Enantiomerically Enriched 1,2-P,N-Bidentate Ferrocenyl Ligands for 1,3-Dipolar Cycloaddition and Transfer Hydrogenation Reactions

Abstract
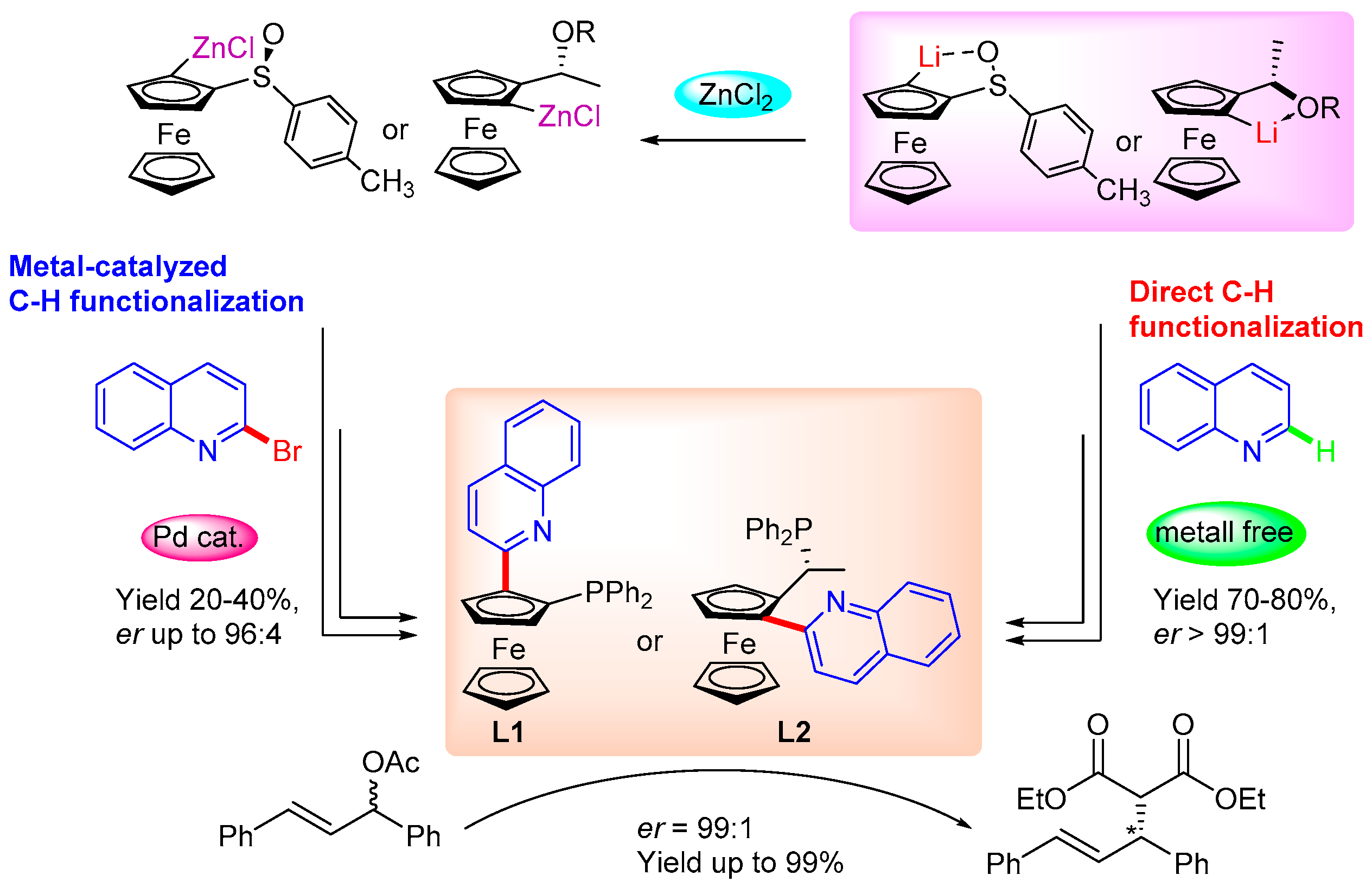
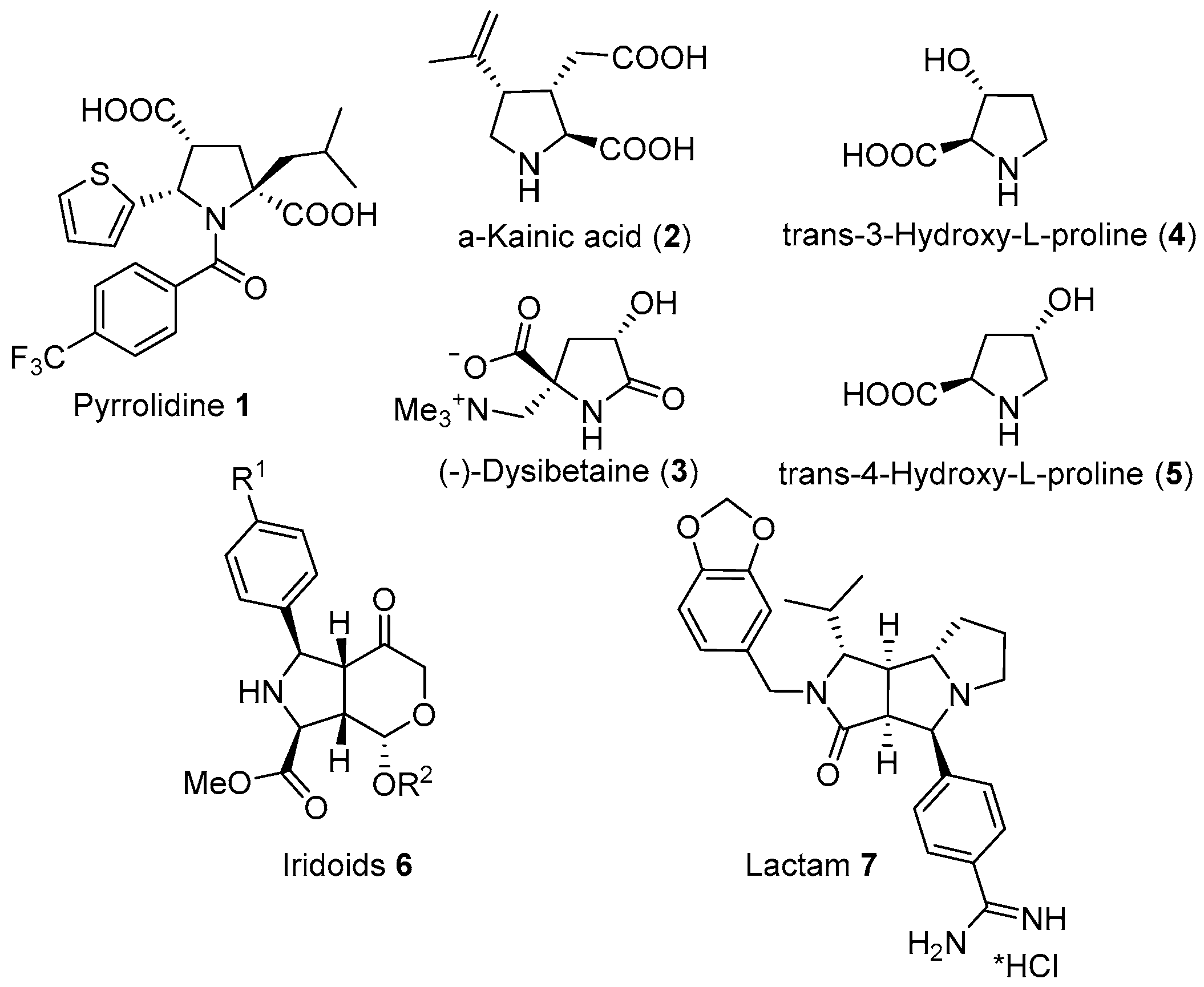
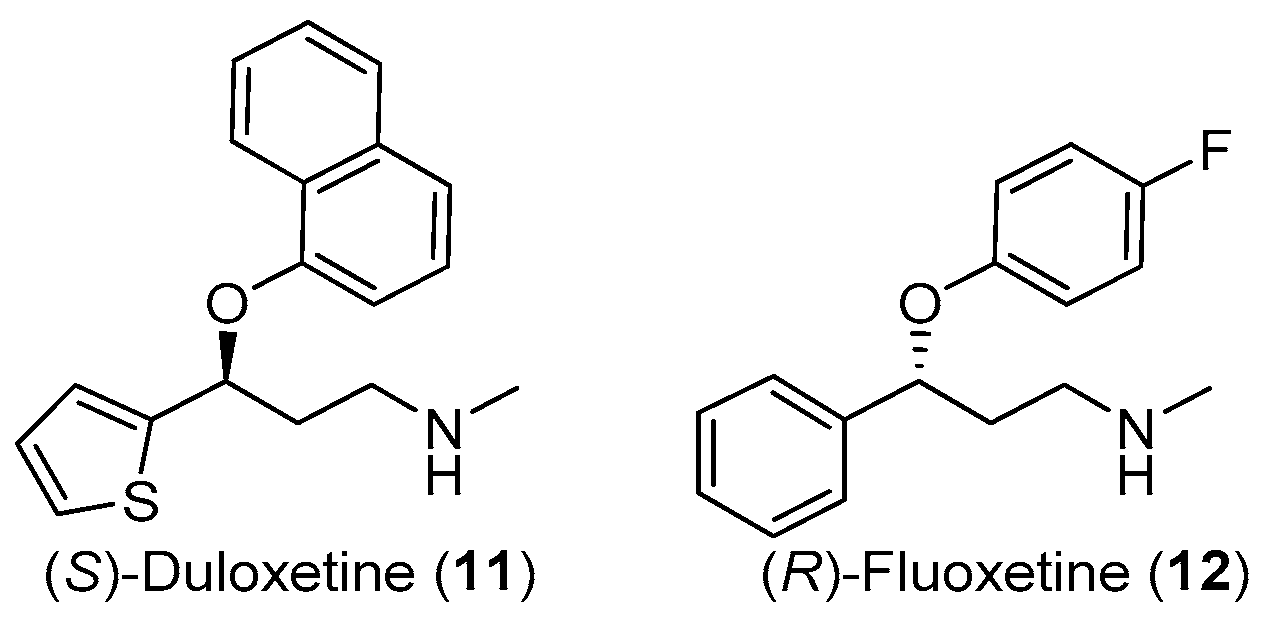
:1. Introduction
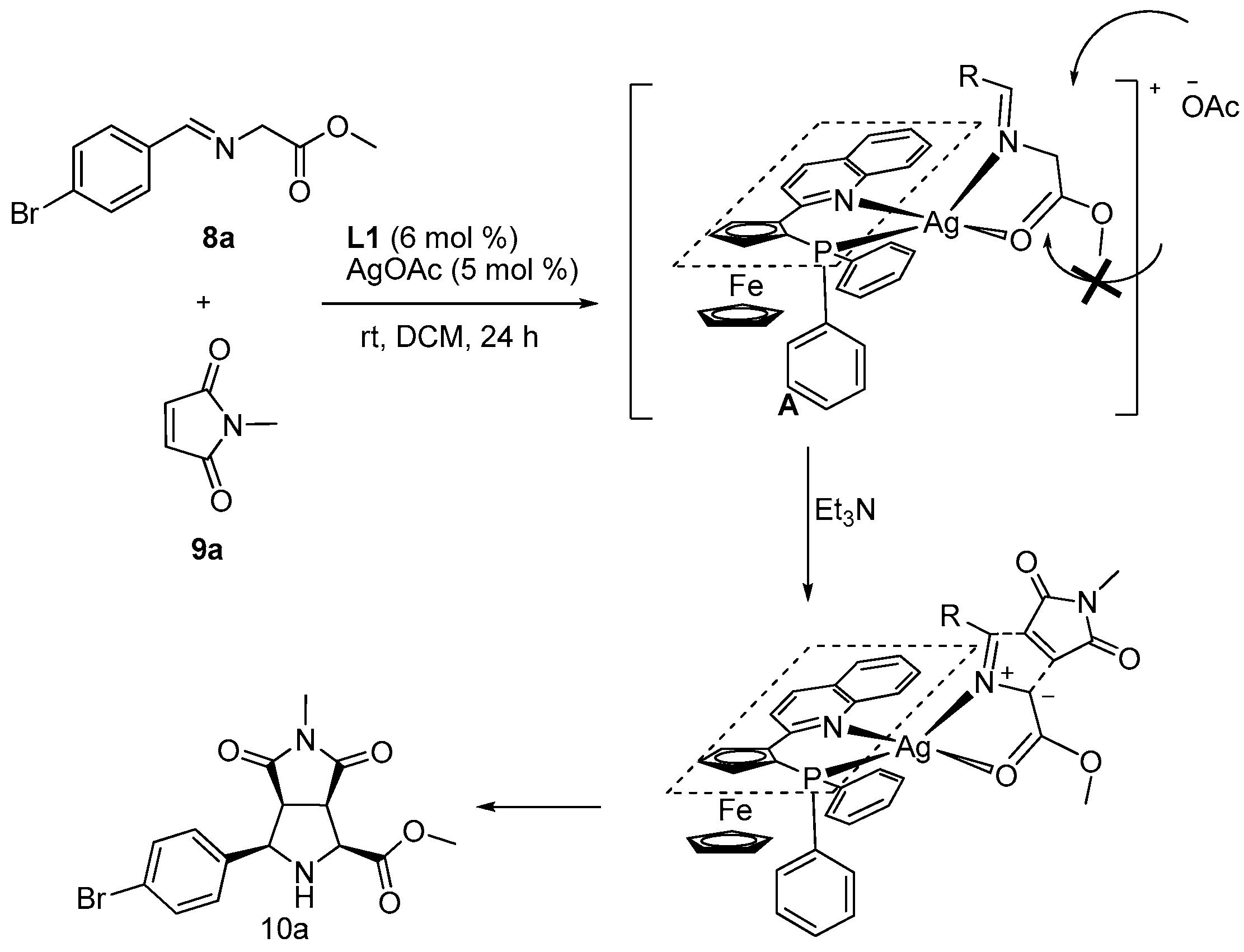
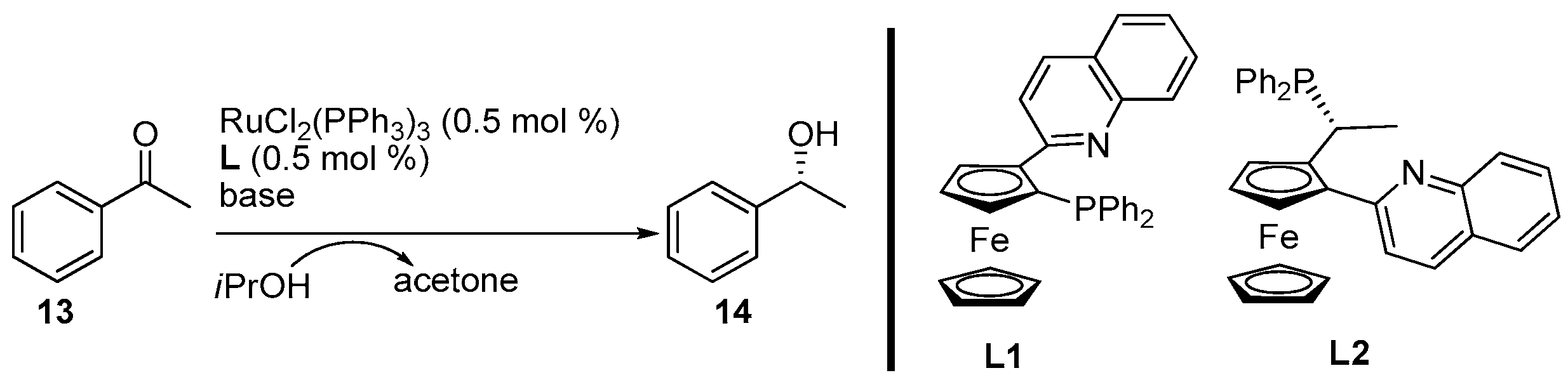
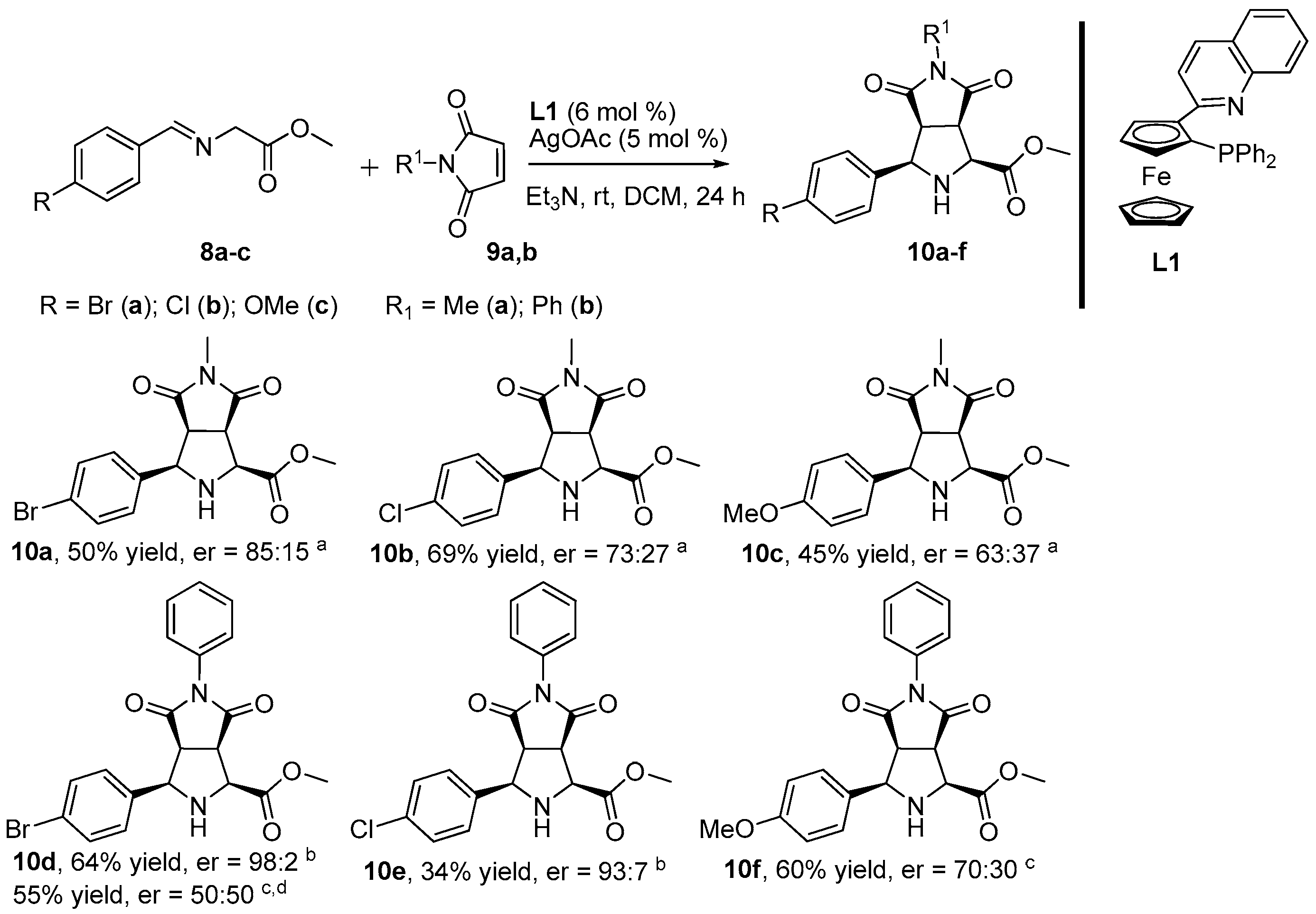
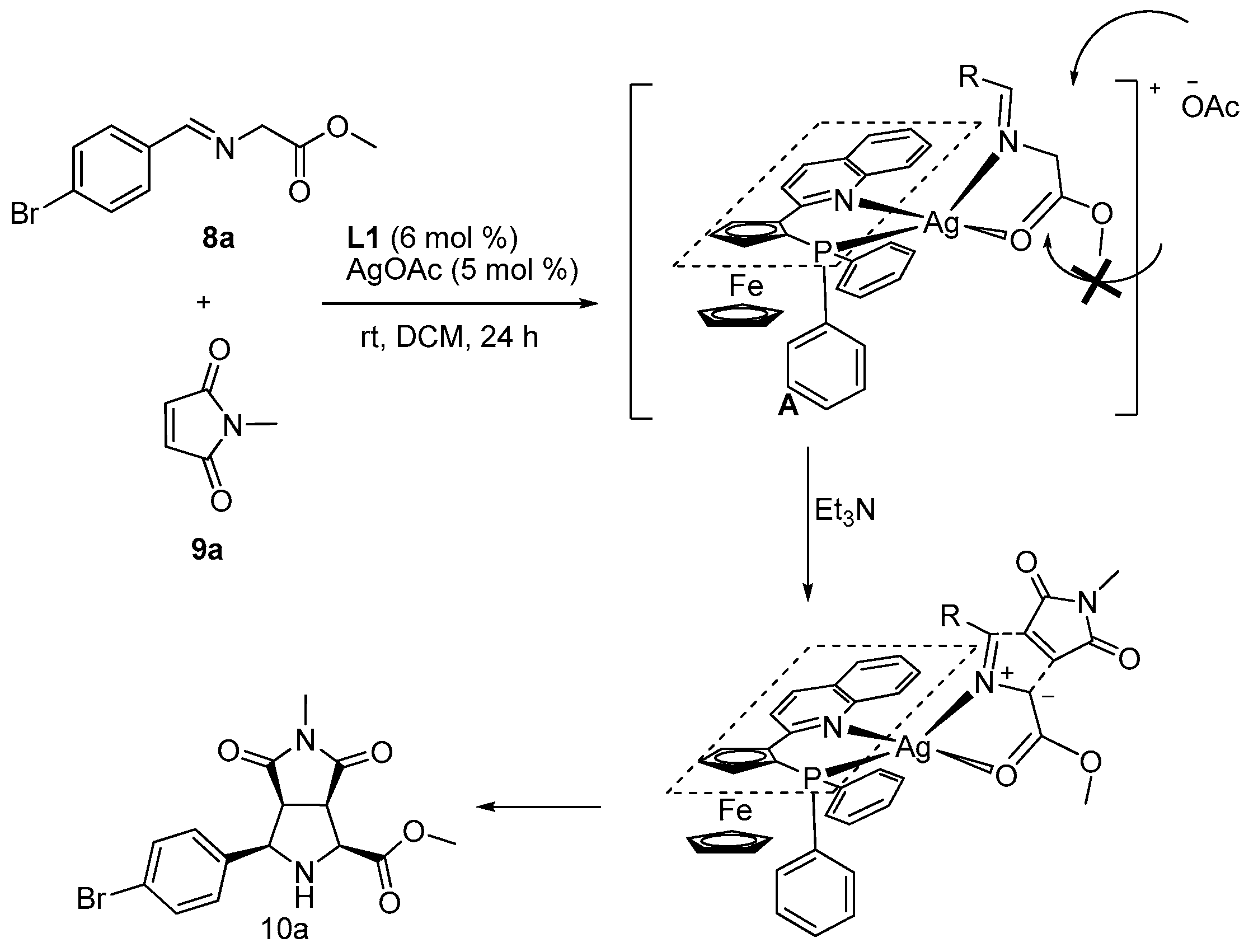
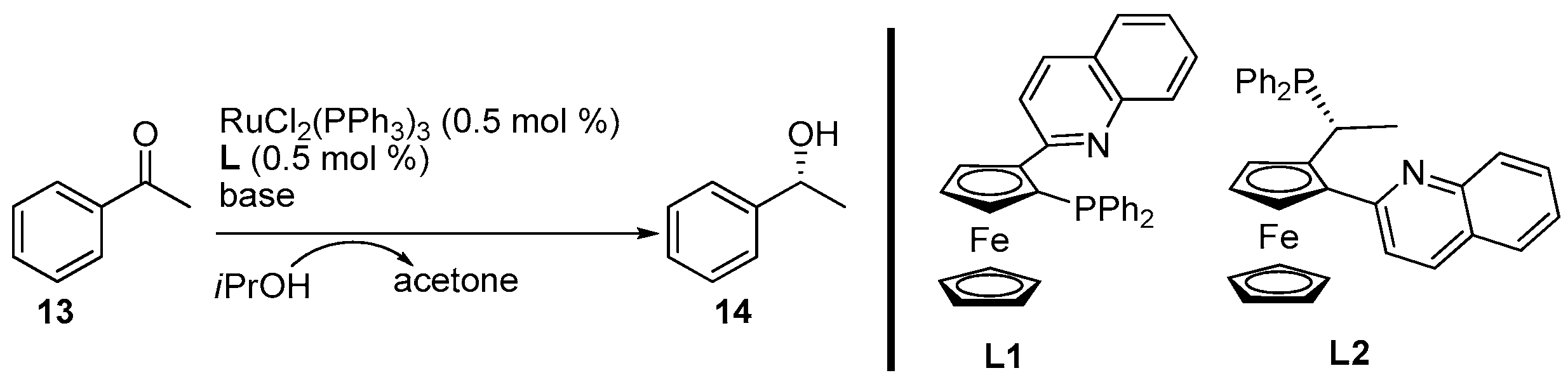
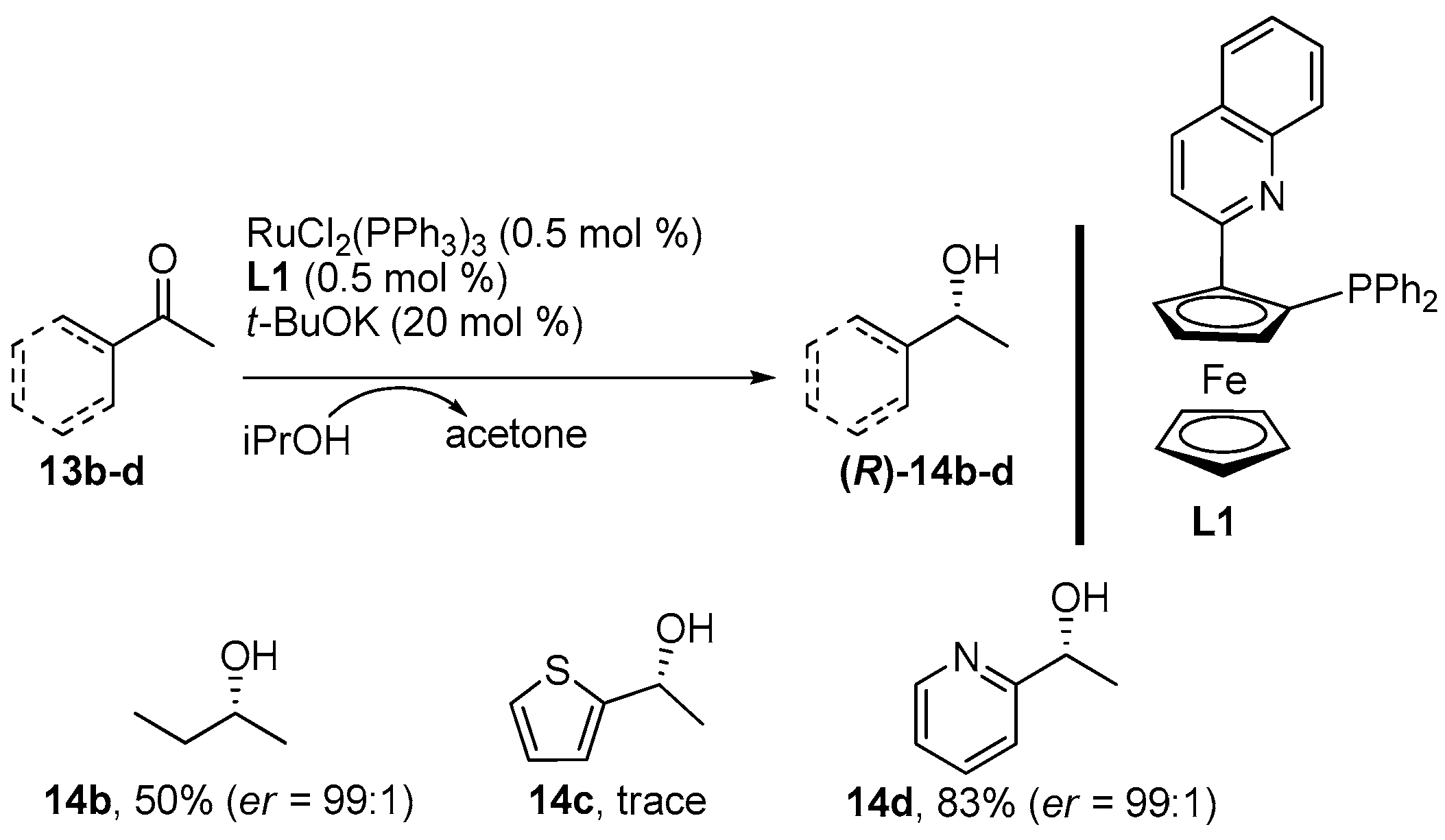
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Products 10
3.3. General Procedure for the Synthesis of Products 14
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dai, L.-X.; Hou, X.-L. (Eds.) Chiral Ferrocenes in Asymmetric Catalysis; Wiley-VCH: Weinheim, Germany, 2010; ISBN 9783527628841. [Google Scholar]
- Xue, Z.-Y.; Liu, T.-L.; Lu, Z.; Huang, H.; Tao, H.-Y.; Wang, C.-J. exo-Selective asymmetric 1,3-dipolar cycloaddition of azomethine ylides with alkylidene malonates catalyzed by AgOAc/TF-BiphamPhos. Chem. Commun. 2010, 46, 1727–1729. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-J.; Xue, Z.-Y.; Liang, G.; Lu, Z. Highly enantioselective 1,3-dipolar cycloaddition of azomethine ylides catalyzed by AgOAc/TF-BiphamPhos. Chem. Commun. 2009, 45, 2905–2907. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Tong, M.-C.; Wang, C.-J. Silver acetate/TF-BiphamPhos-catalyzed endo-selective enantioselective 1,3-dipolar cycloaddition of azomethine ylides with vinyl phenyl sulfone. Adv. Synth. Catal. 2009, 351, 3101–3106. [Google Scholar] [CrossRef]
- Wang, C.-J.; Liang, G.; Xue, Z.-Y.; Gao, F.J. Highly enantioselective 1,3-dipolar cycloaddition of azomethine ylides catalyzed by copper(I)/TF-BiphamPhos complexes. J. Am. Chem. Soc. 2008, 130, 17250–17251. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Torbio, J.; Arrayás, R.G.; Martín-Mature, B.; Carretero, J.C. Catalytic asymmetric 1,3-dipolar cycloaddition of azomethine ylides with α,β-unsaturated ketones. Org. Lett. 2009, 11, 393–396. [Google Scholar] [CrossRef] [PubMed]
- López-Pérez, A.; Adrio, J.; Carretero, J.C. The phenylsulfonyl group as a temporal regiochemical controller in the catalytic asymmetric 1,3-dipolar cycloaddition of azomethine ylides. Angew. Chem. Int. Ed. 2009, 48, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Longmire, J.M.; Wang, B.; Zhang, X. Highly enantioselective Ag(I)-catalyzed [3+2] cycloaddition of azomethine ylides. J. Am. Chem. Soc. 2002, 124, 13400–13401. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Zhang, Z.-M.; Xu, S.; Liu, B.; Xiao, Y.; Zhang, J. Copper(I)/Ming-Phos-catalyzed asymmetric intermolecular [3+2] cycloaddition of azomethine ylides with α-trifluoromethyl α,β-unsaturated esters. ACS Catal. 2017, 7, 210–214. [Google Scholar] [CrossRef]
- Chen, C.; Li, X.; Schreiber, S.L. Catalytic asymmetric [3+2] cycloaddition of azomethine ylides. Development of a versatile stepwise, three-component reaction for diversity-oriented synthesis. J. Am. Chem. Soc. 2003, 125, 10174–10175. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Zhou, Y.-G. Bifunctional AgOAc-catalyzed asymmetric [3+2] cycloaddition of azomethine ylides. Org. Lett. 2005, 7, 5055–5058. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Zhang, X.; Raghunath, M. Cu(I)-Catalyzed highly exo-selective and enantioselective [3+2] cycloaddition of azomethine ylides with acrylates. Org. Lett. 2005, 7, 4241–4244. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-X.; Peng, Q.; Zhang, Y.; Zhang, K.; Hong, W.; Hou, X.-L.; Wu, Y.-D. A highly enantio- and diastereoselective Cu-catalyzed 1,3-dipolar cycloaddition of azomethine ylides with nitroalkenes. Angew. Chem. Int. Ed. 2006, 45, 1979–1983. [Google Scholar] [CrossRef] [PubMed]
- Martín-Rodríguez, M.; Nájera, C.; Sansano, J.M.; Costa, P.R.R.; de Lima, E.C.; Dias, A.G. BINAP–AgSbF6 vs. BINAP–AgClO4 complexes as catalysts for the enantioselective 1,3-dipolar cycloaddition of azomethine ylides and alkenes. Synlett 2010, 962–966. [Google Scholar] [CrossRef]
- Nájera, C.; de Gracia Retamosa, M.; Sansano, J.M.; de Cózar, A.; Cossío, F.P. Enantioselective synthesis of polysubstituted prolines by Binap-silver-catalyzed 1,3-dipolar cycloadditions. Tetrahedron: Asymmetry 2008, 19, 2913–2923. [Google Scholar] [CrossRef]
- Nájera, C.; de Gracia Retamosa, M.; Sansano, J.M. Recoverable (R)- and (S)-Binap-Ag(I) complexes for the enantioselective 1,3-dipolar cycloaddition reaction of azomethine ylides. Org. Lett. 2007, 9, 4025–4028. [Google Scholar] [CrossRef] [PubMed]
- Mancebo-Aracil, J.; Martín-Rodríguez, M.; Nájera, C.; Sansano, J.M.; Costa, P.R.R.; de Lima, E.C.; Dias, A.G. Binap-silver salts as chiral catalysts for the enantioselective 1,3-dipolar cycloaddition of azomethine ylides and alkenes. Tetrahedron Asymmetry 2012, 23, 1596–1606. [Google Scholar] [CrossRef] [Green Version]
- Pandey, G.; Banerjee, P.; Gadre, S.R. Construction of enantiopure pyrrolidine ring system via asymmetric [3+2]-cycloaddition of azomethine ylides. Chem. Rev. 2006, 106, 4484–4517. [Google Scholar] [CrossRef] [PubMed]
- Stanley, L.M.; Sibi, M.P. Enantioselective copper-catalyzed 1,3-dipolar cycloadditions. Chem. Rev. 2008, 108, 2887–2902. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.; Potowski, M.; Jia, Z.-J.; Antonchick, A.P.; Waldmann, H. Catalytic enantioselective 1,3-dipolar cycloadditions of azomethine ylides for biology-oriented synthesis. Acc. Chem. Res. 2014, 47, 1296–1310. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Tada, A.; Tokoro, Y.; Fukuzawa, S.-I. Bifunctional AgOAc/ThioClickFerrophos complex-catalyzed asymmetric 1,3-dipolar cycloaddition of azomethine ylides with aryl- and alkylidene malonates. Tetrahedron Lett. 2014, 55, 1306–1309. [Google Scholar] [CrossRef]
- Wang, H.; Deng, Q.; Zhou, Z.; Hu, S.; Liu, Z.; Zhou, L.-Y. Ag2CO3/CA-AA-AmidPhos multifunctional catalysis in the enantioselective 1,3-dipolar cycloaddition of azomethine ylides. Org. Lett. 2016, 18, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Takayama, H.; Jia, Z.-J.; Kremer, L.; Bauer, J.O.; Strohmann, C.; Ziegler, S.; Antonchick, A.P.; Waldmann, H. Discovery of inhibitors of the Wnt and Hedgehog signaling pathways through the catalytic enantioselective synthesis of an iridoid-inspired compound collection. Angew. Chem. Int. Ed. 2013, 52, 12404–12408. [Google Scholar] [CrossRef] [PubMed]
- Potowski, M.; Merten, C.; Antonchick, A.P.; Waldmann, H. Catalytic aerobic oxidation and tandem enantioselective cycloaddition in cascade multicomponent synthesis. Chem. Eur. J. 2015, 21, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Potowski, M.; Golz, C.; Strohmann, C.; Antonchick, A.P.; Waldmann, H. Biology-oriented synthesis of benzopyrano[3,4-c]pyrrolidines. Bioorg. Med. Chem. 2015, 23, 2895–2903. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.-H.; Wang, C.-Y.; Reddy, G.M.; Tsai, Y.-L.; Lin, W.J. Enantioselective synthesis of polysubstituted benzopyrano[3,4-c]pyrrolidine frameworks via [3+2] cycloaddition of azomethine ylides and coumarin derivatives. Org. Chem. 2016, 81, 10071–10080. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.-F.; Zhang, J.; Xia, C.-G.; Xu, J.-X.; Xu, L.-W. N-tert-Butanesulfinyl imine and aromatic tertiary amide derived non-biaryl atropisomers as chiral ligands for silver-catalyzed endoselective [3+2] cycloaddition of azomethine ylides with maleimides. Tetrahedron 2016, 72, 2690–2699. [Google Scholar] [CrossRef]
- Landert, H.; Spindler, F.; Wyss, A.; Blaser, H.-U.; Pugin, B.; Ribourduoille, Y.; Gschwend, B.; Ramalingam, B.; Pfaltz, A. Chiral mixed secondary phosphine-oxide–phosphines: High-performing and easily accessible ligands for asymmetric hydrogenation. Angew. Chem. Int. Ed. 2010, 49, 6873–6876. [Google Scholar] [CrossRef] [PubMed]
- Baratta, W.; Chelucci, G.; Magnolia, S.; Siega, K.; Rigo, P. Highly productive CNN pincer ruthenium catalysts for the asymmetric reduction of alkyl aryl ketones. Chem. Eur. J. 2009, 15, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Mejía, E.; Aardoom, R.; Togni, A. Asymmetric transfer hydrogenation of ketones catalyzed by rhenium complexes with chiral ferrocenylphosphane ligands. Eur. J. Inorg. Chem. 2012, 5021–5032. [Google Scholar] [CrossRef]
- Utepova, I.A.; Chupakhin, O.N.; Serebrennikova, P.O.; Musikhina, A.A.; Charushin, V.N. Two approaches in the synthesis of planar chiral azinylferrocenes. J. Org. Chem. 2014, 79, 8659–8667. [Google Scholar] [CrossRef] [PubMed]
- Ali Döndas, H.; de Gracia Retamosa, M.; Sansano, J.M. Current trends towards the synthesis of bioactive heterocycles and natural products using 1,3-dipolar cycloadditions (1,3-DC) with azomethine ylides. Synthesis 2017, 49, 2819–2851. [Google Scholar] [CrossRef]
- Gothelf, A.S.; Gothelf, K.V.; Hazell, R.G.; Jørgensen, K.A. Catalytic asymmetric 1,3-dipolar cycloaddition reactions of azomethine ylides—A simple approach to optically active highly functionalized proline derivatives. Angew. Chem. Int. Ed. 2002, 41, 4236–4238. [Google Scholar] [CrossRef]
- Galliford, C.V.; Scheidt, K.A. Pyrrolidinyl-spirooxindole natural products as inspirations for the development of potential therapeutic agents. Angew. Chem. Int. Ed. 2007, 46, 8748–8758. [Google Scholar] [CrossRef] [PubMed]
- Nájera, C.; Sansano, J.M. 1,3-Dipolar cycloadditions: Applications to the synthesis of antiviral agents. Org. Biomol. Chem. 2009, 7, 4567–4581. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Tokumitsu, C.; Miyazaki, T.; Kuwano, S.; Awata, A. Catalytic asymmetric [3+2]-cycloaddition for stereodivergent synthesis of chiral indolyl-pyrrolidines. Org. Biomol. Chem. 2016, 14, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Pyrrole, pyrrolidine, pyridine, piperidine and tropane alkaloids. Nat. Prod. Rep. 2000, 17, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Gruttadauria, M.; Giacalone, F.; Noto, R. Supported proline and proline-derivatives as recyclable organocatalysts. Chem. Soc. Rev. 2008, 37, 1666–1688. [Google Scholar] [CrossRef] [PubMed]
- Guillena, G.; Nájera, C.; Ramón, D.J. Enantioselective direct aldol reaction: The blossoming of modern organocatalysis. Tetrahedron Asymmetry 2007, 18, 2249–2293. [Google Scholar] [CrossRef]
- Pellisser, H. Asymmetric organocatalysis. Tetrahedron 2007, 63, 9267–9331. [Google Scholar] [CrossRef]
- Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2005; ISBN 978-3-527-30517-9.
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric enamine catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef] [PubMed]
- Dalko, P.I. (Ed.) Enantioselective Organocatalysis: Reactions and Experimental Procedures; Wiley-VCH: Weinheim, Germany, 2007; ISBN 9783527610945. [Google Scholar]
- Erkkilä, A.; Majander, I.; Pihko, P.M. Iminium catalysis. Chem. Rev. 2007, 107, 5416–5470. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.; Ku, T.W.; Carr, T.J.; Kiesow, T.; Sarisky, R.T.; Lin-Goerke, J.; Baker, A.; Earnshaw, D.L.; Hofmann, G.A.; Keenan, R.M.; et al. Identification of small molecule inhibitors of the hepatitis C virus RNA-dependent RNA polymerase from a pyrrolidine combinatorial mixture. Bioorg. Med. Chem. Lett. 2005, 15, 1553–1556. [Google Scholar] [CrossRef] [PubMed]
- Nájera, C.; de Gracia Retamosa, M.; Sansano, J.M.; de Cózar, A.; Cossío, F.P. Diastereoselective 1,3-dipolar cycloaddition reactions between azomethine ylides and chiral acrylates derived from methyl (S)- and (R)-lactate—Synthesis of hepatitis C virus RNA-dependent RNA polymerase inhibitors. Eur. J. Org. Chem. 2007, 5038–5049. [Google Scholar] [CrossRef]
- Farwick, A.; Helmchen, G. Enantioselective total synthesis of (−)-α-kainic acid. Org. Lett. 2010, 12, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Wardrop, D.J.; Burge, M.S. Total synthesis of (−)-dysibetaine via a nitrenium ion cyclization–dienone cleavage strategy. Chem. Commun. 2004, 1230–1231. [Google Scholar] [CrossRef] [PubMed]
- Remuzon, P. Trans-4-hydroxy-l-proline, a useful and versatile chiral starting block. Tetrahedron 1996, 52, 13803–13835. [Google Scholar] [CrossRef]
- Obst, U.; Betchmann, P.; Lerner, C.; Seiler, P.; Diedericn, F.; Gramlich, V.; Weber, L.; Banner, D.W.; Schönholzer, P. Synthesis of novel nonpeptidic thrombin inhibitors. Helv. Chim. Acta 2000, 83, 855–909. [Google Scholar] [CrossRef]
- Zhou, Z.; Zheng, X.; Liu, J.; Li, J.; Wen, P.; Wang, H. L-tret-leucine-derived AmidPhos-Silver(I) chiral complexes for the asymmetric [3+2] cycloaddition of azomethine ylides. Synlett 2017, 28, 999–1003. [Google Scholar] [CrossRef]
- Cayuelas, A.; Larrañaga, O.; Nájera, C.; Sansano, J.M.; de Cózar, A.; Cossío, F.P. Taniaphos AgF-catalyzed enantioselective 1,3-dipolar cycloaddition of stabilized ylides derived from 2,2-dimethoxyacetaldehyde. Tetrahedron 2016, 72, 6043–6051. [Google Scholar] [CrossRef]
- Shi, J.-W.; Zhao, M.-X.; Lei, Z.-Y.; Shi, M. Axially chiral BINIM and Ni(II)-catalyzed highly enantioselective 1,3-dipolar cycloaddition reactions of azomethine ylides and N-arylmaleimides. J. Org. Chem. 2008, 73, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, S.; Özkan, H.; Tok, G.; Dişli, A. Facile method for 1,3-dipolar cycloaddition reaction of azomethine ylides: Highly stereoselective synthesis of substituted pyrrolidine derivatives. J. Heterocycl. Chem. 2013, 50, 1437–1440. [Google Scholar] [CrossRef]
- Träff, A.; Lihammar, R.; Bäckvall, J.-E. Chemoenzymatic dynamic kinetic resolution approach to enantiomerically pure (R)- and (S)-duloxetin. J. Org. Chem. 2011, 76, 3917–3921. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-N.; Fang, Q.; Hu, Y.-H.; Yang, L.-Y.; Wu, F.-F.; Xie, L.-J.; Wu, J.; Li, S. Copper(II)-catalyzed enantioselective hydrosilylation of halo-substituted alkyl aryl and heteroaryl ketones: Asymmetric synthesis of (R)-fluoxetine and (S)-duloxetine. Org. Biomol. Chem. 2014, 12, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Zhang, Z.; Liu, Y.; Imamoto, T.; Zhang, W. ZnCl2-Promoted asymmetric hydrogenation of β-secondary-amino ketones catalyzed by a P-chiral Rh–Bisphosphine complex. Angew. Chem. Int. Ed. 2015, 54, 2260–2264. [Google Scholar] [CrossRef] [PubMed]
- Zirakzadeh, A.; Schuecker, R.; Gorgas, N.; Mereiter, K.; Spindler, F.; Weissensteiner, W. Ruthenium complexes of phosphino-substituted ferrocenyloxazolines in the asymmetric hydrogenation and transfer hydrogenation of ketones: A comparison. Organometallics 2012, 31, 4241–4250. [Google Scholar] [CrossRef]
- Roux, E.L.; Malacea, R.; Manoury, E.; Poli, R.; Gonsalvi, L.; Peruzzini, M. Highly efficient asymmetric hydrogenation of alkyl aryl ketones catalyzed by iridium complexes with chiral planar ferrocenyl phosphino-thioether ligands. Adv. Synth. Catal. 2007, 349, 309–313. [Google Scholar] [CrossRef]
- Zhang, A.-L.; Yu, Z.-D.; Yang, L.-W.; Yang, N.-F. Synthesis of several polyethers derived from BINOL and their application in the asymmetric borane reduction of prochiral ketones. Tetrahedron Asymmetry 2015, 26, 173–179. [Google Scholar] [CrossRef]
- Yang, H.; Huo, N.; Yang, P.; Pei, H.; Lv, H.; Zhang, X. Rhodium catalyzed asymmetric hydrogenation of 2-pyridine ketones. Org. Lett. 2015, 17, 4144–4147. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Ligand | [M]/Base | Yield (%) 10a | er (%) 10a |
|---|---|---|---|---|
| 1 | - | [Cu(CH3CN)4][BF4]/Et3N | 65 | 50:50 |
| 2 | L1 | [Cu(CH3CN)4][BF4]/Et3N | 45 | 76:24 |
| 3 | L1 | AgF/Et3N | 50 | 50:50 |
| 4 | L1 | AgOAc/Et3N | 50 | 85:15 |
| Entry | Ligand | T (°C) | Base (mol %) | Time (h) | Yield (%) 14 | er (%) 1 14 |
|---|---|---|---|---|---|---|
| 1 | - | RT | t-BuOK (2) | 48 | 30 2 | 1:1 |
| 2 | L1 | RT | t-BuOK (2) | 48 | 20 2 | >99:1 |
| 3 | L1 | RT | DMAP (2) | 48 | - | - |
| 4 | L1 | RT | NaH (2) | 48 | 11 3 | >99:1 |
| 5 | L1 | RT | Et3N (20) | 72 | 2 3 | >99:1 |
| 6 | L1 | 80 | Et3N (20) | 20 | 40 3 | >99:1 |
| 7 | L1 | 80 | t-BuOK (20) | 20 | 98 3 | >99:1 |
| 8 | L1 | 80 | t-BuOK (10) | 20 | 97 3 | >99:1 |
| 9 | L1 | 80 | t-BuOK (2) | 20 | 96 3 | >99:1 |
| 10 | L2 | 80 | t-BuOK (20) | 20 | 53 3 | >99:1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Utepova, I.A.; Serebrennikova, P.O.; Streltsova, M.S.; Musikhina, A.A.; Fedorchenko, T.G.; Chupakhin, O.N.; Antonchick, A.P. Enantiomerically Enriched 1,2-P,N-Bidentate Ferrocenyl Ligands for 1,3-Dipolar Cycloaddition and Transfer Hydrogenation Reactions. Molecules 2018, 23, 1311. https://doi.org/10.3390/molecules23061311
Utepova IA, Serebrennikova PO, Streltsova MS, Musikhina AA, Fedorchenko TG, Chupakhin ON, Antonchick AP. Enantiomerically Enriched 1,2-P,N-Bidentate Ferrocenyl Ligands for 1,3-Dipolar Cycloaddition and Transfer Hydrogenation Reactions. Molecules. 2018; 23(6):1311. https://doi.org/10.3390/molecules23061311
Chicago/Turabian StyleUtepova, Irina A., Polina O. Serebrennikova, Marina S. Streltsova, Alexandra A. Musikhina, Tatiana G. Fedorchenko, Oleg N. Chupakhin, and Andrey P. Antonchick. 2018. "Enantiomerically Enriched 1,2-P,N-Bidentate Ferrocenyl Ligands for 1,3-Dipolar Cycloaddition and Transfer Hydrogenation Reactions" Molecules 23, no. 6: 1311. https://doi.org/10.3390/molecules23061311