Synthesis, Design, and Structure–Activity Relationship of the Pyrimidone Derivatives as Novel Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase

Abstract
:
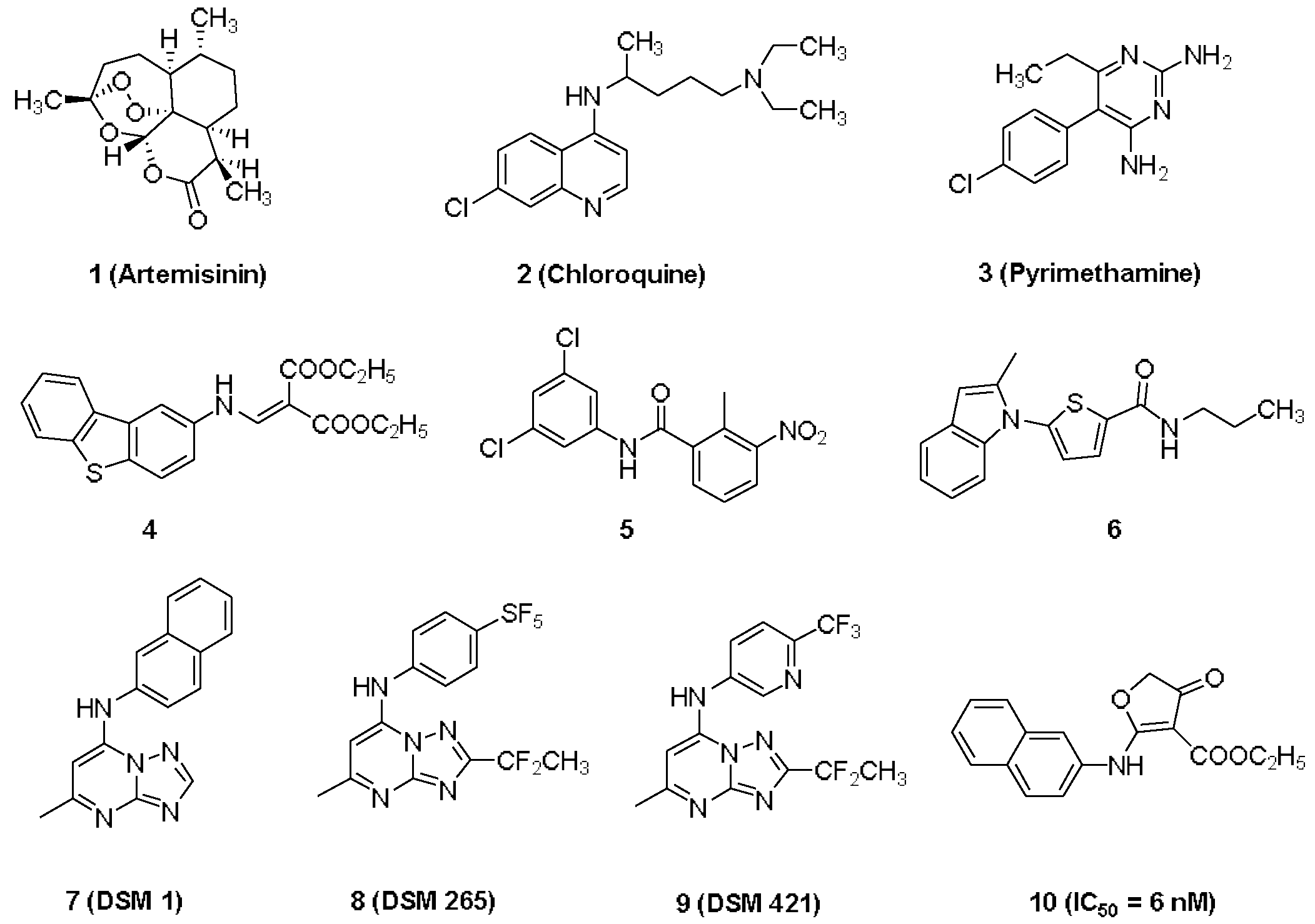
1. Introduction
2. Results and Discussion
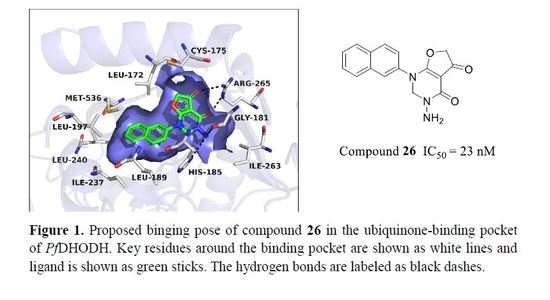
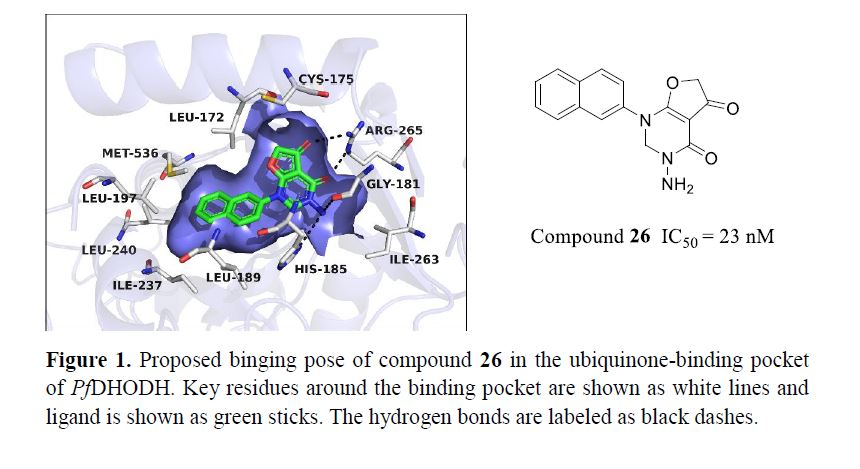
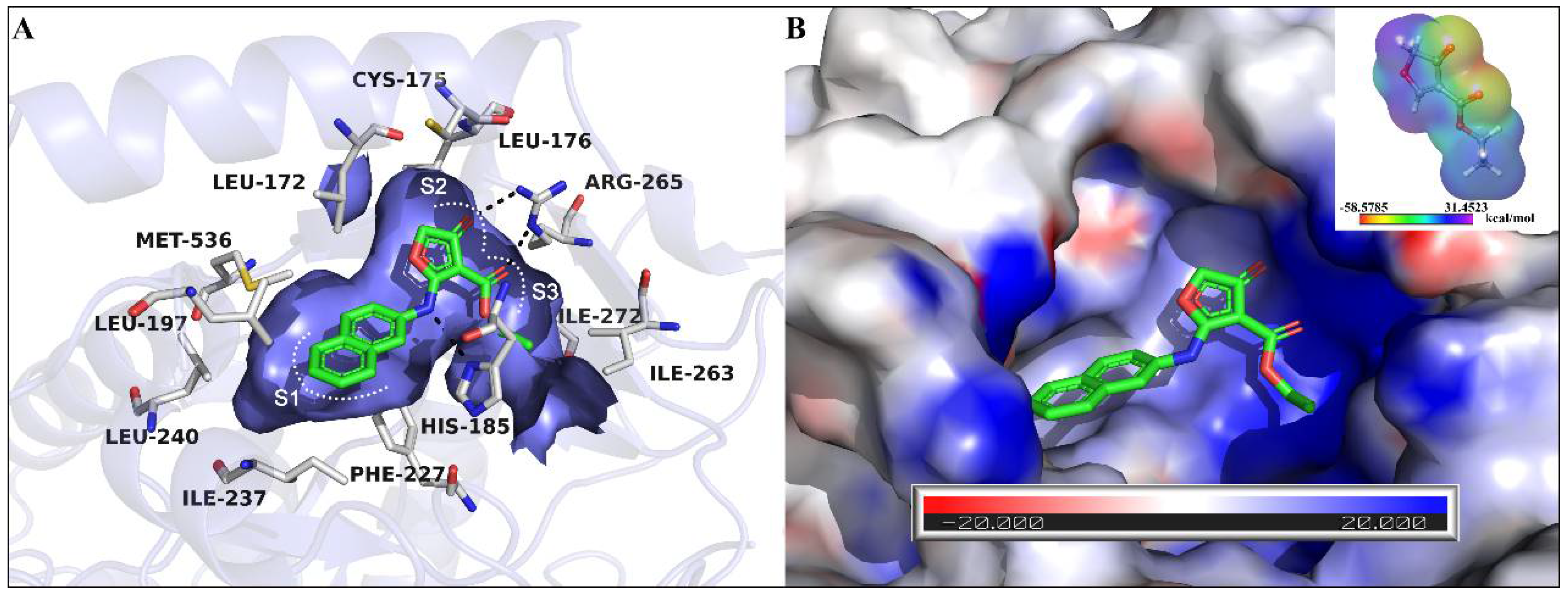
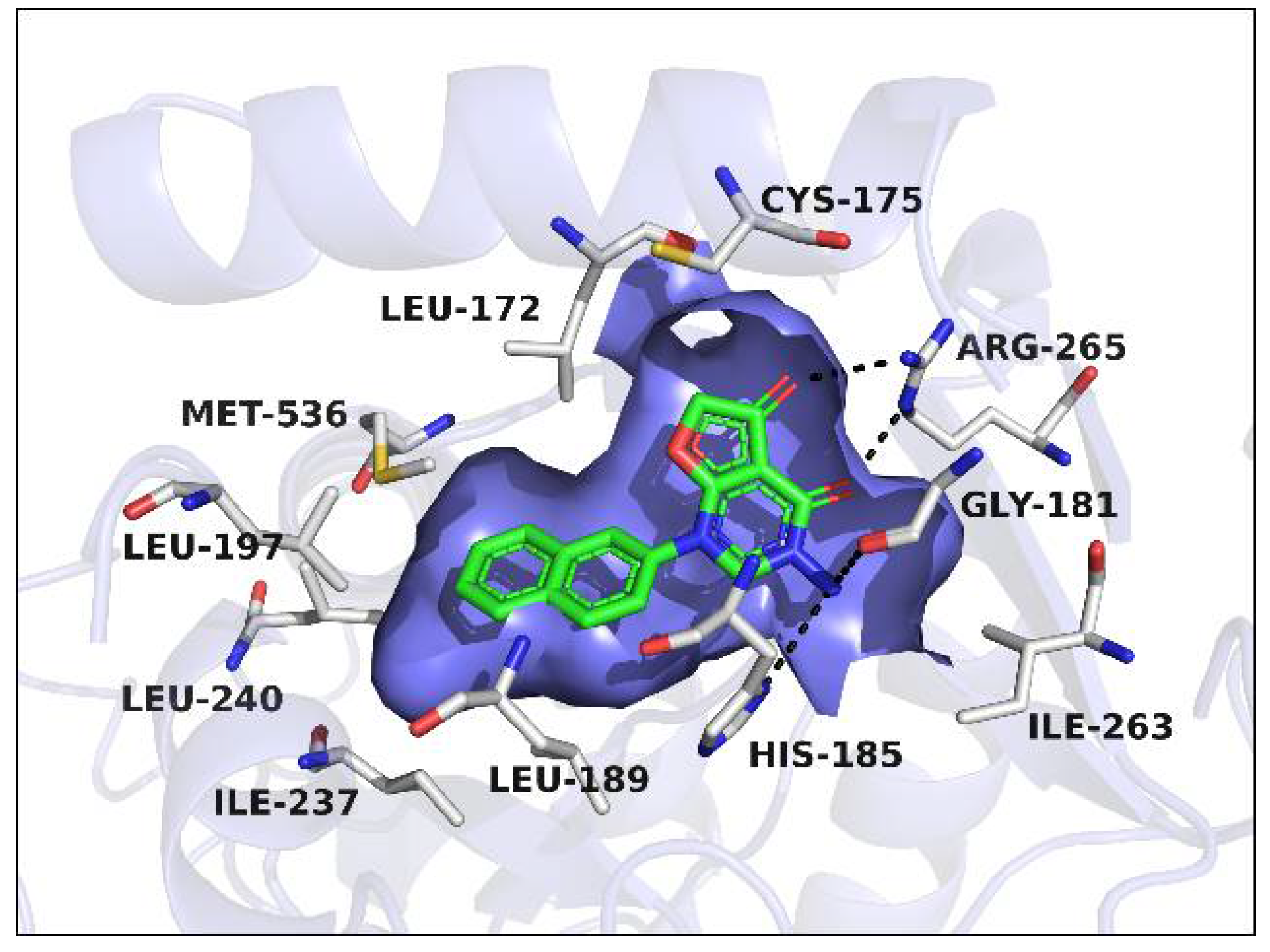
2.1. Proposed Binding Pose of Lead Compound 10 in the Ubiquinone Binding Pocket of PfDHODH
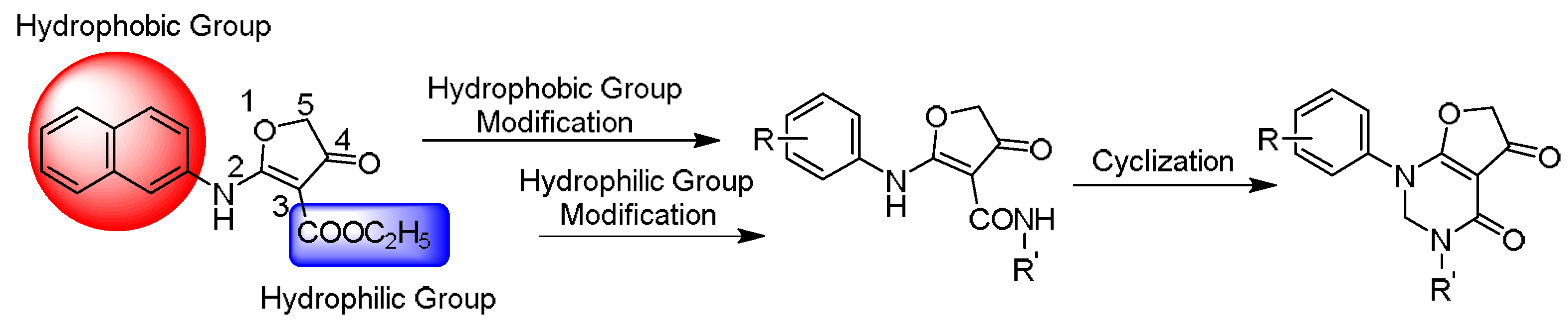
2.2. The Structural Optimization Strategy using the Lead Compound 10
2.3. Chemistry
2.4. Inhibitory Activities against PfDHODH and SAR Study
2.4.1. Hydrophobic Group Modification
2.4.2. Hydrophilic Group Modification
2.4.3. Cyclization Forming a New Scaffold
3. Materials and Methods
3.1. In Vitro Enzyme Assay
3.2. Chemistry Experiment
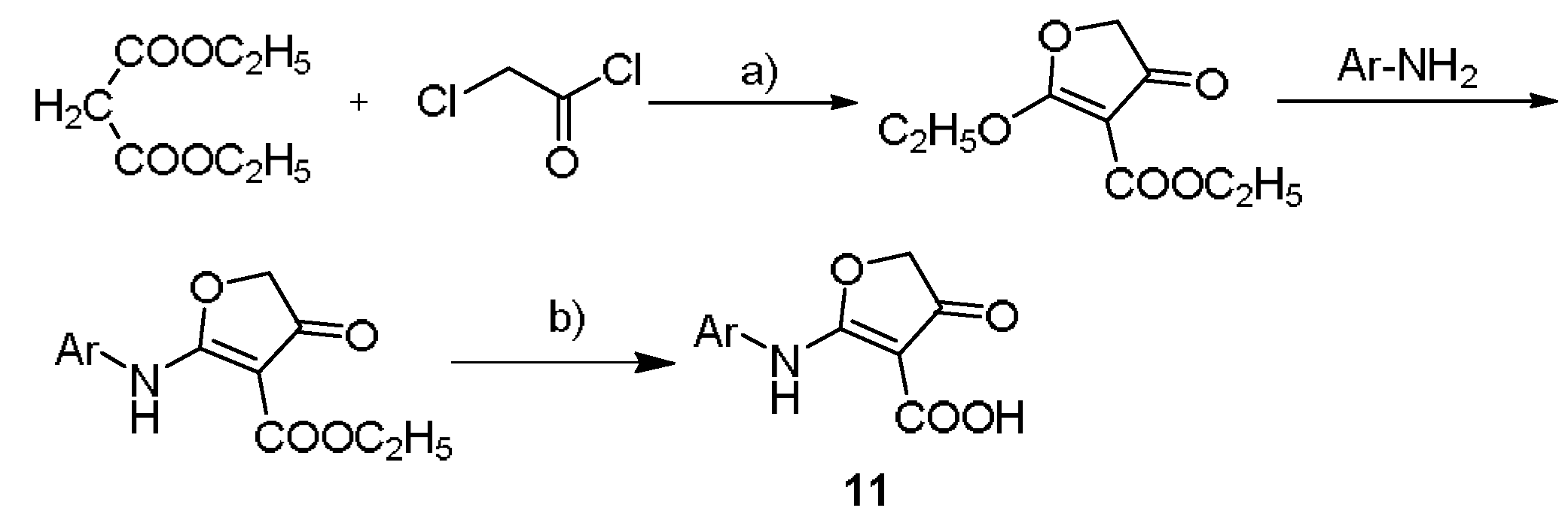
3.2.1. General Procedure for the Synthesis of Intermediates
Synthesis of the ethyl 2-(substituted arylamino)-4-oxo-4,5-dihydrofuran-3 carboxylate
Synthesis of the 2-(substituted arylamino)-4-oxo-4,5 dihydrofuranone-3-carboxylic acid
Synthesis of compound 11

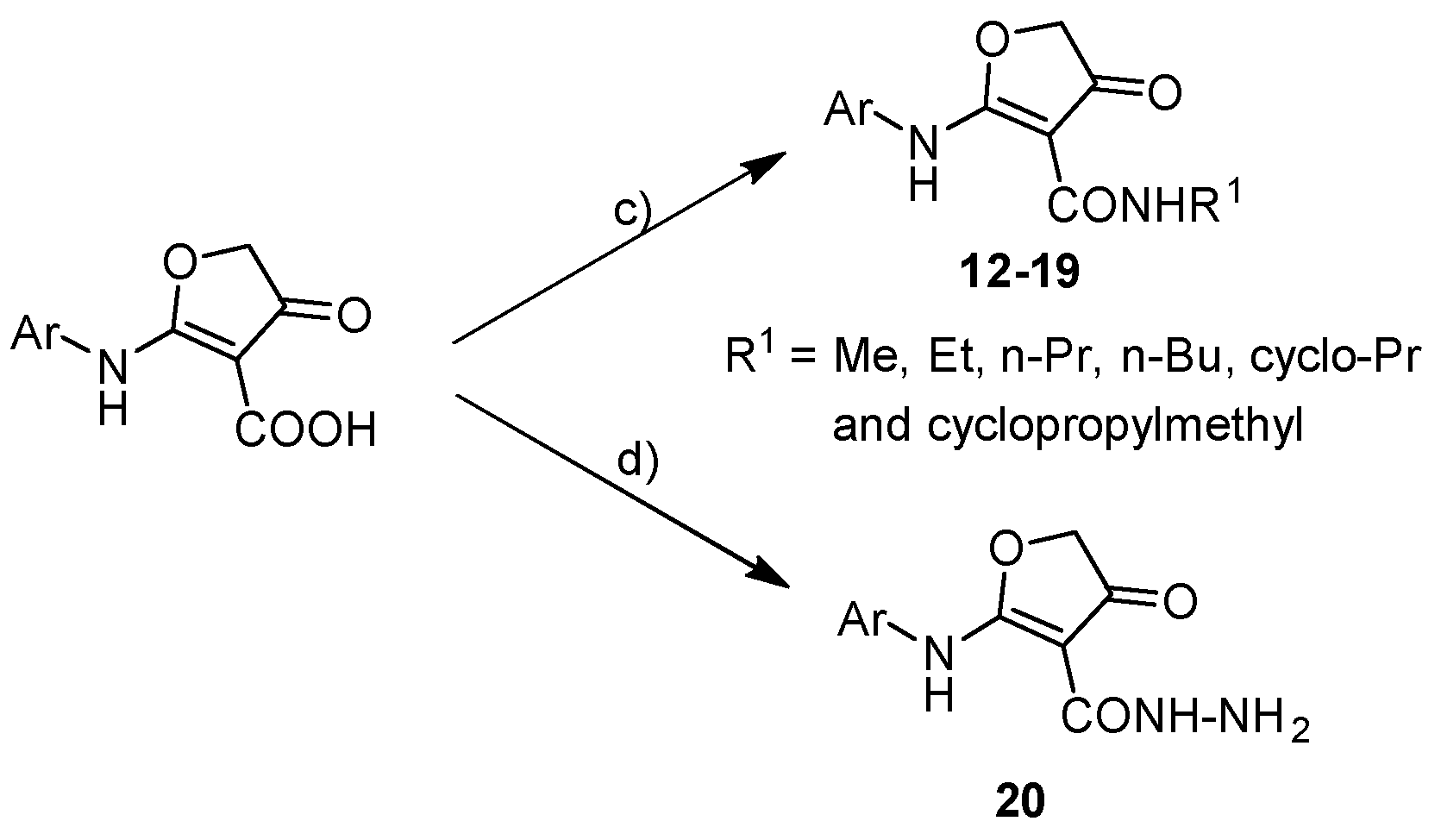
3.2.2. General Procedure for Target Compounds 12–19








Synthesis of Compound 20

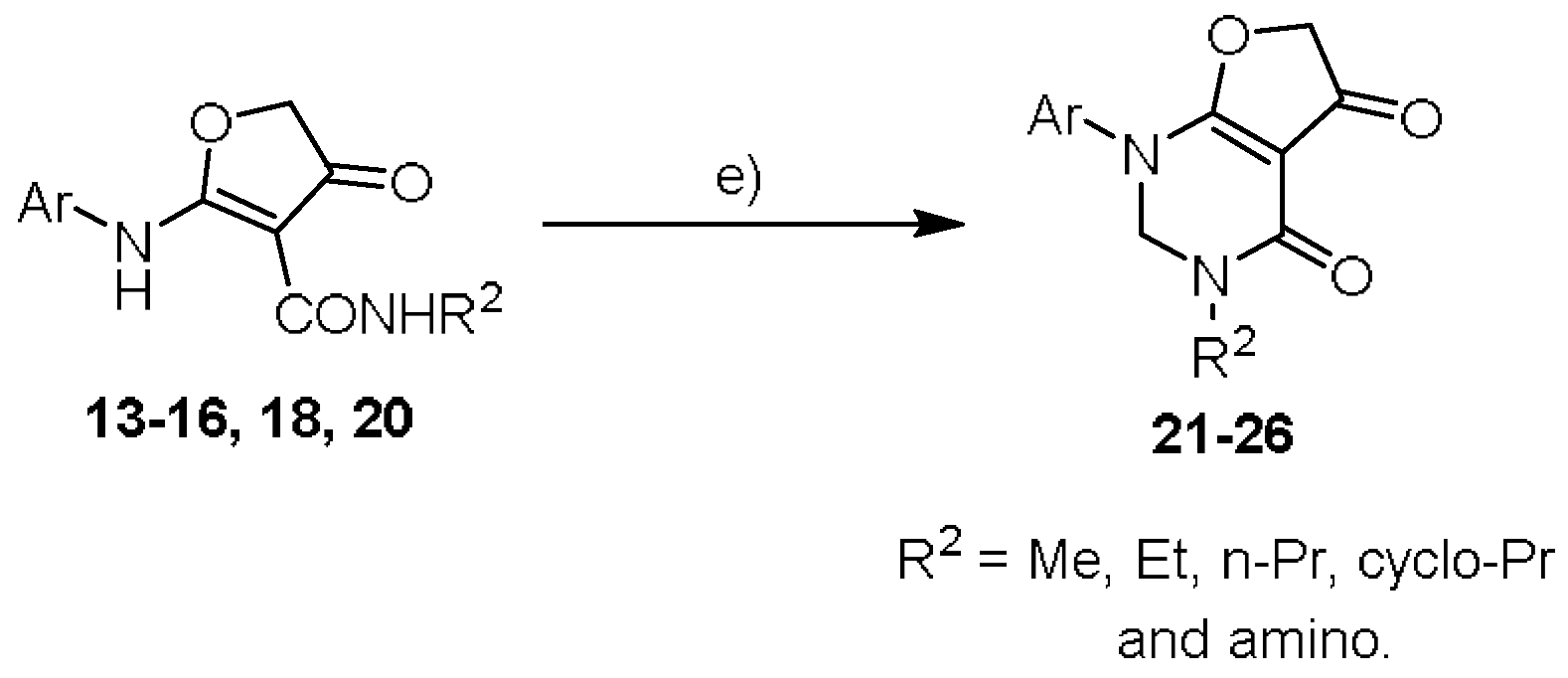
3.2.3. General Procedure for Target Compounds 21–26






4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Miller, L.H.; Ackerman, H.C.; Su, X.Z.; Wellems, T.E. Malaria biology and disease pathogenesis: insights for new treatments. Nat. Med. 2013, 19, 156–167. [Google Scholar] [CrossRef] [PubMed]
- White, N.J.; Pukrittayakamee, S.; Hien, T.T.; Faiz, M.A.; Mokuolu, O.A.; Dondorp, A.M. Malaria. Lancet 2014, 383, 723–735. [Google Scholar] [CrossRef]
- World Health Organization. World Malaria Report 2016; WHO Press: Geneva, Switzerland, 2017. [Google Scholar]
- Dondorp, A.M.; Yeung, S.; White, L.; Nguon, C.; Day, N.P.J.; Socheat, D.; von Seidlein, L. Artemisinin Resistance: Current Status and Scenarios for Containment. Nat. Rev. Microbiol. 2010, 8, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Bray, P.G.; Martin, R.E.; Tilley, L.; Ward, S.A.; Kirk, K.; Fidock, D.A. Defining the Role of PfCRT in Plasmodium falciparum Chloroquine Resistance. Mol. Microbiol. 2005, 56, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.; Ashley, E.A.; Ferreira, P.E.; Zhu, L.; Lin, Z.; Yeo, T.; Chotivanich, K.; Imwong, M.; Pukrittayakamee, S.; Dhorda, M.; et al. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science 2015, 347, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Miotto, O.; Amato, R.; Ashley, E.A.; MacInnis, B.; Almagro-Garcia, J.; Amaratunga, C.; Lim, P.; Mead, D.; Oyola, S.O.; Dhorda, M.; et al. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat. Genet. 2015, 47, 226. [Google Scholar] [CrossRef] [PubMed]
- Menard, D.; Ariey, F. Towards real-time monitoring of artemisinin resistance. Lancet Infect. Dis. 2015, 15, 367–368. [Google Scholar] [CrossRef]
- Hyde, J.E. Mechanisms of Resistance of Plasmodium falciparum to Antimalarial Drugs. Microbes Infect. 2002, 4, 165–174. [Google Scholar] [CrossRef]
- McKie, J.H.; Douglas, K.T.; Chan, C.; Roser, S.A.; Yates, R.; Read, M.; Hyde, J.E.; Dascombe, M.J.; Yuthavong, Y.; Sirawaraporn, W. Rational Drug Design Approach for Overcoming Drug Resistance: Application to Pyrimethamine Resistance in Malaria. J. Med. Chem. 1998, 41, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Barnett, D.S.; Guy, R.K. Antimalarials in development in 2014. Chem. Rev. 2014, 114, 11221–11241. [Google Scholar] [CrossRef] [PubMed]
- Wells, N.C.; Van Huijsduijnen, R.H.; Van Voorhis, W.C. Malaria medicines: A glass half full? Nat. Rev. Drug Discov. 2015, 15, 424–442. [Google Scholar] [CrossRef] [PubMed]
- Burrows, N.; Burlot, E.; Campo, B.; Cherbuin, S.; Jeanneret, S.; Leroy, D. Antimalarial drug discovery the path towards eradication. Parasitology 2014, 141, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Baragana, B.; Hallyburton, I.; Lee, M.C.S.; Norcross, N.R.; Grimaldi, R.; Otto, T.D. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 2015, 522, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: fi nal results of a phase 3, individually randomised, controlled trial. Lancet 2015, 386, 31–45.
- Fagan, R.L.; Nelson, M.N.; Pagano, P.M.; Palfey, B.A. Mechanism of Flavin Reduction in Class 2 Dihydroorotate Dehydrogenases. Biochemistry 2006, 45, 14926–14932. [Google Scholar] [CrossRef] [PubMed]
- Palfey, B.A.; Björnberg, O.; Jensen, K.F. Specific Inhibition of a Family 1A Dihydroorotate Dehydrogenase by Benzoate Pyrimidine Analogues. J. Med. Chem. 2001, 44, 2861–2864. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.A.; Rathod, P.K. Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect. Disord. Drug Targets 2010, 10, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Nagle, S.; Aavait, K.; Chatterjee, A. Road Towards New Antimalarials-Overview of the Strategies and their Chemical Progress. Curr. Med. Chem. 2011, 18, 853–871. [Google Scholar] [CrossRef] [PubMed]
- Munier-Lehmann, H.; Vidalain, P.-O.; Tangy, F.; Janin, Y.L. On Dihydroorotate Dehydrogenases and Their Inhibitors and Uses. J. Med. Chem. 2013, 56, 3148–3167. [Google Scholar] [CrossRef] [PubMed]
- Coteron, J.M.; Marco, M.A.; Esquivias, J.; Deng, X.; White, K.L.; White, J.; Koltun, M.; El Mazouni, F.; Kokkonda, S.; Katneni, K.; et al. Structure-Guided Lead Optimization of Triazolopyrimidine-Ring Substituents Identifies Potent Plasmodium falciparum Dihydroorotate Dehydrogenase Inhibitors with Clinical Candidate Potential. J. Med. Chem. 2011, 54, 5540–5561. [Google Scholar] [CrossRef] [PubMed]
- Malmquist, N.A.; Gujjar, R.; Rathod, P.K.; Phillips, M.A. Analysis of Flavin Oxidation and Electron-Transfer Inhibition in Plasmodium falciparum Dihydroorotate Dehydrogenase. Biochemistry 2008, 47, 2466–2475. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.L.; Schleyerbach, R.; Kirschbaum, B.J. Leflunomide: An Immunomodulatory Drug for the Treatment of Rheumatoid Arthritis and Other Autoimmune Diseases. Immunopharmacology 2000, 47, 273–289. [Google Scholar] [CrossRef]
- Baumgartner, R.; Walloschek, M.; Kralik, M.; Gotschlich, A.; Tasler, S.; Mies, J.; Leban, J. Dual Binding Mode of a Novel Series of DHODH Inhibitors. J. Med. Chem. 2006, 49, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, O.P.; Sayyed, S.G.; Kantner, C.; Ryu, M.; Schnurr, M.; Sardy, M.; Leban, J.; Jankowsky, R.; Ammendola, A.; Doblhofer, R.; et al. 4SC-101, A Novel Small Molecule Dihydroorotate Dehydrogenase Inhibitor, Suppresses Systemic Lupus Erythematosus in MRL-(Fas)lpr Mice. Am. J. Pathol. 2010, 176, 2840–2847. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, J.; Michnoff, C.H.; Malmquist, N.A.; White, J.; Roth, M.G.; Rathod, P.K.; Phillips, M.A. High-throughput Screening for Potent and Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. J. Biol. Chem. 2005, 280, 21847–21853. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Booker, M.; Kramer, M.; Ross, L.; Celatka, C.A.; Kennedy, L.M.; Dvorin, J.D.; Duraisingh, M.T.; Sliz, P.; Wirth, D.F. Identification and Characterization of Small Molecule Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. J. Biol. Chem. 2008, 283, 35078–35085. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.A.; Gujjar, R.; Malmquist, N.A.; White, J.; El Mazouni, F.; Baldwin, J.; Rathod, P.K. Triazolopyrimidine-Based Dihydroorotate Dehydrogenase Inhibitors with Potent and Selective Activity against the Malaria Parasite Plasmodium falciparum. J. Med. Chem. 2008, 51, 3649–3653. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Diaz, M.B.; Mulet, T.; Viera, S.; Gomez, V.; Garuti, H.; Ibanez, J.; Alvarez-Doval, A.; Shultz, L.D.; Martinez, A.; Gargallo-Viola, D.; et al. Improved murine model of malaria using Plasmodium falciparum competent strains and non-myelodepleted NOD-scid IL2Rgammanull mice engrafted with human erythrocytes. Antimicrob. Agents Chemother. 2009, 53, 4533–4536. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.A.; White, K.L.; Kokkonda, S.; Deng, X.; White, J.; El Mazouni, F.; Marsh, K.; Tomchick, D.R.; Manjalanagara, K.; Rudra, K.R.; et al. A Triazolopyrimidine-Based Dihydroorotate Dehydrogenase Inhibitor with Improved Drug-like Properties for Treatment and Prevention of Malaria. ACS Infect. Dis. 2016, 2, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhu, J.; Diao, Y.; Zhou, H.; Ren, X.; Sun, D.; Huang, J.; Han, D.; Zhao, Z.; Zhu, L.; et al. Novel selective and potent inhibitors of malaria parasite dihydroorotate dehydrogenase: discovery and optimization of dihydrothiophenone derivatives. J. Med. Chem. 2013, 56, 7911–7924. [Google Scholar] [CrossRef] [PubMed]
- Woody, S.; Tyler, D.; Matthew, P.J.; Richard, A.F.; Ramy, F. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Le Nours, J.; Johansson, E.; Antal, T.; Ullrich, A.; Löffler, M.; Larsen, S. Inhibitor Binding in a Class 2 Dihydroorotate Dehydrogenase Causes Variations in the Membrane-Associated N-Terminal Domain. Protein Sci. 2004, 13, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.A.; Teffera, Y.; Albrecht, B.K. Discovery of Potent and Selective 8-Fluorotriazolopyridine c-Met Inhibitors. J. Med. Chem. 2015, 58, 2417–2430. [Google Scholar] [CrossRef] [PubMed]
- Knecht, W.; Henseling, J.; Löffler, M. Kinetics of Inhibition of Human and Rat Dihydroorotate Dehydrogenase by Atovaquone, Lawsone Derivatives, Brequinar Sodium and Polyporic Acid. Chem. Biol. Interact. 2000, 124, 61–76. [Google Scholar] [CrossRef]
- McLean, J.E.; Neidhardt, E.A.; Grossman, T.H.; Hedstrom, L. Multiple Inhibitor Analysis of the Brequinar and Leflunomide Binding Sites on Human Dihydroorotate Dehydrogenase. Biochemistry 2001, 40, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 11–26 are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | IC50 (μM) | |||
|---|---|---|---|---|
| Compound | R1 | R2 | PfDHODH a | hDHODH b |
| 11 | 2-Naphthyl | COOH | 0.106 ± 0.009 | >10 |
| 12 | Phenyl | CONHCH2CH3 | >10 | 2.4 ± 0.1 |
| 13 | 5-Aminoindan | CONHCH2CH3 | >10 | >10 |
| 14 | 2-Naphthyl | CONHCH3 | 9 ± 1 | >10 |
| 15 | 2-Naphthyl | CONHCH2CH3 | 4.8 ± 0.4 | >10 |
| 16 | 2-Naphthyl | CONHCH2CH2CH3 | >10 | >10 |
| 17 | 2-Naphthyl | CONHCH2CH2CH2CH3 | >10 | >10 |
| 18 | 2-Naphthyl |  | 6 ± 1 | >10 |
| 19 | 2-Naphthyl |  | >10 | >10 |
| 20 | 2-Naphthyl | CONH–NH2 | 0.07 ± 0.01 | >10 |
 | ||||
| 21 | 5-Aminoindan | CH2CH3 | >10 | >10 |
| 22 | 2-Naphthyl | CH3 | 8 ± 1 | >10 |
| 23 | 2-Naphthyl | CH2CH3 | 4 ± 1 | >10 |
| 24 | 2-Naphthyl | CH2CH2CH3 | 12 ± 6 | >10 |
| 25 | 2-Naphthyl |  | 5 ± 1 | >10 |
| 26 | 2-Naphthyl | NH2 | 0.023 ± 0.001 | >10 |
| DSM1 | — | — | 0.042 ± 0.004 | — |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; Li, W.; Diao, Y.; Sun, H.; Li, H.; Zhu, L.; Zhou, H.; Zhao, Z. Synthesis, Design, and Structure–Activity Relationship of the Pyrimidone Derivatives as Novel Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. Molecules 2018, 23, 1254. https://doi.org/10.3390/molecules23061254
Xu L, Li W, Diao Y, Sun H, Li H, Zhu L, Zhou H, Zhao Z. Synthesis, Design, and Structure–Activity Relationship of the Pyrimidone Derivatives as Novel Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. Molecules. 2018; 23(6):1254. https://doi.org/10.3390/molecules23061254
Chicago/Turabian StyleXu, Le, Wenjie Li, Yanyan Diao, Hongxia Sun, Honglin Li, Lili Zhu, Hongchang Zhou, and Zhenjiang Zhao. 2018. "Synthesis, Design, and Structure–Activity Relationship of the Pyrimidone Derivatives as Novel Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase" Molecules 23, no. 6: 1254. https://doi.org/10.3390/molecules23061254
APA StyleXu, L., Li, W., Diao, Y., Sun, H., Li, H., Zhu, L., Zhou, H., & Zhao, Z. (2018). Synthesis, Design, and Structure–Activity Relationship of the Pyrimidone Derivatives as Novel Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. Molecules, 23(6), 1254. https://doi.org/10.3390/molecules23061254