Synthesis of 2-Deoxybrassinosteroids Analogs with 24-nor, 22(S)-23-Dihydroxy-Type Side Chains from Hyodeoxycholic Acid


Abstract
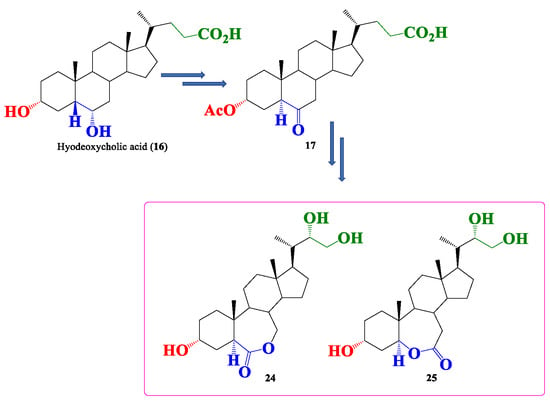
:
1. Introduction
2. Results
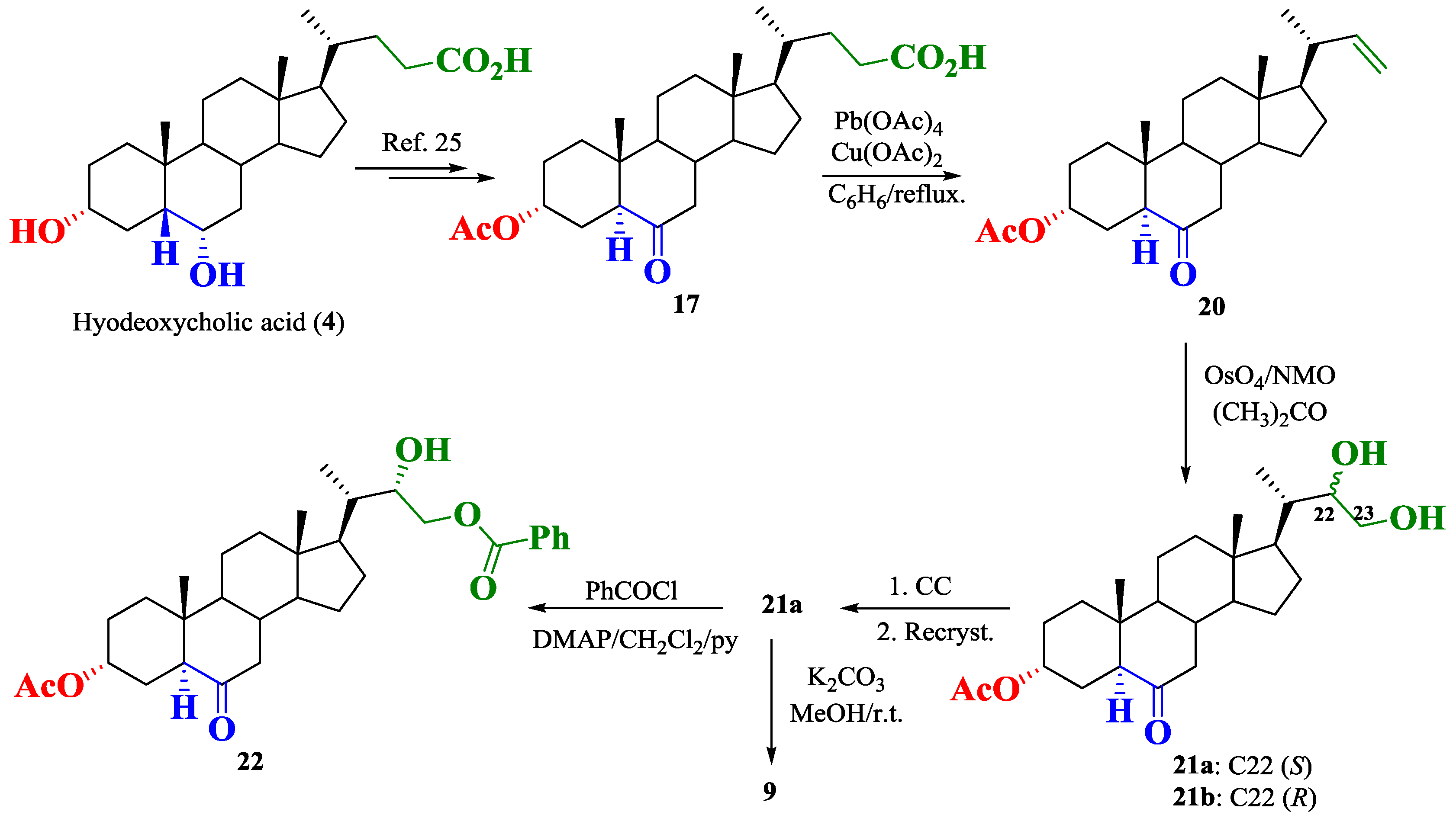
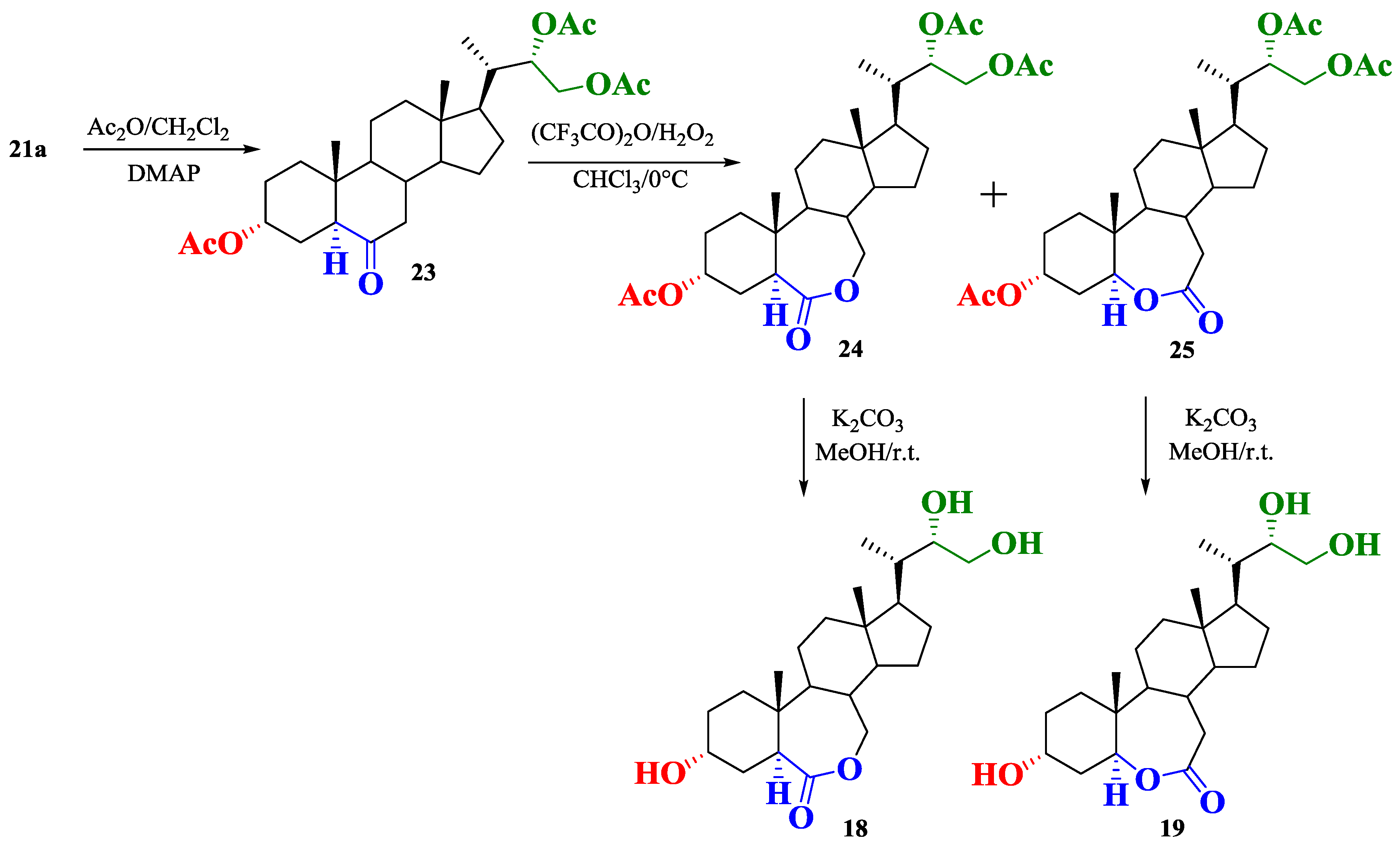
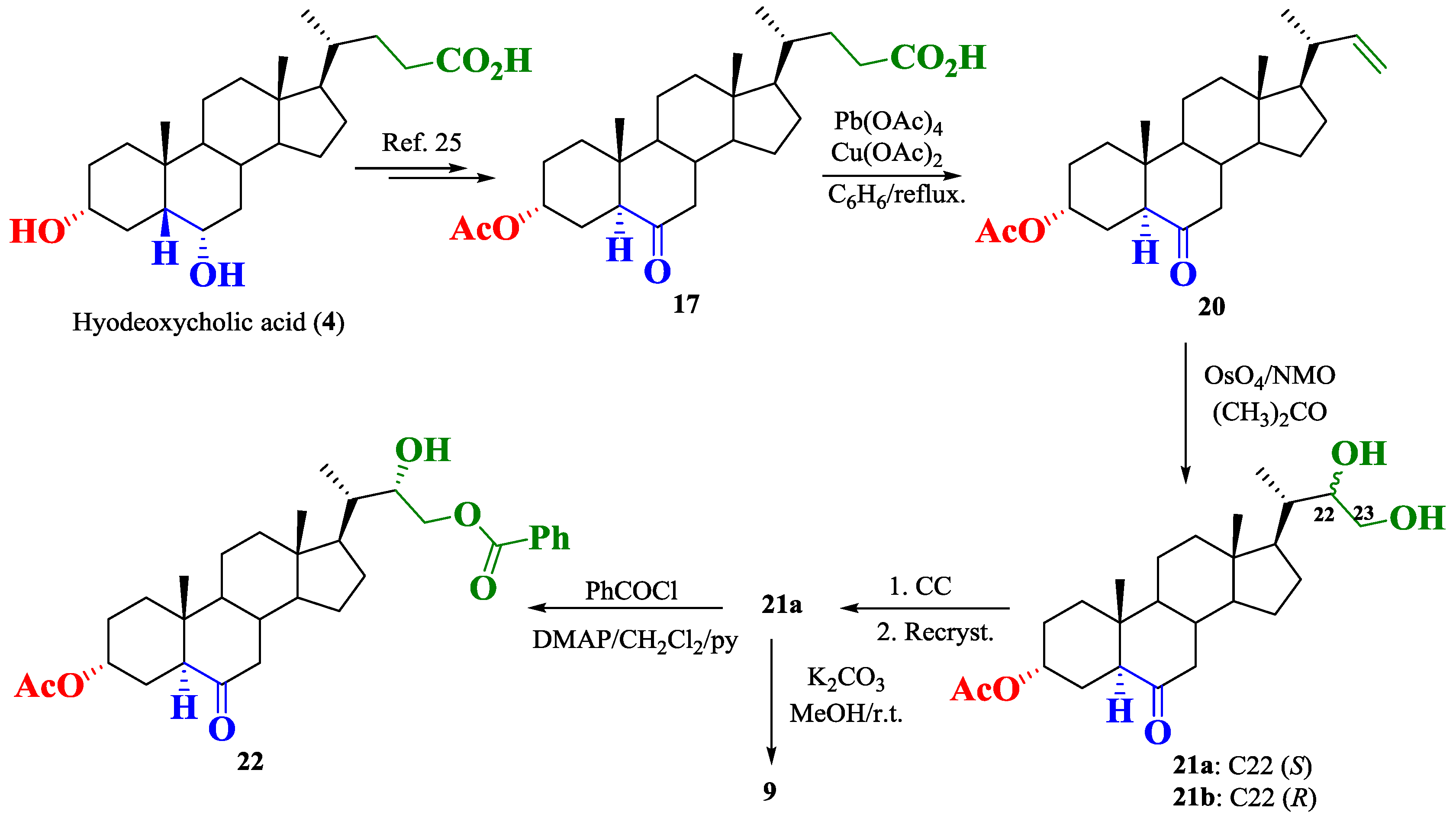
2.1. Synthesis of Brassinosteroids Analogs
2.2. Elucidation of Compound Structures
3. Materials and Methods
3.1. General Experimental Methods
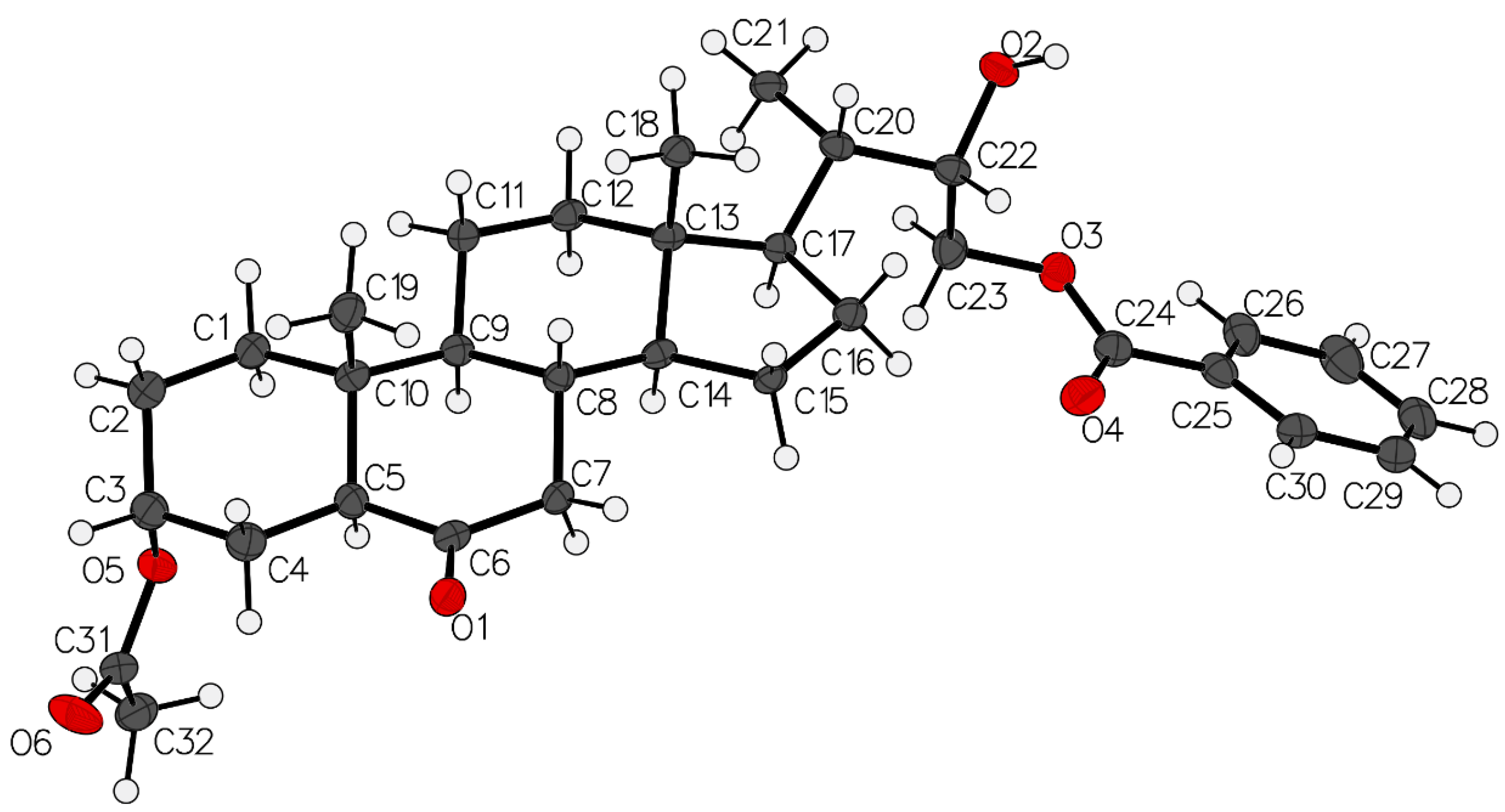
3.2. X-ray Crystal Structure Determination
3.3. Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thompson, M.J.; Meudt, W.J.; Mandava, N.B.; Dutky, S.R.; Lusby, W.R.; Spaulding, D.W. Synthesis of Brassinosteroids and Relationship of Structure to Plant Growth-Promoting Effects. Steroids 1982, 39, 89–105. [Google Scholar] [CrossRef]
- Takatsuto, S.; Yazawa, N.; Ikekawa, N.; Morishita, T.; Abe, H. Synthesis of (24R)-28-Homobrassinolide Analogs and Structure Activity Relationships of Brassinosteroids in the Rice-Lamina Inclination Test. Phytochemistry 1983, 22, 1393–1397. [Google Scholar] [CrossRef]
- Takatsuto, S.; Yazawa, N.; Ikekawa, N.; Takematsu, T.; Takeuchi, Y.; Koguchi, M. Structure Activity Relationship of Brassinosteroids. Phytochemistry 1983, 22, 2437–2441. [Google Scholar] [CrossRef]
- Yokota, T.; Arima, M.; Takahashi, N.; Takatsuto, S.; Ikekawa, N.; Takematsu, T. 2-Deoxycastasterone, a new brassinolide-related bioactive steroid from pinus pollen. Agric. Biol. Chem. 1983, 47, 2419–2420. [Google Scholar] [CrossRef]
- Schneider, J.A.; Yoshihara, K.; Nakanishi, K.; Kato, N. Typhasterol (2-deoxycastasterone)—A new plant-growth regulator from cat-tail pollen. Tetrahedron Lett. 1983, 24, 3859–3860. [Google Scholar] [CrossRef]
- Abe, H.; Morishita, T.; Uchiyama, M.; Takatsuto, S.; Ikekawa, N. A New Brassinolide-Related Steroid in the Leaves of Thea-Sinensis. Agric. Biol. Chem. 1984, 48, 2171–2172. [Google Scholar] [CrossRef]
- Suzuki, Y.; Yamaguchi, I.; Yokota, T.; Takahashi, N. Identification of castasterone, typhasterol and teasterone from the pollen of zea-mays. Agric. Biol. Chem. 1986, 50, 3133–3138. [Google Scholar] [CrossRef]
- Brosa, C.; Capdevila, J.M.; Zamora, I. Brassinosteroids: A new way to define the structural requirements. Tetrahedron 1996, 52, 2435–2448. [Google Scholar] [CrossRef]
- Brosa, C.; Soca, L.; Terricabras, E.; Ferrer, J.C.; Alsina, A. New synthetic brassinosteroids: A 5 alpha-hydroxy-6-ketone analog with strong plant growth promoting activity. Tetrahedron 1998, 54, 12337–12348. [Google Scholar] [CrossRef]
- Iglesias-Arteaga, M.A.; Martinez, C.P.; Manchado, F.C. Spirostanic analogues of castasterone. Steroids 2002, 67, 159–163. [Google Scholar] [CrossRef]
- Romero-Avila, M.; de Dios-Bravo, G.; Mendez-Stivalet, J.M.; Rodriguez-Sotres, R.; Iglesias-Arteaga, M.A. Synthesis and biological activity of furostanic analogues of brassinosteroids bearing the 5 alpha-hydroxy-6-oxo moiety. Steroids 2007, 72, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Rosado-Abon, A.; de Dios-Bravo, G.; Rodriguez-Sotres, R.; Iglesias-Arteaga, M.A. Synthesis and plant growth promoting activity of polyhydroxylated ketones bearing the 5 alpha-hydroxy-6-oxo moiety and cholestane side chain. Steroids 2012, 77, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.S.; Zhou, W.S.; Jiang, B.; Pan, X.F. Studies on Steroidal Plant-Growth Regulator 9. The Preparation of 22R-Penta-Nor-Brassinolides and 22S-24,25,26,27,28-Penta-Nor-Brassinolides. Acta Chim. Sinica 1989, 47, 1017–1021. [Google Scholar]
- Yang, Y.X.; Zheng, L.T.; Shi, J.J.; Gao, B.; Chen, Y.K.; Yang, H.C.; Chen, H.L.; Li, Y.C.; Zhen, X.C. Synthesis of 5 alpha-cholestan-6-one derivatives and their inhibitory activities of NO production in activated microglia: Discovery of a novel neuroinflammation inhibitor. Bioorg. Med. Chem. Lett. 2014, 24, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, L.; Cortes, M. Synthesis and biological activities of two new brassinosteroids functionalized in ring C. Bol. Soc. Chil. Quim. 2002, 47, 335–347. [Google Scholar]
- Espinoza, L. Synthesis of Four New Brassinosteroids Analogues 11-Oxo-Functionalized on C Ring, with 24-Nor Side Chain and Containing 5â-Cholanic Acid Skeleton. Org. Chem. Curr. Res. 2015, 4, 1–6. [Google Scholar]
- Espinoza, L.; Cortes, M. Synthesis and biological activity of brassinosteroids analogues. Bol. Soc. Chil. Quim. 2002, 47, 511–516. [Google Scholar]
- Back, T.G.; Blazecka, P.G.; Krishna, M.V. A new synthesis of castasterone and brassinolide from stigmasterol—A concise and stereoselective elaboration of the side-chain from a C-22 aldehyde. Can. J. Chem. 1993, 71, 156–163. [Google Scholar] [CrossRef]
- Back, T.G.; Blazecka, P.G.; Krishna, M.V. A concise synthesis of the brassinolide side-chain. Tetrahedron Lett. 1991, 32, 4817–4818. [Google Scholar] [CrossRef]
- Back, T.G.; Krishna, M.V. Synthesis of castasterone and formal synthesis of brassinolide from stigmasterol via a selenosulfonation approach. J. Org. Chem. 1991, 56, 454–457. [Google Scholar] [CrossRef]
- Zhou, W.S.; Shen, Z.W. Study on the Synthesis of Brassinolide and Related-Compounds 15. Formal Synthesis of Brassinolide Via Stereoselective Sulfenate Sulfoxide Transformation. J. Chem. Soc. Perkin Trans. 1 1991, 2, 2827–2830. [Google Scholar] [CrossRef]
- Tsubuki, M.; Keino, K.; Honda, T. Stereoselective synthesis of plant-growth-regulating steroids—Brassinolide, castasterone, and their 24,25-substituted analogs. J. Chem. Soc. Perkin Trans. 1 1992, 2643–2649. [Google Scholar] [CrossRef]
- Furuta, T.; Yamamoto, Y. Stereocontrol of three contiguous chiral centers of brassinosteroid side-chain using alpha-alkoxy organoleads. J. Org. Chem. 1992, 57, 2981–2982. [Google Scholar] [CrossRef]
- Nakamura, T.; Tanino, K.; Kuwajima, I. Highly stereoselective chelation controlled ene-reaction of 2-(alkylthio)allyl silyl ethers. Tetrahedron Lett. 1993, 34, 477–480. [Google Scholar] [CrossRef]
- Brosa, C.; Peracaula, R.; Puig, R.; Ventura, M. Use of dihydroquinidine 9-o-(9′-phenanthryl) ether in osmium-catalyzed asymmetric dihydroxylation in the synthesis of brassinosteroids. Tetrahedron Lett. 1992, 33, 7057–7060. [Google Scholar] [CrossRef]
- Hazra, B.G.; Pore, V.S.; Joshi, P.L. Short-step stereoselective synthesis of 2-alpha,3-alpha,22-triacetoxy-23,24-dinor-5-alpha-cholan-6-one—Key intermediate for the preparation of 24(1)-norbrassinolide, dolicholide and dolichosterone. J. Chem. Soc. Perkin Trans. 1 1993, 15, 1819–1822. [Google Scholar] [CrossRef]
- Zhou, W.S.; Huang, L.F. Studies on steroidal plant-growth regulator. 25. Concise stereoselective construction of side-chain of brassinosteroid from the intact side-chain of hyodeoxycholic acid—Formal syntheses of brassinolide, 25-methylbrassinolide, 26,27-bisnorbrassinolide and their related-compounds. Tetrahedron 1992, 48, 1837–1852. [Google Scholar]
- Zhou, W.S.; Huang, L.F.; Sun, L.Q.; Pan, X.F. Studies on Steroidal Plant-Growth Regulator. 21. A Stereoselective Construction of Brassinosteroid Side-Chain—A New Practical Synthesis of Brassinolide and Its Analogs. Tetrahedron Lett. 1991, 32, 6745–6748. [Google Scholar] [CrossRef]
- Zhou, W.S.; Huang, L.F.; Sun, L.Q.; Pan, X.F. Studies on A Steroidal Plant-Growth Regulator. Part 26. Stereoselective Construction of the Brassinolide Side-Chain: New Practical Syntheses of Brassinolide Analogs from Hyodeoxycholic Acid. J. Chem. Soc. Perkin Trans. 1 1992, 15, 2039–2043. [Google Scholar] [CrossRef]
- Huang, L.F.; Zhou, W.S.; Sun, L.Q.; Pan, X.F. Studies on steroidal plant-growth regulators. Part 29. Osmium tetroxide-catalyzed asymmetric dihydroxylation of the (22E,24R) and the (22E,24S)-24-alkyl steroidal unsaturated side-chain. J. Chem. Soc. Perkin Trans. 1 1993, 14, 1683–1686. [Google Scholar] [CrossRef]
- Hazra, B.G.; Joshi, P.L.; Bahule, B.B.; Argade, N.P.; Pore, V.S.; Chordia, M.D. Stereoselective synthesis of (22r,23r,24s)-3-beta-hydroxy-5-ene-22,23-dihydroxy-24-methyl-cholestane—A brassinolide intermediate from 16-dehydropregnenolone acetate. Tetrahedron 1994, 50, 2523–2532. [Google Scholar] [CrossRef]
- Bacha, J.D.; Kochi, J.K. Alkenes from acids by oxidative decarboxylation. Tetrahedron 1968, 24, 2215–2216. [Google Scholar] [CrossRef]
- Vaydia, A.S.; Dixit, S.M.; Rao, A.S. Degradation of bile acid side chain with lead tetraacetate. Tetrahedron 1968, 50, 5173–5174. [Google Scholar] [CrossRef]
- Zhou, W.S.; Tian, W.S. Studies on Steroidal Plant-Growth Hormones. 2. Stereoselective Synthesis of (22S, 23S)-Typhasterol from Hyodeoxycholic Acid. Acta Chim. Sinica 1985, 43, 1060–1067. [Google Scholar]
- Zhou, W.S.; Jiang, B.; Pan, X.F. Stereoselective Synthesis of the Brassinolide Side-Chain—Novel Syntheses of Brassinolide and Related-Compounds. Tetrahedron 1990, 46, 3173–3188. [Google Scholar] [CrossRef]
- Wada, K.; Marumo, S. Synthesis and Plant Growth-Promoting Activity of Brassinolide Analogs. Agric. Biol. Chem. 1981, 45, 2579–2585. [Google Scholar]
- Takatsuto, S.; Yazawa, N.; Ishiguro, M.; Morisaki, M.; Ikekawa, N. Stereoselective Synthesis of Plant Growth-Promoting Steroids, Brassinolide, Castasterone, Typhasterol, and Their 28-Nor Analogs. J. Chem. Soc. Perkin Trans. 1 1984, 139–146. [Google Scholar] [CrossRef]
- Takatsuto, S. Synthesis of teasterone and typhasterol, brassinolide-related steroids with plant-growth-promoting activity. J. Chem. Soc. Perkin Trans. 1 1986, 1833–1836. [Google Scholar] [CrossRef]
- Takatsuto, S.; Ikekawa, N. Synthesis of homodolichosterone and related 2-deoxysteroids. Chem. Pharm. Bull. 1987, 35, 829–832. [Google Scholar] [CrossRef]
- Takatsuto, S.; Ikekawa, N.; Morishita, T.; Abe, H. Structure Activity Relationship of Brassinosteroids with Respect to the A/B-Ring Functional-Groups. Chem. Pharm. Bull. 1987, 35, 211–216. [Google Scholar] [CrossRef]
- Zhou, W.S.; Tian, W.S. Study on the Synthesis of Brassinolide and Related-Compounds. 3. Stereoselective Synthesis of Typhasterol from Hyodeoxycholic Acid. Tetrahedron 1987, 43, 3705–3712. [Google Scholar] [CrossRef]
- Aburatani, M.; Takeuchi, T.; Mori, K. Facile Syntheses of Brassinosteroids: Brassinolide, Castasterone, Teasterone and Typhasterol. Agric. Biol. Chem. 1987, 51, 1909–1913. [Google Scholar] [CrossRef]
- Huang, L.F.; Zhou, W.S. Studies on Steroidal Plant-Growth Regulators. Part 33. Novel Method for Construction of the Side-Chain of 23-Arylbrassinosteroids Via Heck Arylation and Asymmetric Dihydroxylation As Key Steps. J. Chem. Soc. Perkin Trans. 1 1994, 3579–3585. [Google Scholar] [CrossRef]
- Schmidt, J.; Voigt, B.; Adam, G. 2-Deoxybrassinolide—A naturally-occurring brassinosteroid from apium-graveolens. Phytochemistry 1995, 40, 1041–1043. [Google Scholar] [CrossRef]
- Voigt, B.; Schmidt, J.; Adam, G. Synthesis of 24-epiteasterone, 24-epityphasterol and their B-homo-6a-oxalactones from ergosterol. Tetrahedron 1996, 52, 1997–2004. [Google Scholar] [CrossRef]
- Herrera, H.; Carvajal, R.; Olea, A.F.; Espinoza, L. Structural modifications of deoxycholic acid to obtain three known brassinosteroid analogues and full NMR spectroscopic characterization. Molecules 2016, 21, 1139. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, M.; Kajikawa, A.; Haruyama, T.; Ogura, Y.; Okubayashi, M.; Morisaki, M.; Ikekawa, N. Synthetic studies of withanolides. Part 1. Synthesis of 5,6beta-epoxy-4beta-hydroxy-5beta-cholest-2-en-1-one and related compounds. J. Chem. Soc. Perkin Trans. 1 1975, 2295–2302. [Google Scholar] [CrossRef]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Cryist. B 2013, 69, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooft, R.W.W.; Straver, L.H.; Spek, A.L. Determination of absolute structure using Bayesian statistics on Bijvoet differences. J. Appl. Crystallogr. 2008, 41, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Takatsuto, S.; Ikekawa, N. Remote Substituent Effect on the Regioselectivity in the Baeyer-Villiger Oxidation of 5-Alpha-Cholestan-6-One Derivatives. Tetrahedron Lett. 1983, 24, 917–920. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment-Olex2 dissected. Acta Cryst. A 2015, 71, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 9, 17–20, 21a, 22–25 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
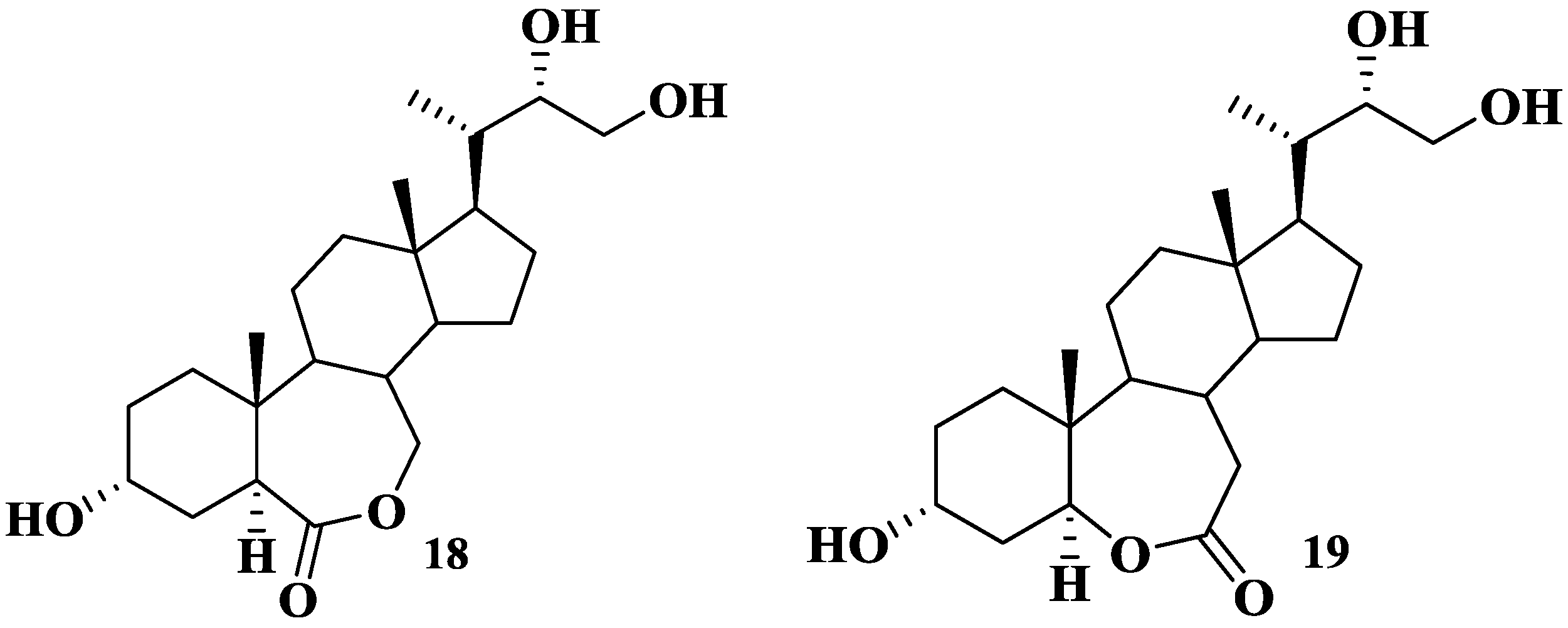{kind=link}
{kind=link}
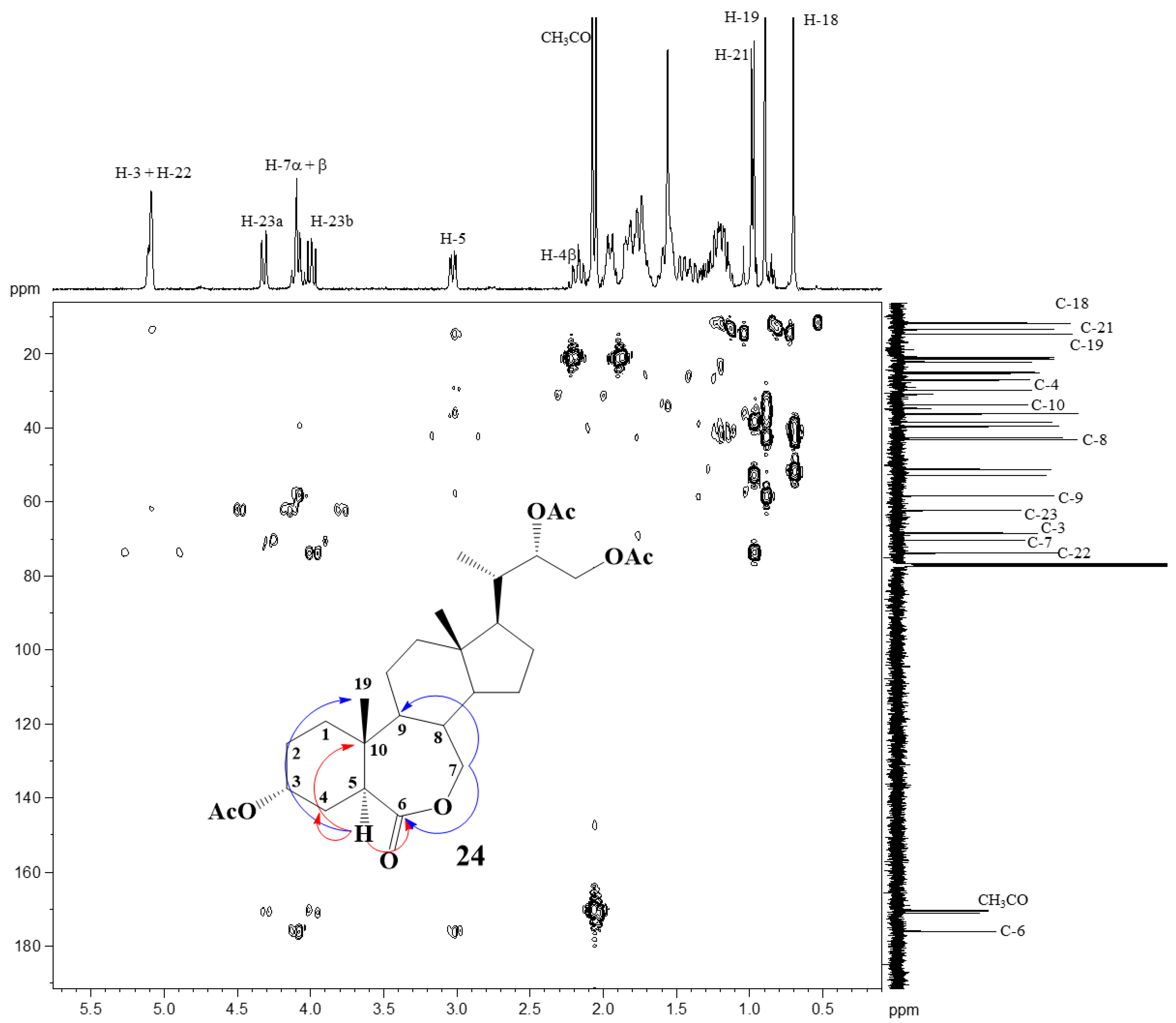{kind=link}
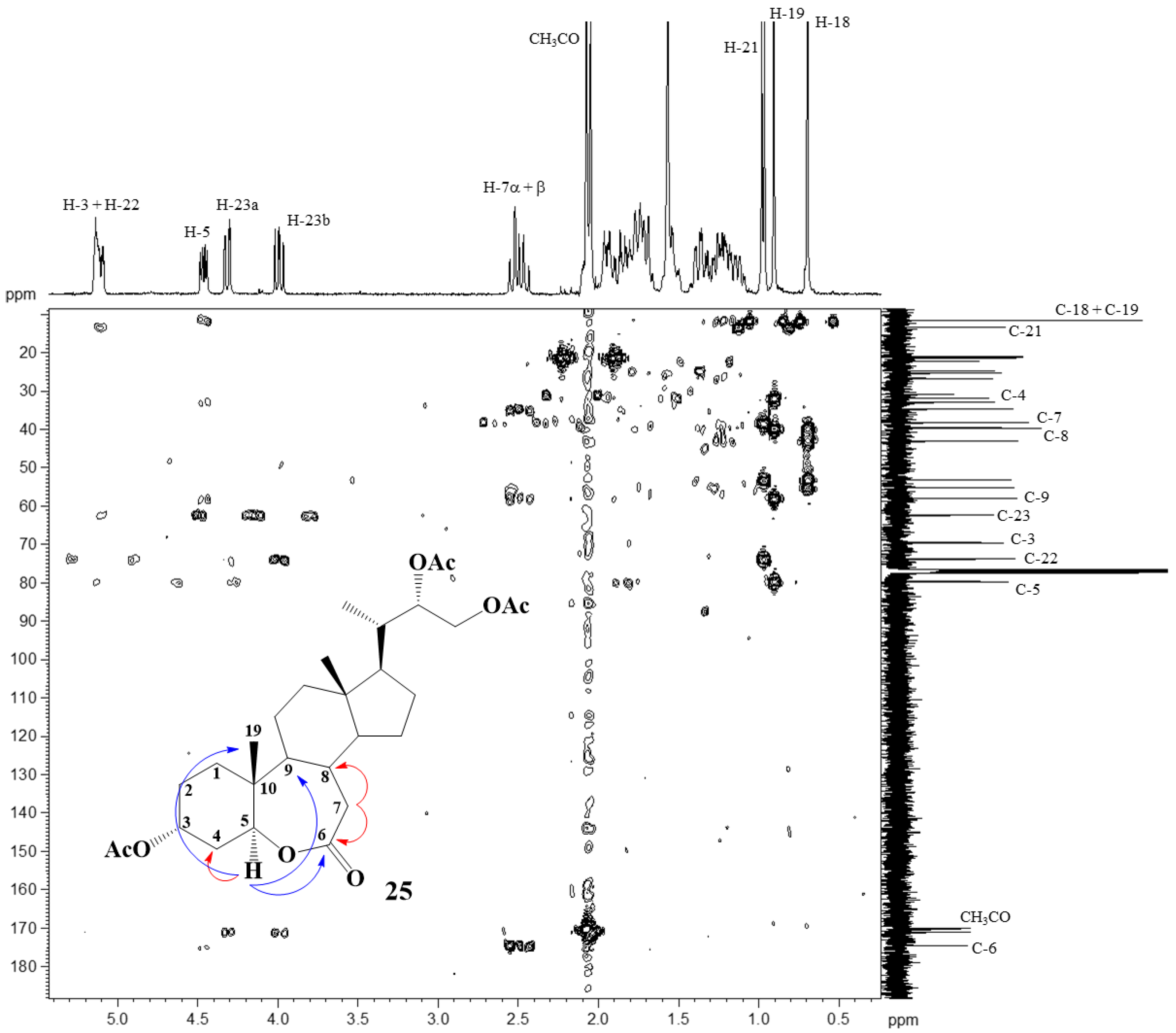{kind=link}
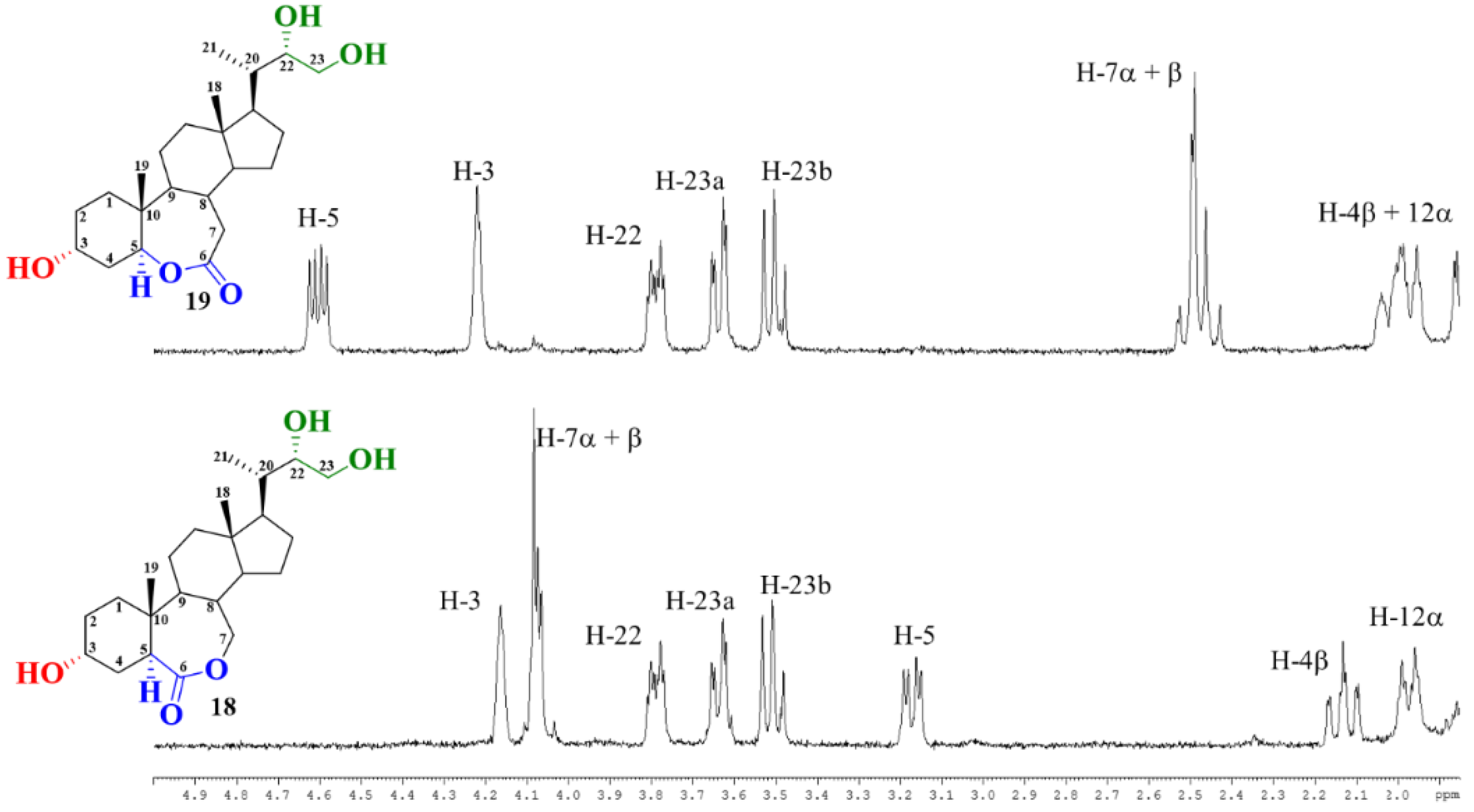{kind=link}
{kind=link}
{kind=link}
| C | 20 | 21a | 22 | 9 * |
|---|---|---|---|---|
| 1 | 32.38 | 32.17 | 32.36 | 32.88 |
| 2 | 28.19 | 27.20 | 27.41 | 28.73 |
| 3 | 68.85 | 68.70 | 68.81 | 66.02 |
| 4 | 25.27 | 25.05 | 25.26 | 28.53 |
| 5 | 52.58 | 52.38 | 52.58 | 52.84 |
| 6 | 211.81 | 211.72 | 211.62 | 214.49 |
| 7 | 46.75 | 46.49 | 46.69 | 47.61 |
| 8 | 37.92 | 37.74 | 37.90 | 39.45 |
| 9 | 53.79 | 52.68 | 52.90 | 54.19 |
| 10 | 41.28 | 41.07 | 41.23 | 42.62 |
| 11 | 21.06 | 20.87 | 21.06 | 22.19 |
| 12 | 39.36 | 39.19 | 39.41 | 40.76 |
| 13 | 42.94 | 43.16 | 43.41 | 44.46 |
| 14 | 55.41 | 53.51 | 53.72 | 55.08 |
| 15 | 25.01 | 24.78 | 25.00 | 28.45 |
| 16 | 23.91 | 23.85 | 24.02 | 25.07 |
| 17 | 56.80 | 56.15 | 56.41 | 57.56 |
| 18 | 12.18 | 11.56 | 11.81 | 12.14 |
| 19 | 12.41 | 12.20 | 12.40 | 12.67 |
| 20 | 41.10 | 39.87 | 40.33 | 42.01 |
| 21 | 20.04 | 12.86 | 12.90 | 13.42 |
| 22 | 144.89 | 73.69 | 71.77 | 75.19 |
| 23 | 111.84 | 62.22 | 66.39 | 63.19 |
| CH3CO | 170.26 | 170.15 | 170.27 | - |
| CH3CO | 21.41 | 21.21 | 21.41 | - |
| CH3CO | - | - | - | - |
| CH3CO | - | - | - | - |
| CH3CO | - | - | - | - |
| CH3CO | - | - | - | - |
| COAr | - | - | 167.01 | - |
| 1’ | - | - | 129.86 | - |
| 2’ | - | - | 129.63 | - |
| 3’ | - | - | 128.45 | - |
| 4’ | - | - | 133.22 | - |
| C | 23 | 24 | 25 | 18 * | 19 * |
|---|---|---|---|---|---|
| 1 | 32.35 | 33.66 | 31.84 | 32.78 | 31.06 |
| 2 | 27.01 | 26.95 | 26.65 | 24.84 | 25.25 |
| 3 | 68.79 | 68.38 | 69.56 | 64.32 | 65.60 |
| 4 | 25.24 | 29.74 | 32.95 | 32.50 | 35.39 |
| 5 | 52.55 | 42.57 | 79.68 | 41.67 | 80.04 |
| 6 | 211.49 | 175.97 | 174.64 | 177.44 | 176.08 |
| 7 | 46.63 | 70.31 | 38.12 | 70.43 | 37.88 |
| 8 | 37.85 | 39.42 | 38.31 | 39.54 | 39.89 |
| 9 | 52.84 | 58.31 | 57.97 | 58.15 | 57.77 |
| 10 | 41.18 | 36.14 | 39.55 | 36.17 | 39.64 |
| 11 | 21.03 | 22.10 | 22.17 | 22.03 | 22.02 |
| 12 | 39.38 | 39.53 | 39.69 | 40.03 | 39.48 |
| 13 | 43.39 | 43.03 | 43.09 | 42.91 | 42.91 |
| 14 | 53.67 | 52.80 | 55.13 | 52.70 | 54.94 |
| 15 | 24.98 | 25.13 | 25.33 | 27.87 | 27.46 |
| 16 | 23.94 | 24.83 | 24.83 | 27.20 | 26.76 |
| 17 | 56.37 | 51.10 | 53.14 | 50.98 | 52.97 |
| 18 | 11.81 | 11.57 | 11.59 | 11.41 | 11.32 |
| 19 | 12.38 | 14.54 | 11.59 | 14.39 | 11.32 |
| 20 | 38.32 | 38.32 | 34.78 | 39.33 | 34.77 |
| 21 | 13.37 | 13.28 | 13.29 | 12.77 | 12.66 |
| 22 | 73.97 | 73.80 | 73.80 | 73.57 | 73.50 |
| 23 | 62.39 | 62.30 | 62.35 | 62.08 | 62.02 |
| CH3CO | 171.09 | 171.07 | 171.12 | - | - |
| CH3CO | 21.39 | 21.36 | 21.32 | - | - |
| CH3CO | 170.42 | 170.40 | 170.41 | - | - |
| CH3CO | 21.23 | 21.21 | 21.24 | - | - |
| CH3CO | 170.25 | 170.28 | 170.17 | - | - |
| CH3CO | 20.86 | 20.85 | 20.88 | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carvajal, R.; González, C.; Olea, A.F.; Fuentealba, M.; Espinoza, L. Synthesis of 2-Deoxybrassinosteroids Analogs with 24-nor, 22(S)-23-Dihydroxy-Type Side Chains from Hyodeoxycholic Acid. Molecules 2018, 23, 1306. https://doi.org/10.3390/molecules23061306
Carvajal R, González C, Olea AF, Fuentealba M, Espinoza L. Synthesis of 2-Deoxybrassinosteroids Analogs with 24-nor, 22(S)-23-Dihydroxy-Type Side Chains from Hyodeoxycholic Acid. Molecules. 2018; 23(6):1306. https://doi.org/10.3390/molecules23061306
Chicago/Turabian StyleCarvajal, Rodrigo, Cesar González, Andrés F. Olea, Mauricio Fuentealba, and Luis Espinoza. 2018. "Synthesis of 2-Deoxybrassinosteroids Analogs with 24-nor, 22(S)-23-Dihydroxy-Type Side Chains from Hyodeoxycholic Acid" Molecules 23, no. 6: 1306. https://doi.org/10.3390/molecules23061306
APA StyleCarvajal, R., González, C., Olea, A. F., Fuentealba, M., & Espinoza, L. (2018). Synthesis of 2-Deoxybrassinosteroids Analogs with 24-nor, 22(S)-23-Dihydroxy-Type Side Chains from Hyodeoxycholic Acid. Molecules, 23(6), 1306. https://doi.org/10.3390/molecules23061306