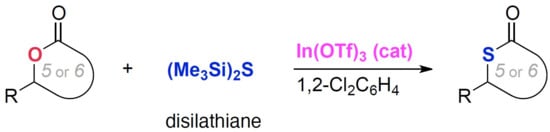
3.4. Product Characterization
Dihydro-5-phenyl-2(3H)-thiophenone (
2a) [
13]. General procedure A was followed with
5-phenyldihydrofuran-2-one (
1a, 80.2 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2a as a colorless oil (73.4 mg, 83%):
1H-NMR (CDCl
3, 500 MHz) δ 2.24–2.32 (m, 1 H, C
H2), 2.59–2.82 (m, 3 H, C
H2, C
H2), 5.01 (dd,
J = 10.0, 5.5 Hz, 1 H, C
H), 7.30–7.44 (m, 5 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 35.0, 42.9, 54.3, 127.4, 128.1, 128.9, 139.4, 207.9; MS (EI)
m/
z (%) 178 (M
+, 78), 117 (100).
Dihydro-5-(2-methylphenyl)-2(3H)-thiophenone (
2b) [
13]. General procedure B was followed with
5-(2-methylphenyl)dihydrofuran-2-one (
1b, 89.9 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2b as a colorless oil (69.7 mg, 73%):
1H-NMR (CDCl
3, 500 MHz) δ 2.26–2.34 (m, 1 H, C
H2), 2.42 (s, 3 H, C
H3), 2.55–2.61 (m, 1 H, C
H2), 2.67–2.74 (m, 1 H, C
H2), 2.78–2.84 (m, 1 H, C
H2), 5.25 (dd,
J = 9.5, 5.5 Hz, 1 H, C
H), 7.19–7.27 (m, 3 H, Ar
H), 7.56 (d,
J = 8.0 Hz, 1 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 19.5, 33.4, 42.7, 50.3, 126.5, 126.7, 127.8, 130.7, 135.7, 137.2, 208.0; MS (EI)
m/
z (%) 192 (M
+, 82), 117 (100).
Dihydro-5-(3-methylphenyl)-2(3H)-thiophenone (
2c) [
13]. General procedure B was followed with
5-(3-methylphenyl)dihydrofuran-2-one (
1c, 92.3 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2c as a dark yellow oil (81.9 mg, 82%):
1H-NMR (CDCl
3, 500 MHz) δ 2.23–2.31 (m, 1 H, C
H2), 2.37 (s, 3 H, C
H3), 2.57–2.81 (m, 3 H, C
H2, C
H2), 4.97 (dd,
J = 10.0, 5.5 Hz, 1 H, C
H), 7.13 (d,
J = 7.5 Hz, 1 H, Ar
H), 7.21–7.28 (m, 3 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 21.4, 35.0, 42.9, 54.3, 124.4, 128.1, 128.7, 128.9, 138.6, 139.3, 208.0; MS (EI)
m/
z (%) 192 (M
+, 100).
Dihydro-5-(4-methylphenyl)-2(3H)-thiophenone (
2d) [
13]. General procedure A was followed with
5-(4-methylphenyl)dihydrofuran-2-one (
1d, 87.5 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2d as a colorless oil (61.3 mg, 64%):
1H-NMR (CDCl
3, 500 MHz) δ 2.21–2.29 (m, 1 H, C
H2), 2.35 (s, 3 H, C
H3), 2.56–2.78 (m, 3 H, C
H2, C
H2), 4.96 (dd,
J = 10.0, 5.5 Hz, 1 H, C
H), 7.17 (d,
J = 8.0 Hz, 2 H, Ar
H), 7.30 (d,
J = 8.0 Hz, 2 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 21.0, 35.0, 42.9, 54.1, 127.2, 129.5, 136.4, 137.9, 208.0; MS (EI)
m/
z (%) 192 (M
+, 82), 117 (100).
Dihydro-5-(2,5-dimethylphenyl)-2(3H)-thiophenone (
2e) [
13]. General procedure A was followed with
5-(2,5-dimethylphenyl)dihydrofuran-2-one (
1e, 94.5 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2e as a yellow oil (66.6 mg, 65%):
1H-NMR (CDCl
3, 500 MHz) δ 2.24–2.31 (m, 1 H, C
H2), 2.33 (s, 3 H, C
H3), 2.36 (s, 3 H, C
H3), 2.53–2.58 (m, 1 H, C
H2), 2.65–2.73 (m, 1 H, C
H2), 2.76–2.82 (m, 1 H, C
H2), 5.22 (dd,
J = 10.0, 5.5 Hz, 1 H, C
H), 7.01 (d,
J = 7.5 Hz, 1 H, Ar
H), 7.07 (d,
J = 7.5 Hz, 1 H, Ar
H), 7.36 (s, 1 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 19.0, 21.0, 33.4, 42.7, 50.3, 127.1, 128.5, 130.6, 132.5, 136.2, 136.9, 208.1; MS (EI)
m/
z (%) 206 (M
+, 85), 131 (100).
Dihydro-5-([1,1′-biphenyl]-4-yl)-2(3H)-thiophenone (2f). General procedure A was followed with 5-[1,1′-Biphenyl]-4-yldihydro-2(3H)-furanone (1f, 96.2 mg). Column chromatography (10/1 hexane/EtOAc) and GPC afforded 2f as a colorless solid (61.6 mg, 61%): m.p. 120–121 °C; 1H-NMR (CDCl3, 500 MHz) δ 2.78–2.36 (m, 1 H, CH2), 2.63–2.83 (m, 3 H, CH2, CH2), 5.05 (dd, J = 10.0, 5.5 Hz, 1 H, CH), 7.35–7.61 (m, 9 H, ArH); 13C-NMR (CDCl3, 126 MHz) δ 35.0, 42.9, 54.1, 127.0, 127.5, 127.6, 127.9, 128.8, 138.4, 140.4, 141.1, 207.8; MS (EI) m/z (%) 254 (M+, 100); HRMS (EI) calcd for [M]+ (C16H14OS) m/z 254.0765, found 254.0771.
Dihydro-5-(3-methoxyphenyl)-2(3H)-thiophenone (
2g) [
13]. General procedure B was followed with
5-(3-methoxyphenyl)dihydrofuran-2-one (
1g, 100.9 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2g as a yellow oil (86.4 mg, 79%):
1H-NMR (CDCl
3, 500 MHz) δ 2.21–2.30 (m, 1 H, C
H2), 2.59–2.78 (m, 3 H, C
H2, C
H2), 3.81 (s, 3 H, C
H3), 4.96 (dd,
J = 10.0, 5.5 Hz, 1 H, C
H), 6.85 (dd,
J = 8.0, 3.0 Hz, 1 H, Ar
H), 6.97 (s, 1 H, Ar
H), 7.00 (d,
J = 8.0 Hz, 1 H, Ar
H), 7.28 (dd,
J = 8.0, 8.0 Hz, 1 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 34.8, 42.7, 54.2, 55.2, 113.1, 113.3, 119.6, 129.8, 140.9, 159.8, 207.8; MS (EI)
m/
z (%) 208 (M
+, 100).
Dihydro-5-(4-methoxyphenyl)-2(3H)-thiophenone (
2h) [
13]. General procedure B was followed with
5-(4-methoxyphenyl)dihydrofuran-2-one (
1h, 97.7 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2h as a colorless solid (23.2 mg, 22%): m.p. 74–75 °C;
1H-NMR (CDCl
3, 500 MHz) δ 2.21–2.29 (m, 1 H, C
H2), 2.56–2.59 (m, 1 H, C
H2), 2.65–2.80 (m, 2 H, C
H2, C
H2), 3.81 (s, 3 H, C
H3), 4.97 (dd,
J = 10.5, 5.5 Hz, 1 H, C
H), 6.90 (d,
J = 8.0 Hz, 2 H, Ar
H), 7.35 (d,
J = 8.0 Hz, 2 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 35.2, 43.1, 54.0, 55.3, 114.2, 128.6, 131.3, 159.4, 208.1; MS (EI)
m/
z (%) 208 (M
+, 79), 147 (100).
Dihydro-5-(4-fluorophenyl)-2(3H)-thiophenone (
2i) [
13]. General procedure B was followed with
5-(4-fluorophenyl)dihydrofuran-2-one (
1i, 96.3 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2i as a pale green oil (82.3 mg, 79%):
1H-NMR (CDCl
3, 297 MHz) δ 2.17–2.29 (m, 1 H, C
H2), 2.56–2.82 (m, 3 H, C
H2, C
H2), 4.99 (dd,
J = 9.8, 5.0 Hz, 1 H, C
H), 7.03–7.08 (m, 2 H, Ar
H), 7.38–7.42 (m, 2 H, Ar
H);
13C-NMR (CDCl
3, 75 MHz) δ 35.1, 42.8, 53.5, 115.7 (d,
JC–F = 20.9 Hz), 129.0 (d,
JC–F = 9.0 Hz), 135.2 (d,
JC–F = 3.7 Hz), 162.3 (d,
JC–F = 246.8 Hz), 207.4; MS (EI)
m/
z (%) 196 (M
+, 72), 135 (100).
Dihydro-5-(4-chlorophenyl)-2(3H)-thiophenone (
2j) [
13]. General procedure A was followed with
5-(4-chlorophenyl)dihydrofuran-2-one (
1j, 98.1 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2j as a colorless solid (85.5 mg, 87%): m.p. 45–48 °C;
1H-NMR (CDCl
3, 500 MHz) δ 2.18–2.26 (m, 1 H, C
H2), 2.59–2.80 (m, 3 H, C
H2, C
H2), 4.97 (dd,
J = 10.0, 5.5 Hz, 1 H, C
H), 7.34–7.35 (m, 4 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 34.9, 42.7, 53.5, 128.7, 129.0, 133.8, 138.0, 207.2; MS (EI)
m/
z (%) 214 (M
++2, 23), 212 (M
+, 63), 117 (100).
Dihydro-5-(4-bromophenyl)-2(3H)-thiophenone (
2k) [
13]. General procedure A was followed with
5-(4-bromophenyl)dihydrofuran-2-one (
1k, 120.1 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2k as a colorless solid (96.1 mg, 74%): m.p. 58–60 °C;
1H-NMR (CDCl
3, 500 MHz) δ 2.18–2.26 (m, 1 H, C
H2), 2.58–2.80 (m, 3 H, C
H2, C
H2), 4.95 (dd,
J = 10.0, 5.5 Hz, 1 H, C
H), 7.30 (d,
J = 7.0 Hz, 2 H, Ar
H), 7.50 (d,
J = 7.0 Hz, 2 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 34.9, 42.7, 53.6, 122.0, 129.1, 132.0, 138.6, 207.2; MS (EI)
m/
z (%) 258 (M
+ + 2, 59), 256 (M
+, 58), 117 (100).
Dihydro-5-(5,6,7,8-tetrahydronaphthalen-2-yl)-2(3H)-thiophenone (
2l) [
13]. General procedure B was followed with
5-(5,6,7,8-Tetrahydronaphthalen-2-yl)dihydrofuran-2(3H)-one (
1l, 96.8 mg). Column chromatography (10/1 hexane/EtOAc) and GPC afforded
2l as a colorless oil (37.4 mg, 36%):
1H-NMR (CDCl
3, 500 MHz) δ 1.78–1.81 (m, 4 H, C
H2), 2.23–2.31 (m, 1 H, C
H2), 2.55–2.80 (m, 7 H, C
H2, C
H2), 4.94 (dd,
J = 10.5, 5.5 Hz, 1 H, C
H), 7.07 (d,
J = 8.0 Hz 1 H, Ar
H) 7.12–7.15 (m, 2 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 23.0, 23.1, 29.1, 29.4, 35.0, 43.0, 54.3, 124.4, 128.1, 129.6, 136.4, 137.3, 137.7, 208.3; MS (EI)
m/
z (%) 232 (M
+, 100).
Dihydro-5-(thiophen-2-yl)-2(3H)-thiophenone (2m). General procedure B was followed with dihydro-5-(3-thienyl)-2(3H)-furanone (1m, 89.0 mg). Column chromatography (10/1 hexane/EtOAc) afforded 2m as a light green oil (22.3 mg, 23%): 1H-NMR (CDCl3, 500 MHz) δ 2.34–2.42 (m, 1 H, CH2), 2.65–2.85 (m, 3 H, CH2, CH2), 5.29 (dd, J = 7.5, 5.5 Hz, 1 H, CH), 6.97 (dd, J = 5.0, 3.5 Hz, 1 H, ArH), 7.07 (d, J = 3.5 Hz, 1 H, ArH), 7.27 (dd, J = 5.0, 3.5 Hz, 1 H, ArH); 13C-NMR (CDCl3, 126 MHz) δ 35.6, 42.3, 49.1, 125.3, 125.6, 126.9, 143.7, 207.0; MS (EI) m/z (%) 184 (M+, 64), 123 (100); HRMS (EI) calcd for [M]+ (C8H8OS2) m/z 184.0017, found 184.0011.
Thiophthalide (
2o) [
16]. General procedure A was followed with phthalide (
1o, 67.6 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2o as a colorless solid (26.8 mg, 18%): m.p. 68–70 °C;
1H-NMR (CDCl
3, 500 MHz) δ 4.48 (s, 2 H, C
H2), 7.48 (dd,
J = 7.5, 7.5 Hz, 1 H, Ar
H), 7.55 (d,
J = 7.0 Hz, 1 H, Ar
H), 7.63 (dd,
J = 7.5, 7.5 Hz, 1 H, Ar
H), 7.85 (d,
J = 7.5 Hz, 1 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 34.6, 123.9, 126.3, 128.0, 133.1, 135.8, 147.0, 198.0; MS (EI)
m/
z (%) 150 (M
+, 89), 121 (100).
3-Phenyl-benzo[c]thiophen-1(3H)-one (
2p) [
13]. General procedure A was followed with
3-phenylisobenzofuran-1-one (
1p, 105.9 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2p as a pale yellow solid (7.5 mg, 85%): m.p. 87–88 °C;
1H-NMR (CDCl
3, 500 MHz) δ 5.91 (s, 1 H, C
H), 7.25–7.36 (m, 6 H, Ar
H), 7.48 (dd,
J = 7.5, 7.5 Hz, 1 H, Ar
H), 7.56 (dd,
J = 7.5, 7.5 Hz, 1 H, Ar
H), 7.86 (d,
J = 7.5 Hz, 1 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 54.6, 123.6, 126.6, 128.29, 128.33, 128.4, 129.1, 133.6, 135.7, 138.8, 151.2, 197.2; MS (EI)
m/
z (%) 226 (M
+, 100).
Tetrahydro-6-phenyl-2H-benzothiopyran-2-one (
2q) [
13]. General procedure A was followed with
tetrahydro-6-phenyl-2H-pyran-2-one (
1q, 89.3 mg). Colum chromatography (10/1 hexane/EtOAc) afforded
2q as an orange oil (9.7 mg, 10%):
1H-NMR (CDCl
3, 500 MHz) δ 1.94–2.10 (m, 2 H, C
H2), 2.14–2.19 (m, 1 H, C
H2), 2.37–2.41 (m, 1 H, C
H2), 2.57–2.43 (m, 1 H, C
H2), 2.59–2.78 (m, 1 H, C
H2), 4.65 (dd,
J = 11.0, 4.0 Hz, 1 H, C
H), 7.29–7.32 (m, 1 H, Ar
H), 7.35–7.39 (m, 4 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 22.7, 32.4, 40.5, 50.5, 127.7, 128.1, 128.9, 140.4, 201.3; MS (EI)
m/
z (%) 192 (M
+, 59), 104 (100).
Tetrahydro-6-(4-methylphenyl)-2H-thiopyran-2-one (2r). General procedure A was followed with tetrahydro-6-(4-methylphenyl)-2H-Pyran-2-one (1r, 92.3 mg). Column chromatography (10/1 hexane/EtOAc) and GPC afforded 2r as a colorless oil (9.5 mg, 9%): 1H-NMR (CDCl3, 500 MHz) δ 1.93–2.07 (m, 2 H, CH2), 2.15–2.17 (m, 1 H, CH2), 2.35 (s, 3 H, CH3), 2.37–2.44 (m, 1 H, CH2), 2.72–2.76 (m, 1 H, CH2), 4.61 (dd, J = 11.0, 3.5 Hz, 1 H, CH), 7.17 (d, J = 7.5 Hz, 2 H, ArH), 7.26 (d, J = 7.5 Hz, 2 H, ArH); 13C-NMR (CDCl3, 126 MHz) δ 21.1, 22.7, 32.4, 40.5, 50.2, 127.5, 129.5, 137.4, 137.9, 201.6; MS (EI) m/z (%) 206 (M+, 62), 118 (100); HRMS (EI) calcd for [M]+ (C12H14OS) m/z 206.0765, found 206.0766.
6-(4-Chlorophenyl)tetrahydro-2H-thiopyran-2-one (2s). General procedure A was followed with 6-(4-chlorophenyl)tetrahydro-2H-pyran-2-one (1s, 107.5 mg). Column chromatography (10/1 hexane/EtOAc) and GPC afforded 2s as a colorless oil (4.8 mg, 4%): 1H-NMR (CDCl3, 500 MHz) δ 1.93–2.02 (m, 2 H, CH2), 2.04–2.19 (m, 1 H, CH2), 2.34–2.39 (m, 1 H, CH2), 2.56–2.63 (m, 1 H, CH2), 2.73–2.78 (m, 1 H, CH2), 4.62 (dd, J = 11.0, 4.5 Hz, 1 H, CH), 7.30–7.35 (m, 4 H, ArH); 13C-NMR (CDCl3, 126 MHz) δ 22.6, 32.4, 40.5, 49.7, 129.0, 129.1, 133.9, 139.0, 200.8; MS (EI) m/z (%) 226 (M++2, 20), 210 (M+, 23), 138 (100); HRMS (EI) calcd for [M]+ (C11H11OSCl) m/z 226.0219, found 226.0241.
1,4-Dihydro-3H-2-benzothiopyran-3-one (
2t) [
13]. General procedure A was followed with
1,4-dihydro-3H-2-benzopyran-3-one (
1t, 74.3 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2t as a pale yellow solid (40.9 mg, 50%): m.p. 90–93 °C;
1H-NMR (CDCl
3, 500 MHz) δ 3.79 (s, 2 H, C
H2), 4.22 (s, 2 H, C
H2), 7.21–7.32 (m, 4 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 34.2, 49.2, 126.6, 127.4, 128.0, 128.7, 133.7, 134.2, 202.9; MS (EI)
m/
z (%) 164 (M
+, 14), 104 (100).
Phthalic thioanhydride (
2u) [
17]. General procedure A was followed with
phthalic anhydride (
1u, 77.3 mg). Column chromatography (10/1 hexane/EtOAc) afforded
2u as a yellow solid (56.1 mg, 74%): m.p. 68–70 °C;
1H-NMR (CDCl
3, 500 MHz) δ 7.81–7.83 (m, 2 H, Ar
H), 7.97–7.99 (m, 2 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 123.8, 135.0, 138.7, 189.8; MS (EI)
m/
z (%) 164 (M
+, 100).
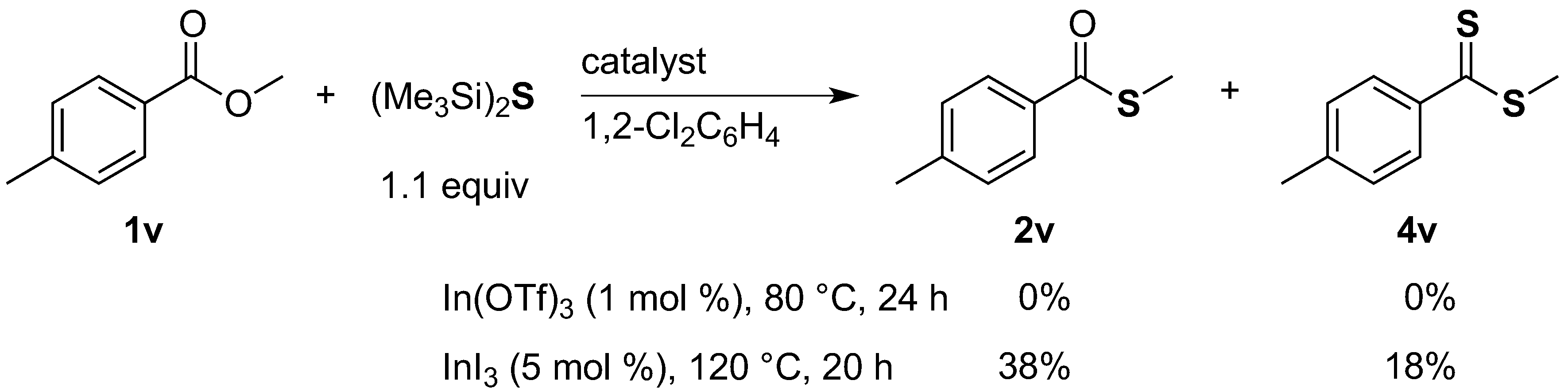
S-Methyl 4-methylbenzothioate (2v). General procedure A was followed with methyl 4-methylbenzoate (1v, 75.0 mg). Column chromatography (100/1 hexane/EtOAc) afforded 2v as a red oil (31.8 mg, 38%): 1H-NMR (CDCl3, 500 MHz) δ 2.40 (s, 3 H, CH3), 2.46 (s, 3 H, CH3), 7.24 (d, J = 14.5 Hz, 2 H, ArH), 7.87 (d, J = 14.5 Hz, 2 H, ArH); 13C-NMR (CDCl3, 126 MHz) δ 11.6, 21.7, 127.2, 129.2, 134.5, 144.1, 192.1; MS (EI) m/z (%) 166 (M+, 5), 119 (100).
Methyl 4-methylbenzodithioate (
4v) [
18]. General procedure A was followed with
methyl 4-methylbenzoate (
1v, 75.0 mg). Column chromatography (100/1 hexane/EtOAc) afforded
4v as an orange oil (15.9 mg, 18%):
1H-NMR (CDCl
3, 500 MHz) δ 2.38 (s, 3 H, C
H3), 2.77 (s, 3 H, C
H3), 7.18 (d,
J = 13.5 Hz, 2 H, Ar
H), 7.94 (d,
J = 13.5 Hz, 2 H, Ar
H);
13C-NMR (CDCl
3, 126 MHz) δ 20.5, 21.5, 126.8, 129.0, 142.6, 143.2, 228.8; MS (EI)
m/
z (%) 182 (M
+, 22), 135(100).
5-[1,1′-Biphenyl]-4-yldihydro-2(3H)-furanone (1f). A colorless solid: m.p. 100–102 °C; 1H-NMR (CDCl3, 500 MHz) δ 2.19–2.29 (m, 1 H, CH2), 2.66–2.72 (m, 3 H, CH2), 5.54–5.57 (m, 1 H, CH), 7.35–7.46 (m, 5 H, ArH), 7.58–7.62 (m, 4 H, ArH); 13C-NMR (CDCl3, 126 MHz) δ 29.0, 30.9, 81.0, 125.8, 127.1, 127.45, 127.53, 128.8, 138.3, 140.4, 141.4, 176.9; MS (EI) m/z (%) 238 (M+, 100); HRMS (EI) calcd for [M]+ (C16H14O2) m/z 238.0994, found 238.1002.
Tetrahydro-6-(4-methylphenyl)-2H-pyran-2-one (1r). A colorless solid: m.p. 81–83 °C; 1H-NMR (CDCl3, 500 MHz) δ 1.80–1.88 (m, 1 H, CH2), 1.92–1.98 (m, 2 H, CH2), 2.09–2.14 (m, 1 H, CH2), 2.34 (s, 3 H, CH3), 2.51–2.56 (m, 1 H, CH2), 2.57–2.71 (m, 1 H, CH2), 5.30 (dd, J = 10.5, 3.5 Hz, 1 H, CH), 7.17 (d, J = 7.5 Hz, 2 H, ArH), 7.22 (d, J = 7.5 Hz, 2 H, ArH); 13C-NMR (CDCl3, 126 MHz) δ 18.4, 21.0, 29.3, 30.3, 81.5, 125.5, 129.1, 136.6, 137.8, 171.4; MS (EI) m/z (%) 190 (M+, 42), 118 (100); HRMS (EI) calcd for [M]+ (C12H14O2) m/z 190.0994, found 190.0995.
Tetrahydro-6-(4-chlorophenyl)-2H-pyran-2-one (1s). An orange solid: m.p. 91–98 °C; 1H-NMR (CDCl3, 500 MHz) δ 1.77–1.85 (m, 1 H, CH2), 1.96–2.01 (m, 2 H, CH2), 2.12–2.16 (m, 1 H, CH2), 2.53–2.60 (m, 1 H, CH2), 2.67–2.73 (m, 1 H, CH2), 5.32 (dd, J = 10.5, 3.0 Hz, 1 H, CH), 7.28 (d, J = 8.5 Hz, 2 H, ArH), 7.34 (d, J = 8.5 Hz, 2 H, ArH); 13C-NMR (CDCl3, 126 MHz) δ 18.4, 29.3, 30.4, 80.8, 127.0, 128.6, 133.9, 138.2, 171.0; MS (EI) m/z (%) 212 (M++2, 7), 210 (M+, 23), 70 (100); HRMS (EI) calcd for [M]+ (C11H11O2Cl) m/z 210.0448, found 210.0449.







{kind=link}
{kind=link}
{kind=link}
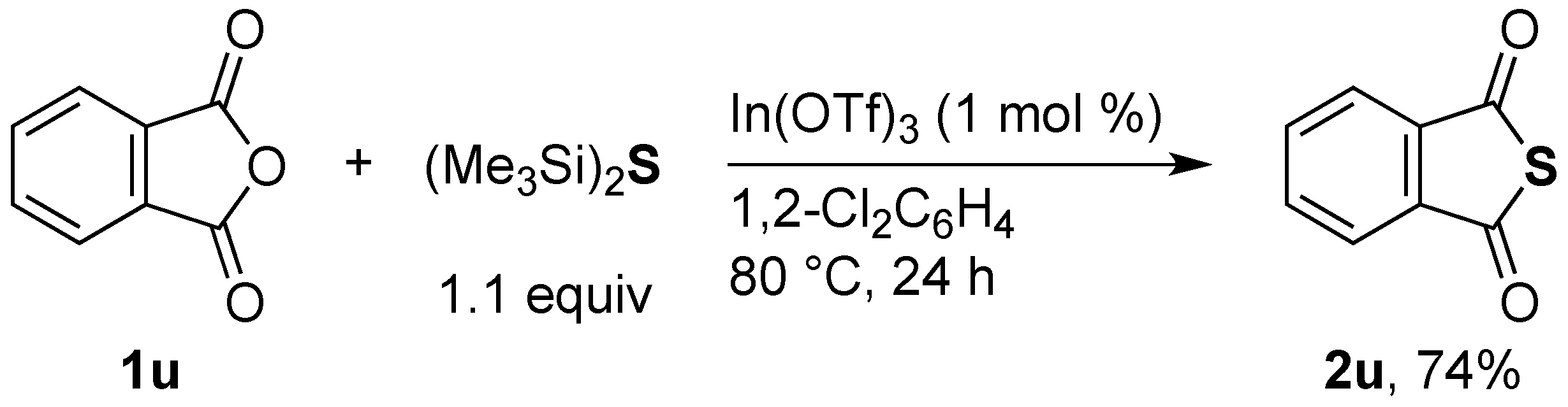{kind=link}
{kind=link}



















