
Development of a Mouse Model of Prostate Cancer Using the Sleeping Beauty Transposon and Electroporation

Abstract
:1. Introduction
2. Results
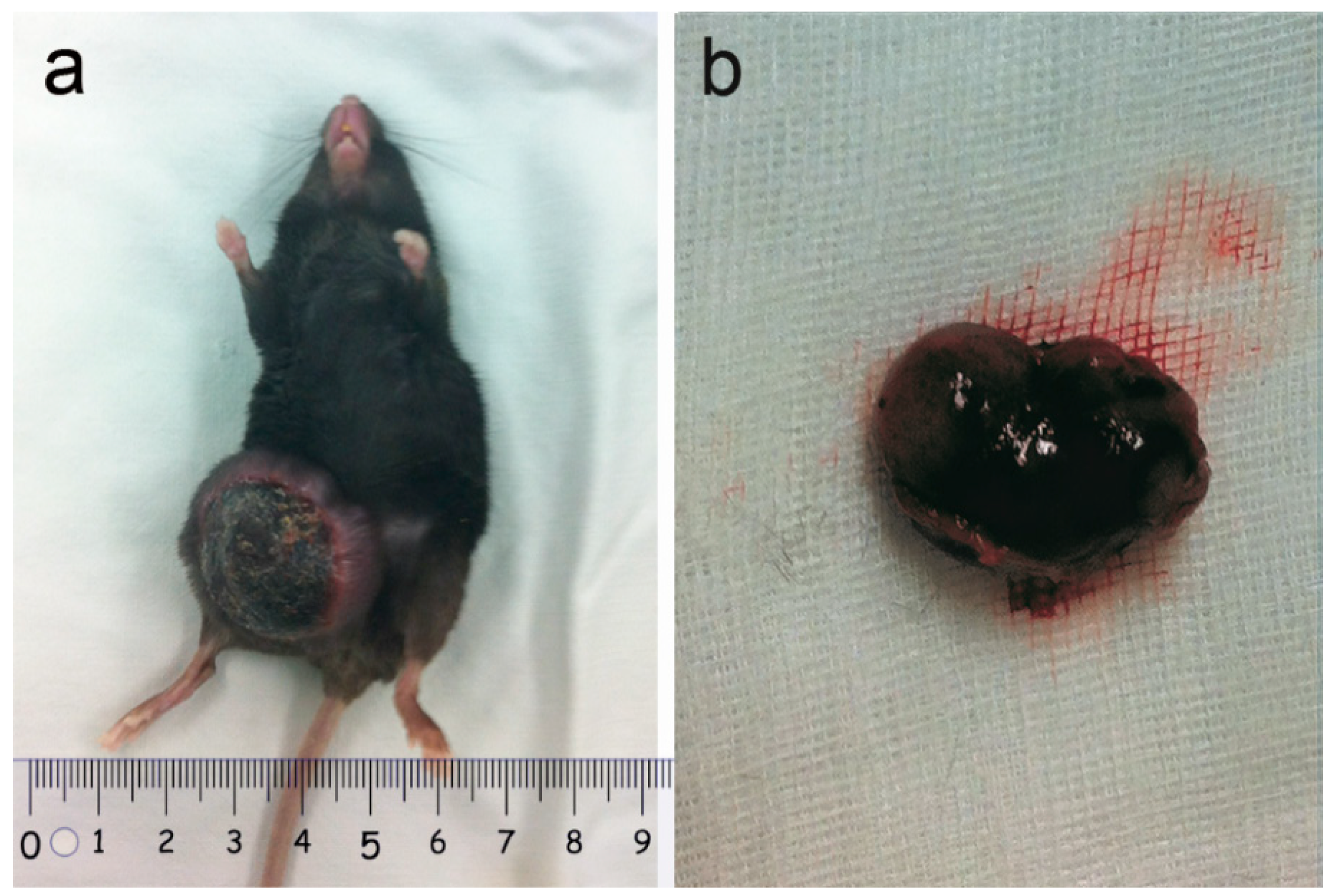
2.1. Tumor Observation
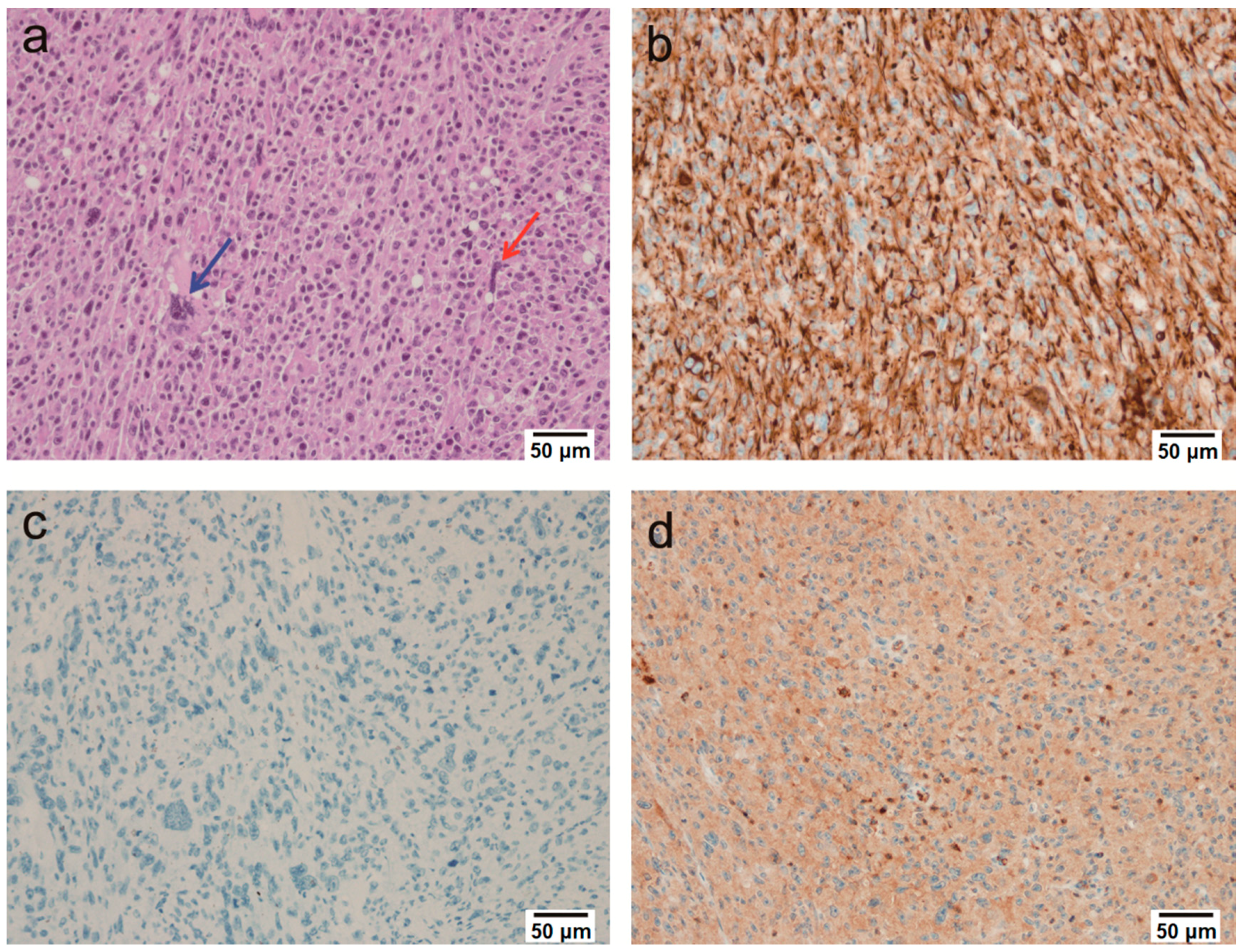
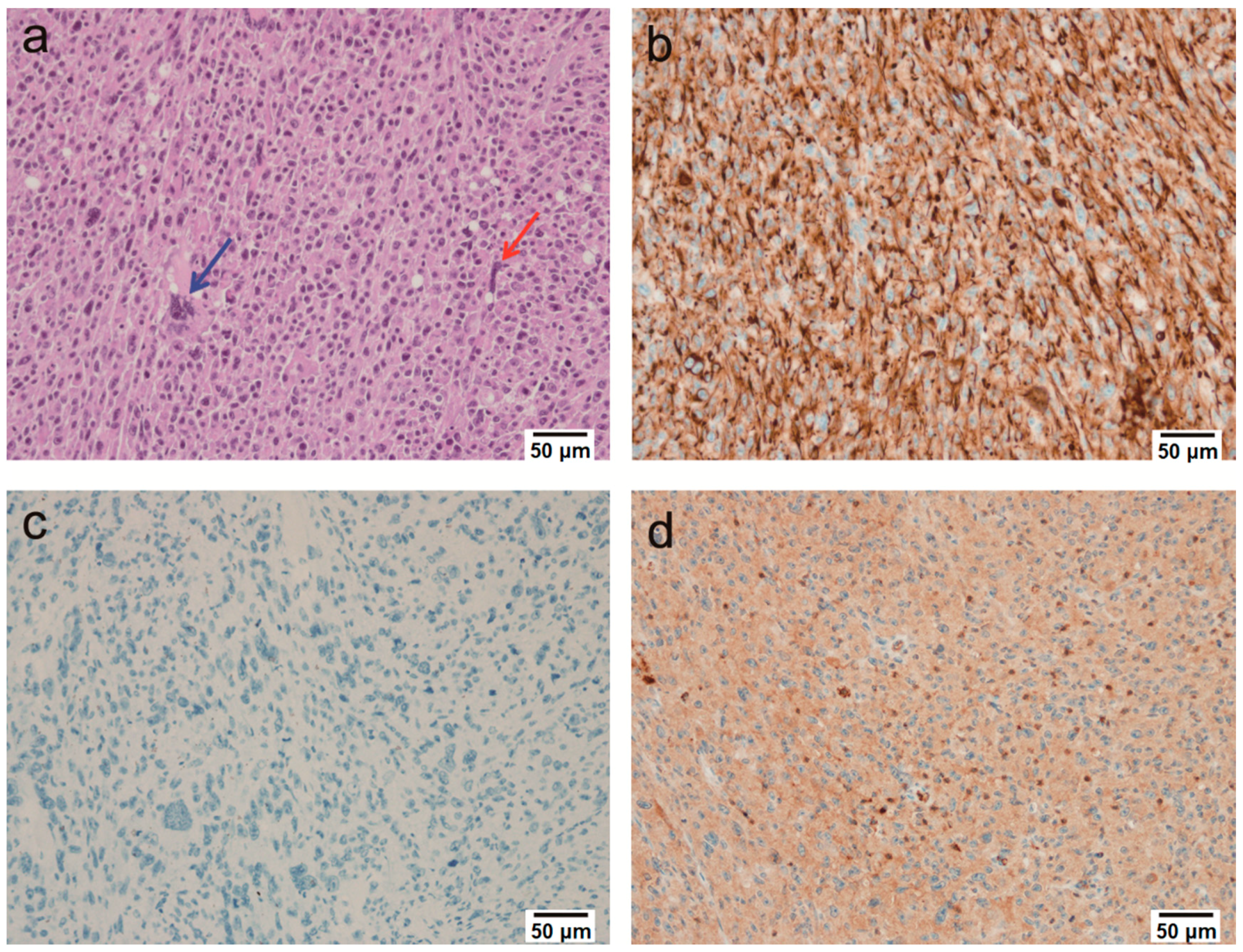
2.2. Microscopic Findings
2.3. Immunohistochemical Findings
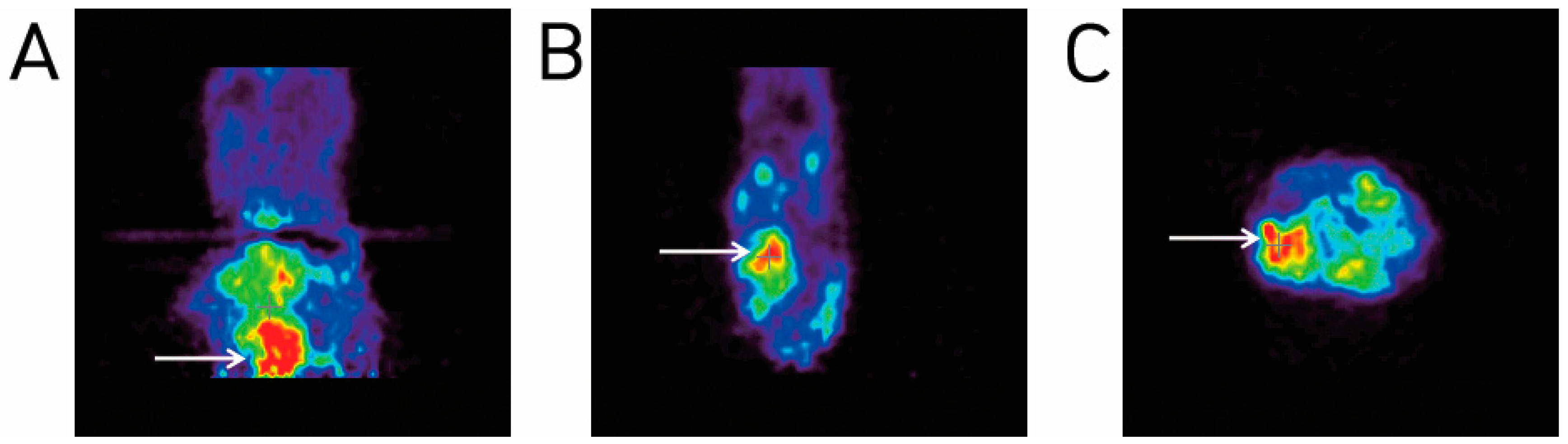
2.4. Animal PET Imaging
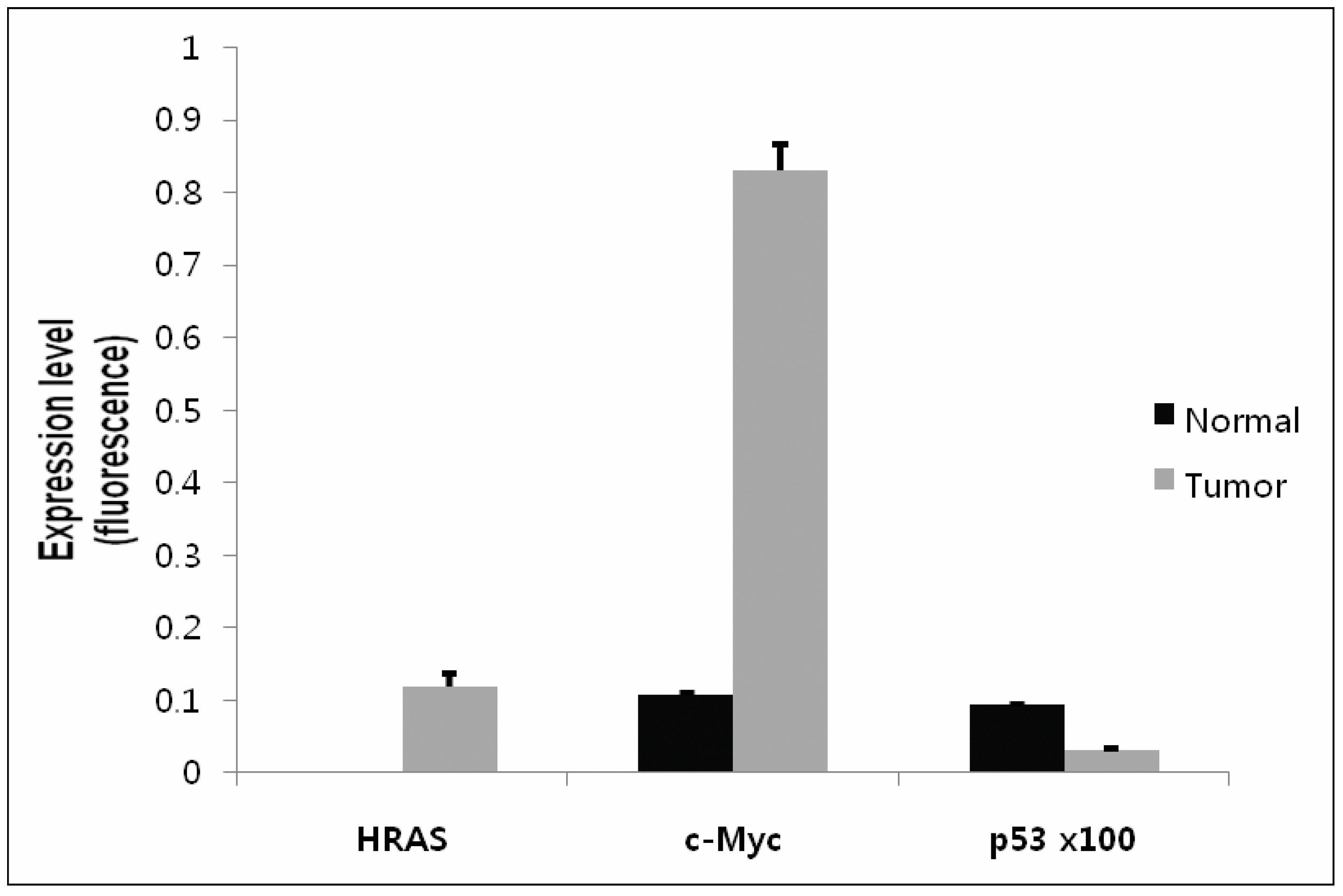
2.5. Confirmation of Gene Expression Using Real-Time qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animals
4.3. Plasmid Construction
4.4. DNA Plasmid Injection
4.5. In Vivo Electroporation
4.6. Radiopharmaceutical Preparation and Animal PET Imaging
4.7. Histology and Immunohistochemistry
4.8. Detection of Gene Expression Using Quantitative Real Time-qRT-PCR
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shappell, S.B.; Thomas, G.V.; Roberts, R.L.; Herbert, R.; Ittmann, M.M.; Rubin, M.A.; Humphrey, P.A.; Sundberg, J.P.; Rozengurt, N.; Barrios, R.; et al. Prostate pathology of genetically engineered mice: Definitions and classification. The consensus report from the bar harbor meeting of the mouse models of human cancer consortium prostate pathology committee. Cancer Res. 2004, 64, 2270–2305. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; Williams, B.O. Mouse models of prostate cancer. Prostate Cancer 2011, 2011, 895238. [Google Scholar] [CrossRef] [PubMed]
- Suwa, T.; Nyska, A.; Haseman, J.K.; Mahler, J.F.; Maronpot, R.R. Spontaneous lesions in control b6c3f1 mice and recommended sectioning of male accessory sex organs. Toxicol. Pathol. 2002, 30, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvak, Z. Molecular reconstruction of sleeping beauty, a tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef]
- Dupuy, A.J. Transposon-based screens for cancer gene discovery in mouse models. Semin. Cancer Biol. 2010, 20, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivics, Z.; Izsvak, Z. The expanding universe of transposon technologies for gene and cell engineering. Mob. DNA 2010, 1, 25. [Google Scholar] [CrossRef] [PubMed]
- Belur, L.R.; Podetz-Pedersen, K.M.; Sorenson, B.S.; Hsu, A.H.; Parker, J.B.; Carlson, C.S.; Saltzman, D.A.; Ramakrishnan, S.; McIvor, R.S. Inhibition of angiogenesis and suppression of colorectal cancer metastatic to the liver using the sleeping beauty transposon system. Mol. Cancer 2011, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Howell, V.M. Sleeping beauty—A mouse model for all cancers? Cancer Lett. 2012, 317, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Aronovich, E.L.; McIvor, R.S.; Hackett, P.B. The sleeping beauty transposon system: A non-viral vector for gene therapy. Hum. Mol. Genet. 2011, 20, R14-20. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.M.; Frandsen, J.L.; Kirchhof, N.; McIvor, R.S.; Largaespada, D.A. Somatic integration of an oncogene-harboring sleeping beauty transposon models liver tumor development in the mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 17059–17064. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jeong, M.; Oh, J.; Cho, Y.; Shen, X.; Stone, J.; Yan, J.; Rothkopf, Z.; Khan, A.S.; Cho, B.M. Preclinical evaluation of multi antigenic hcv DNA vaccine for the prevention of hepatitis c virus infection. Sci. Rep. 2017, 7, 43531. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, B.H.; Park, S.G.; Jung, S.Y.; Lee, D.H.; Son, W.C. Induction of rat liver tumor using the sleeping beauty transposon and electroporation. Biochem. Biophys. Res. Commun. 2013, 434, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Di Matteo, M.; Belay, E.; Chuah, M.K.; Vandendriessche, T. Recent developments in transposon-mediated gene therapy. Expert Opin. Biol. Ther. 2012, 12, 841–858. [Google Scholar] [CrossRef] [PubMed]
- Izsvak, Z.; Hackett, P.B.; Cooper, L.J.; Ivics, Z. Translating sleeping beauty transposition into cellular therapies: Victories and challenges. Bioessays 2010, 32, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Podetz-Pedersen, K.M.; Bell, J.B.; Steele, T.W.; Wilber, A.; Shier, W.T.; Belur, L.R.; McIvor, R.S.; Hackett, P.B. Gene expression in lung and liver after intravenous infusion of polyethylenimine complexes of sleeping beauty transposons. Hum. Gene Ther. 2010, 21, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Bigey, P.; Bureau, M.F.; Scherman, D. In vivo plasmid DNA electrotransfer. Curr. Opin. Biotechnol. 2002, 13, 443–447. [Google Scholar] [CrossRef]
- Jung, S.; Choi, H.J.; Park, H.K.; Jo, W.; Jang, S.; Ryu, J.E.; Kim, W.J.; Yu, E.S.; Son, W.C. Electroporation markedly improves sleeping beauty transposon-induced tumorigenesis in mice. Cancer Gene Ther. 2014, 21, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.K.; Scott, P.M.; Marsh, B.M.; Zhao, L.; Than, B.L.; O’Sullivan, M.G.; Sarver, A.L.; Dupuy, A.J.; Largaespada, D.A.; Cormier, R.T. A sleeping beauty transposon-mediated screen identifies murine susceptibility genes for adenomatous polyposis coli (apc)-dependent intestinal tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 5765–5770. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.M.; Ward, J.M.; Yew, C.C.; Kovochich, A.; Dawson, D.W.; Black, M.A.; Brett, B.T.; Sheetz, T.E.; Dupuy, A.J.; Chang, D.K.; et al. Sleeping beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 5934–5941. [Google Scholar] [CrossRef] [PubMed]
- Quintana, R.M.; Dupuy, A.J.; Bravo, A.; Casanova, M.L.; Alameda, J.P.; Page, A.; Sanchez-Viera, M.; Ramirez, A.; Navarro, M. A transposon-based analysis of gene mutations related to skin cancer development. J. Investig. Dermatol. 2013, 133, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Ro, S.W.; Jung, G.; Ju, H.L.; Yu, E.S.; Son, W.C. Sleeping beauty transposon system harboring hras, c-myc and shp53 induces sarcomatoid carcinomas in mouse skin. Oncol. Rep. 2013, 29, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Rahrmann, E.P.; Collier, L.S.; Knutson, T.P.; Doyal, M.E.; Kuslak, S.L.; Green, L.E.; Malinowski, R.L.; Roethe, L.; Akagi, K.; Waknitz, M.; et al. Identification of pde4d as a proliferation promoting factor in prostate cancer using a sleeping beauty transposon-based somatic mutagenesis screen. Cancer Res. 2009, 69, 4388–4397. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E.; Schaefer-Ridder, M.; Wang, Y.; Hofschneider, P.H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1982, 1, 841–845. [Google Scholar] [PubMed]
- Heller, L.C.; Heller, R. In vivo electroporation for gene therapy. Hum. Gene Ther. 2006, 17, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Hargrave, B.; Downey, H.; Strange, R., Jr.; Murray, L.; Cinnamond, C.; Lundberg, C.; Israel, A.; Chen, Y.J.; Marshall, W., Jr.; Heller, R. Electroporation-mediated gene transfer directly to the swine heart. Gene Ther. 2013, 20, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Abate-Shen, C.; Shen, M.M. Molecular genetics of prostate cancer. Genes Dev. 2000, 14, 2410–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, A.A.; Foster, B.A.; Allison, J.P.; Greenberg, N.M.; Kwon, E.D. The tramp mouse as a model for prostate cancer. Curr. Protoc. Immunol. 2001. [Google Scholar] [CrossRef]
- Richmond, A.; Su, Y. Mouse xenograft models vs gem models for human cancer therapeutics. Dis. Model. Mech. 2008, 1, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Frese, K.K.; Tuveson, D.A. Maximizing mouse cancer models. Nat. Rev. Cancer 2007, 7, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Copeland, N.G.; Jenkins, N.A. Harnessing transposons for cancer gene discovery. Nat. Rev. Cancer 2010, 10, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Yant, S.R.; Park, J.; Huang, Y.; Mikkelsen, J.G.; Kay, M.A. Mutational analysis of the n-terminal DNA-binding domain of sleeping beauty transposase: Critical residues for DNA binding and hyperactivity in mammalian cells. Mol. Cell. Biol. 2004, 24, 9239–9247. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Oh, S.; Ericson, K.; Demorest, Z.L.; Vengco, I.; Gharagozlou, S.; Chen, W.; Low, W.C.; Ohlfest, J.R. Transposon-based interferon gamma gene transfer overcomes limitations of episomal plasmid for immunogene therapy of glioblastoma. Cancer Gene Ther. 2007, 14, 550–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the SB transposon vectors and SB transposase are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue Marker | IHC Antibody | Type | Dilution | Reactivity |
|---|---|---|---|---|
| Epithelial tissue marker | Pan-cytokeratin | M | 1:3000 | + |
| Cytokeratin 7 | P | 1:1000 | − | |
| Cytokeratin 20 | M | 1:1000 | − | |
| Muscular tissue marker | Myogenin | M | 1:1000 | − |
| Desmin | M | 1:4000 | − | |
| α-smooth muscle actin | M | 1:1000 | − | |
| Hematopoietic cell marker | CD45 | M | 1:2000 | − |
| CD163 | M | 1:2000 | − | |
| CD68 | M | 1:2000 | − | |
| Melanoma marker | Melanoma | M | 1:100 | − |
| S100 | M | 1:400 | − | |
| Adipose tissue marker | CDK4 | M | 1:1000 | − |
| MDM2 | M | 1:1000 | − | |
| Prostate marker | PSA | P | 1:100 | + |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.-J.; Lee, H.-B.; Jung, S.; Park, H.-K.; Jo, W.; Cho, S.-M.; Kim, W.-J.; Son, W.-C. Development of a Mouse Model of Prostate Cancer Using the Sleeping Beauty Transposon and Electroporation. Molecules 2018, 23, 1360. https://doi.org/10.3390/molecules23061360
Choi H-J, Lee H-B, Jung S, Park H-K, Jo W, Cho S-M, Kim W-J, Son W-C. Development of a Mouse Model of Prostate Cancer Using the Sleeping Beauty Transposon and Electroporation. Molecules. 2018; 23(6):1360. https://doi.org/10.3390/molecules23061360
Chicago/Turabian StyleChoi, Hyun-Ji, Han-Byul Lee, Sunyoung Jung, Hyun-Kyu Park, Woori Jo, Sung-Min Cho, Woo-Jin Kim, and Woo-Chan Son. 2018. "Development of a Mouse Model of Prostate Cancer Using the Sleeping Beauty Transposon and Electroporation" Molecules 23, no. 6: 1360. https://doi.org/10.3390/molecules23061360
APA StyleChoi, H.-J., Lee, H.-B., Jung, S., Park, H.-K., Jo, W., Cho, S.-M., Kim, W.-J., & Son, W.-C. (2018). Development of a Mouse Model of Prostate Cancer Using the Sleeping Beauty Transposon and Electroporation. Molecules, 23(6), 1360. https://doi.org/10.3390/molecules23061360