | 0.8 |
| SiFH3-C6H6 | 3.253 (−0.05) | −3.7 | −3.7 | 0.1 | 1.3 |
| SiFH3-C5H5- | 2.477 (−0.82) | −27.5 | −18.1 | 9.4 | 2.0 |
| GeFH3-C2H2 | 3.285 (−0.02) | −2.6 | −2.6 | 0.1 | 1.1 |
| GeFH3-C2H4 | 3.253 (−0.05) | −2.9 | −2.9 | 0.1 | 1.5 |
| GeFH3-C6H6 | 3.203 (−0.10) | −4.3 | −4.2 | 0.1 | 2.7 |
| GeFH3-C5H5− | 2.525 (−0.78) | −29.3 | −21.0 | 8.3 | 4.4 |
| SnFH3-C2H2 | 3.325 (−0.13) | −3.4 | −3.3 | 0.1 | 1.3 |
| SnFH3-C2H4 | 3.280 (−0.17) | −3.8 | −3.7 | 0.2 | 1.8 |
| SnFH3-C6H6 | 3.183 (−0.27) | −5.5 | −5.2 | 0.3 | 3.2 |
| SnFH3-C5H5− | 2.519 (−0.93) | −41.9 | −30.6 | 11.4 | 5.2 |
| PbFH3-C2H2 | 3.323 (−0.18) | −3.5 | −3.4 | 0.1 | 2.2 |
| PbFH3-C2H4 | 3.267 (−0.23) | −3.8 | −3.7 | 0.1 | 3.1 |
| PbFH3-C6H6 | 3.148 (−0.35) | −5.8 | −5.6 | 0.3 | 5.7 |
| PbFH3-C5H5− | 2.624 (−0.88) | −39.3 | −31.8 | 7.6 | 8.3 |
| Complex | ZF% | Angle% | ENBO1 | ENBO2 | El-Trans | Z-Charge a |
|---|---|---|---|---|---|---|
| CFH3-C2H2 | 0.14 | 0.00 | 0.5 | 0.0 | −0.001 | −0.158 |
| CFH3-C2H4 | 0.14 | 0.00 | 0.6 | 0.0 | −0.002 | −0.158 |
| CFH3-C6H6 | 0.29 | 0.09 | 0.4 | 0.8 | −0.004 | −0.156 |
| CFH3-C5H5- | 1.95 | 0.28 | 1.5 | 2.4 | −0.030 | −0.378 |
| SiFH3-C2H2 | 0.25 | 0.65 | 1.7 | 0.3 | −0.014 | 1.227 |
| SiFH3-C2H4 | 0.31 | 0.74 | 2.3 | 0.4 | −0.021 | 1.218 |
| SiFH3-C6H6 | 0.31 | 0.74 | 1.8 | 1.0 | −0.019 | 1.221 |
| SiFH3-C5H5− | 4.46 | 9.06 | 24.5 | 20.9 | −0.250 | 1.086 |
| GeFH3-C2H2 | 0.40 | 0.85 | 3.1 | 0.6 | −0.019 | 1.025 |
| GeFH3-C2H4 | 0.52 | 0.85 | 4.1 | 0.7 | −0.027 | 1.015 |
| GeFH3-C6H6 | 0.52 | 0.85 | 3.2 | 1.6 | −0.025 | 1.021 |
| GeFH3-C5H5− | 5.18 | 8.95 | 33.4 | 22.1 | −0.250 | 0.906 |
| SnFH3-C2H2 | 0.46 | 1.34 | 4.1 | 1.2 | −0.024 | 1.246 |
| SnFH3-C2H4 | 0.57 | 1.43 | 5.4 | 1.9 | −0.035 | 1.232 |
| SnFH3-C6H6 | 0.62 | 1.72 | 4.8 | 2.9 | −0.035 | 1.240 |
| SnFH3-C5H5− | 5.11 | 11.45 | 37.9 | 40.5 | −0.289 | 1.148 |
| PbFH3-C2H2 | 0.59 | 0.99 | 4.9 | 1.0 | −0.027 | 1.085 |
| PbFH3-C2H4 | 0.78 | 1.28 | 7.0 | 2.4 | −0.039 | 1.070 |
| PbFH3-C6H6 | 0.83 | 1.68 | 6.1 | 3.9 | −0.044 | 1.080 |
| PbFH3-C5H5− | 5.44 | 8.78 | 36.5 | 31.0 | −0.289 | 1.013 |
| Complex | ΔEPauli | ΔEelstat | ΔEorb | ΔEdisp | ΔEint |
|---|---|---|---|---|---|
| CFH3-C2H2 | 2.4 | −1.3 | −0.8 | −1.4 | −1.1 |
| CFH3-C2H4 | 2.7 | −1.4 | −0.9 | −1.6 | −1.2 |
| CFH3-C6H6 | 4.7 | −2.4 | −1.5 | −3.6 | −2.8 |
| CFH3-C5H5− | 8.9 | −11.0 | −5.6 | −4.2 | −11.9 |
| SiFH3-C2H2 | 5.6 | −3.4 | −2.7 | −2.2 | −2.6 |
| SiFH3-C2H4 | 7.3 | −4.3 | −3.5 | −2.8 | −3.2 |
| SiFH3-C6H6 | 8.2 | −4.1 | −3.3 | −4.5 | −3.8 |
| SiFH3-C5H5− | 54.2 | −43.4 | −34.9 | −5.6 | −29.8 |
| GeFH3-C2H2 | 7.1 | −4.4 | −3.2 | −2.7 | −3.2 |
| GeFH3-C2H4 | 9.1 | −5.5 | −4.1 | −3.4 | −3.8 |
| GeFH3-C6H6 | 10.6 | −5.6 | −4.1 | −5.8 | −5.0 |
| GeFH3-C5H5− | 56.1 | −47.7 | −32.8 | −6.3 | −30.7 |
| SnFH3-C2H2 | 8.7 | −5.8 | −3.8 | −3.0 | −3.9 |
| SnFH3-C2H4 | 11.6 | −7.3 | −5.0 | −3.9 | −4.6 |
| SnFH3-C6H6 | 14.0 | −7.8 | −5.6 | −6.9 | −6.2 |
| SnFH3-C5H5− | 75.3 | −68.1 | −41.8 | −6.6 | −41.2 |
| PbFH3-C2H2 | 9.2 | −6.3 | −3.8 | −2.8 | −3.6 |
| PbFH3-C2H4 | 12.9 | −8.3 | −5.1 | −3.7 | −4.2 |
| PbFH3-C6H6 | 17.9 | −10.0 | −6.6 | −7.4 | −6.2 |
| PbFH3-C5H5− | 71.4 | −65.9 | −37.2 | −6.6 | −38.4 |
| Complex | ρBCP | ∇2ρBCP | HBCP | BP-Type |
|---|---|---|---|---|
| CFH3-C2H2 | 0.005 | 0.020 | 0.001 | C···C |
| CFH3-C2H4 | 0.005 | 0.018 | 0.001 | C···C |
| CFH3-C6H6 | 0.007 | 0.022 | 0.001 | (C)H···C |
| CFH3-C5H5− | 0.011 | 0.038 | 0.001 | (C)H···C |
| SiFH3-C2H2 | 0.007 | 0.022 | 0.001 | Si···C |
| SiFH3-C2H4 | 0.008 | 0.023 | 0.001 | (Si)H···C |
| SiFH3-C6H6 | 0.008 | 0.025 | 0.001 | (Si)H···C |
| SiFH3-C5H5− | 0.036 | 0.012 | −0.012 | Si···C |
| GeFH3-C2H2 | 0.009 | 0.027 | 0.001 | Ge···NNA(CC) |
| GeFH3-C2H4 | 0.010 | 0.028 | 0.001 | Ge···BCP(CC) |
| GeFH3-C6H6 | 0.010 | 0.028 | 0.001 | Ge···C |
| - | 0.007 | 0.024 | 0.001 | (Ge)H···C |
| GeFH3-C5H5− | 0.037 | 0.045 | −0.008 | Ge···C |
| SnFH3-C2H2 | 0.010 | 0.025 | 0.001 | Sn···NNA(CC) |
| SnFH3-C2H4 | 0.011 | 0.029 | 0.001 | Sn···BCP(CC) |
| SnFH3-C6H6 | 0.012 | 0.029 | 0.001 | Sn···C |
| - | 0.007 | 0.022 | 0.001 | (Sn)H···C |
| SnFH3-C5H5− | 0.045 | 0.066 | −0.011 | Sn···C |
| PbFH3-C2H2 | 0.011 | 0.034 | 0.001 | Pb···NNA(CC) |
| PbFH3-C2H4 | 0.013 | 0.036 | 0.001 | Pb···BCP(CC) |
| PbFH3-C6H6 | 0.014 | 0.039 | 0.001 | Pb···C |
| - | 0.009 | 0.026 | 0.001 | (Pb)H···C |
| PbFH3-C5H5− | 0.042 | 0.074 | −0.008 | Pb···C |
| - | 0.015 | 0.041 | 0.001 | (Pb)H···C |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabowski, S.J. Tetrel Bonds with π-Electrons Acting as Lewis Bases—Theoretical Results and Experimental Evidences. Molecules 2018, 23, 1183. https://doi.org/10.3390/molecules23051183
Grabowski SJ. Tetrel Bonds with π-Electrons Acting as Lewis Bases—Theoretical Results and Experimental Evidences. Molecules. 2018; 23(5):1183. https://doi.org/10.3390/molecules23051183
Chicago/Turabian StyleGrabowski, Sławomir J. 2018. "Tetrel Bonds with π-Electrons Acting as Lewis Bases—Theoretical Results and Experimental Evidences" Molecules 23, no. 5: 1183. https://doi.org/10.3390/molecules23051183
APA StyleGrabowski, S. J. (2018). Tetrel Bonds with π-Electrons Acting as Lewis Bases—Theoretical Results and Experimental Evidences. Molecules, 23(5), 1183. https://doi.org/10.3390/molecules23051183