Piplartine Analogues and Cytotoxic Evaluation against Glioblastoma

 ,
, 
Abstract
:1. Introduction
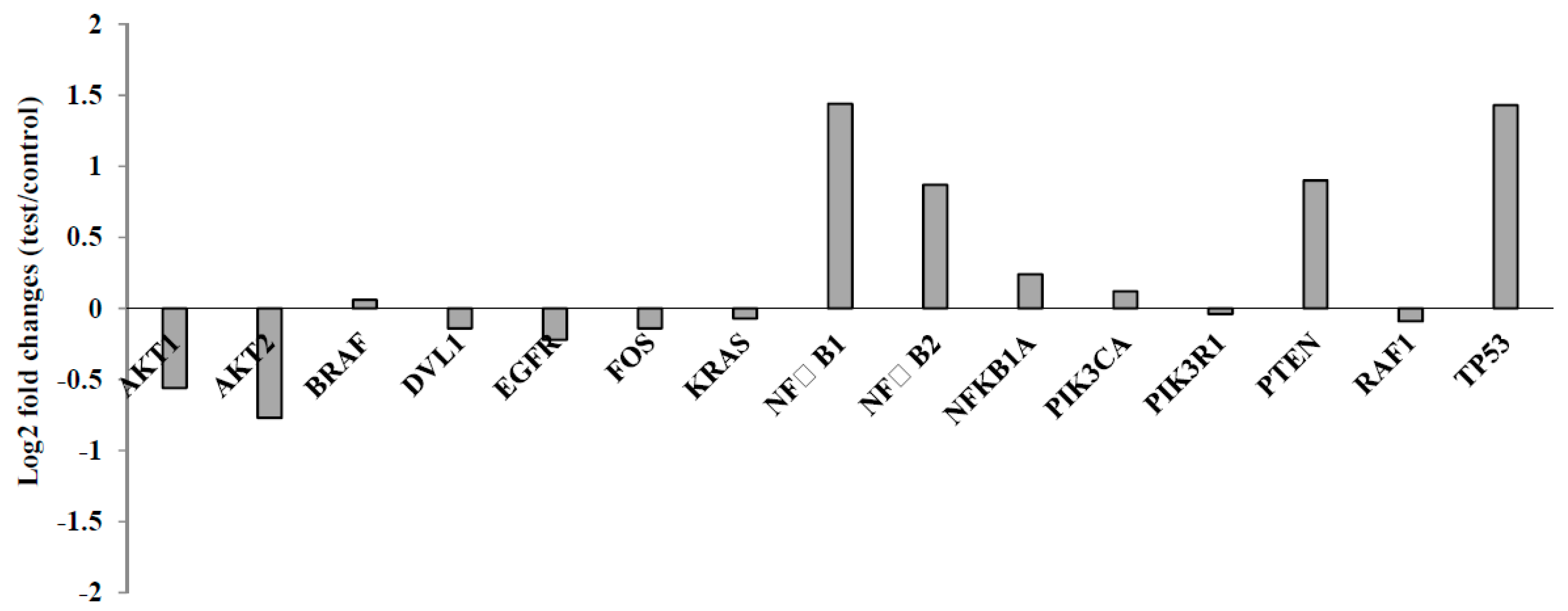
2. Results
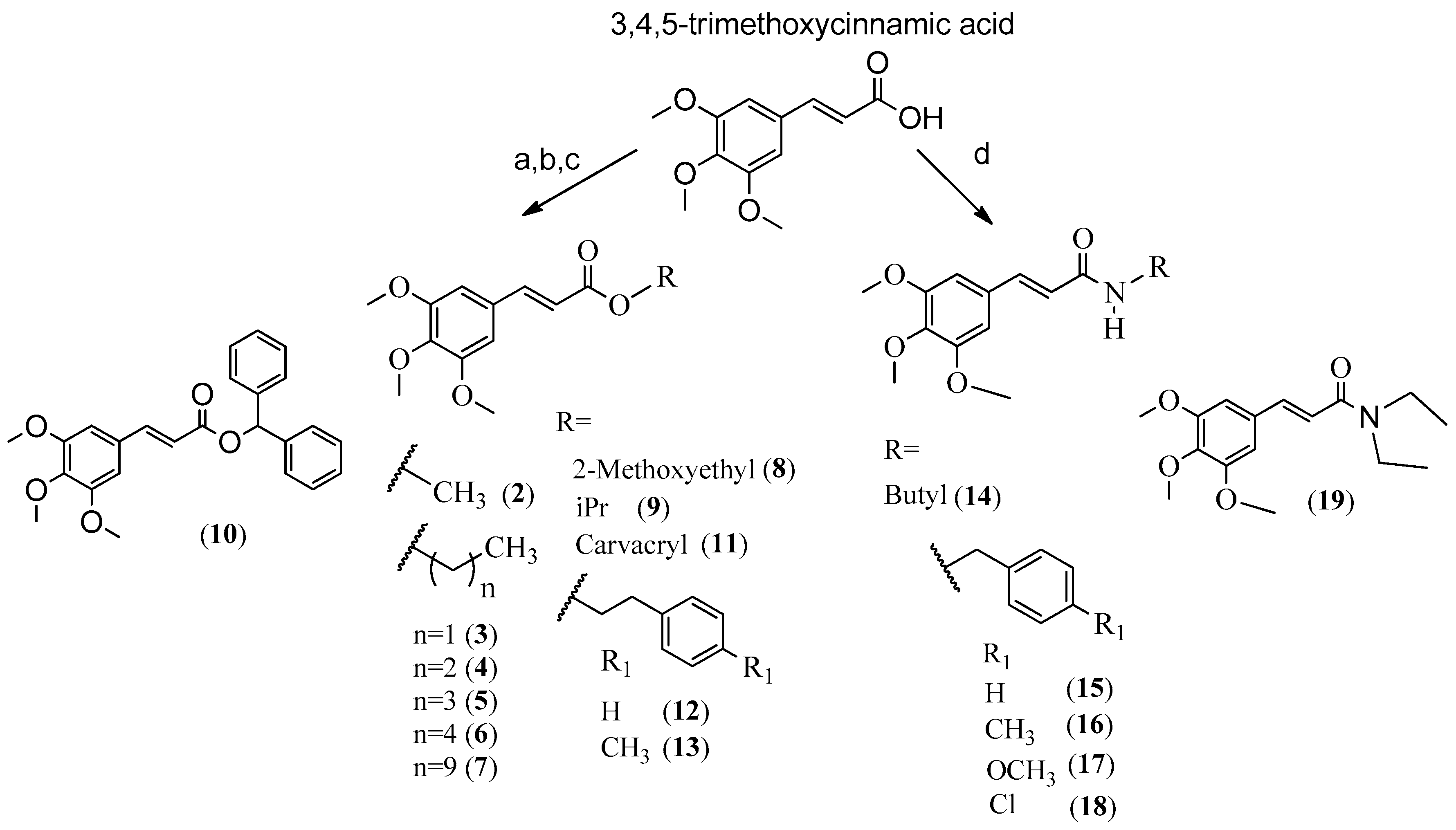
Chemistry
3. Discussion
3.1. Structure Activity Relationship (SAR)
3.2. Biological Activity
4. Materials and Methods
4.1. General Procedure for Synthesis of Compounds 2–5, 8 and 9
4.1.1. (E)-Methyl-3-(3,4,5-trimethoxyphenyl)acrylate (2)
4.1.2. (E)-Ethyl-3-(3,4,5-trimethoxyphenyl)acrylate (3)
4.1.3. (E)-Propyl-3-(3,4,5-trimethoxyphenyl)acrylate (4)
4.1.4. (E)-Butyl-3-(3,4,5-trimethoxyphenyl)acrylate (5)
4.1.5. (E)-2-Methoxyethyl-3-(3,4,5-trimethoxyphenyl)acrylate (8)
4.1.6. (E)-Isopropyl 3-(3,4,5-Trimethoxyphenyl)acrylate (9)
4.2. General Procedure for Synthesis of Compounds 6, 7, 10 and 13
4.2.1. (E)-Pentyl 3-(3,4,5-trimethoxyphenyl)acrylate (6)
4.2.2. (E)-Decyl-3-(3,4,5-trimethoxyphenyl)acrylate (7)
4.2.3. (E)-Benzhydryl 3-(3,4,5-trimethoxyphenyl)acrylate (10)
4.2.4. (E)-4-Methylphenethyl 3-(3,4,5-Trimethoxyphenyl)acrylate (13)
4.3. General Procedure for Synthesis of Compounds 11 and 12
4.3.1. (E)-Carvacryl-3-(3,4,5-trimethoxyphenyl)acrylate (11)
4.3.2. (E)-Phenethyl 3-(3,4,5-trimethoxyphenyl)acrylate (12)
4.4. General Synthesis of Amides
4.4.1. (E)-N-Butyl-3-(3,4,5-trimethoxyphenyl)acrylamide (14)
4.4.2. (E)-N-Benzyl-3-(3,4,5-trimethoxyphenyl)acrylamide (15)
4.4.3. (E)-N-(4-Methylbenzyl)-3-(3,4,5-trimethoxyphenyl)acrylamide (16)
4.4.4. (E)-N-(4-Methoxybenzyl)-3-(3,4,5-trimethoxyphenyl)acrylamide (17)
4.4.5. (E)-N-(4-Chlorobenzyl)-3-(3,4,5-trimethoxyphenyl)acrylamide (18)
4.4.6. (E)-N,N-Diethyl-3-(3,4,5-trimethoxyphenyl)acrylamide (19)
4.5. Cell Cultures and Conditions
4.6. In Vitro Evaluation of Anticancer Activity by MTT and LDH Assays
4.6.1. MTT Assay
4.6.2. LDH Assay
4.7. Total Antioxidant Capacity and Total Oxidant Status Assays
4.8. Apoptosis Detection by Hoechst 33,258 Staining
4.9. RNA Extraction and Quantitative Reverse-Transcription Polymerase Chain Reaction Analyses
4.10. Biosafety Evaluation
4.11. SCE Testing
4.12. Nucleic Acid Oxidation
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-OH-E2 | 4-Hydroxy-Estradiol |
| 8-OH-dG | 8-Hydroxy-2′-deoxyguanosine |
| 18SRNA | 18S ribosomal Ribonucleic Acid |
| AChE | 8-OH-dG-Acetylcholinesterase |
| ATP | Adenosine triphosphate |
| AKT1 | Other denomination of protein kinase B (PKB) type 1 |
| AKT2 | Other denomination of protein kinase B (PKB) type 2 |
| ANOVA | Analysis of variance |
| BpV-pic | Bisperoxovanadium-pic |
| BRAF | B-Raf proto-Oncogene, Serine/Threonine Kinase |
| 3-BTA | (E)-benzhydryl 3-(3,4,5-trimethoxyphenyl)acrylate |
| CT | Cycle Threshold |
| DMSO | Dimethyl sulfoxide |
| cDNA | Complementary Deoxyribonucleic Acid |
| DVL1 | Dishevelled type 1 |
| EGF | Epidermal Growth Factor |
| EGFR | Epidermal Growth Factor Receptor |
| FBS | Fetal Bovine Serum |
| FPG | Fluorescence Plus Giemsa |
| ER | Endoplasmic Reticulum |
| FOS | Finkel Osteosarcoma |
| GBM | Glioblastoma |
| HGG | High-Grade Glioma |
| HHV | Human Herpes virus |
| IC50 | The half maximal inhibitory concentration |
| IGF-1 | Insulin-like Growth Factor-1 |
| KRAS | Kirsten Rat Sarcoma viral oncogene homolog |
| LDH | Lactate Dehydrogenase |
| MMC | Mitomycin C |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NC | Negative Control |
| NFĸB | Necrosis Factor kappa B |
| NFkB1 | Necrosis Factor kappa B1 |
| NFkB1A | Necrosis Factor kappa B inhibitor-α |
| NFkB2 | Necrosis Factor kappa B2 |
| PBS | Phosphate-buffered saline |
| PC | Positive control |
| PCR | Polymerase Chain Reaction |
| PHWB | Peripheral Human Whole Blood |
| PPL | Piplartine |
| PTEN | Phosphatase and Tensin Homolog |
| PIP3K | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| PIK3CA | Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic |
| PIK3R1 | Phosphatidylinositol-3-kinase regulatory subunit 1 |
| PRDX4 | Peroxiredoxin 4 |
| RAF1 | Rapidly Accelerated Fibrosarcoma type 1 |
| ROS | Reactive Oxygen Species |
| RT | Radiotherapy |
| RT-PCR | Real Time-Polymerase Chain Reaction |
| SAR | Structure Activity Relationship |
| SCE | Sister Chromatid Exchange |
| SH-SY5Y | Human Neuroblastoma cell model |
| SV40 | Simian Vacuolating virus 40 |
| TAC | Total Antioxidant Capacity |
| TAS | Total Antioxidant Status |
| TMZ | Temozolomide |
| TOS | Total Oxidant Status |
| TP53 | Tumor Protein p53 |
References
- Ohgaki, H.; Kleihues, P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglialgliomas. J. Neuropathol. Exp. Neurol. 2005, 64, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Gallego, O. Nonsurgical treatment of recurrent glioblastoma. Curr. Oncol. 2015, 22, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, T.; Cantor, K.P.; Zhang, Y.; Chiu, B.C.; Lynch, C.F. Risk of brain glioma not associated with cigarette smoking or use of other tobacco products in Iowa. Cancer Epidemiol. Biomark. Prev. 2001, 10, 413–414. [Google Scholar]
- Huncharek, M.; Kupelnick, B.; Wheeler, L. Dietary cured meat and the risk of adult glioma: A meta-analysis of nine observational studies. J. Environ. Pathol. Toxicol. Oncol. 2003, 22. [Google Scholar] [CrossRef]
- Hardell, L.; Carlberg, M.; Mild, K.H. Epidemiological evidence for an association between use of wireless phones and tumor diseases. Pathophysiology 2009, 16, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, R.A.; Kozinetz, C.A.; Arrington, A.S.; Madden, C.R.; Butel, J.S. Simian virus 40 in human cancers. Am. J. Med. 2003, 114, 675–684. [Google Scholar] [CrossRef]
- Chi, J.; Gu, B.; Zhang, C.; Peng, G.; Zhou, F.; Chen, Y.; Wang, J. Human herpesvirus 6 latent infection in patients with glioma. J. Infect. Dis. 2012, 206, 1394–1398. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Ramiro, A.; Ramírez-Ortega, D.; Pérez de la Cruz, V.; Hérnandez-Pedro, N.Y.; González-Esquivel, D.F.; Sotelo, J.; Pineda, B. Role of redox status in development of glioblastoma. Front. Immunol. 2016, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, M.; Caffo, M.; Minutoli, L.; Marini, H.; Abbritti, R.V.; Squadrito, F.; Trichilo, V.; Valenti, A.; Barresi, V.; Altavilla, D.; et al. ROS and brain gliomas: An overview of potential and innovative therapeutic strategies. Int. J. Mol. Sci. 2016, 17, 984. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Song, J.; Kim, S.H.; Parikh, A.K.; Mo, X.; Palanichamy, K.; Kaur, B.; Yu, J.; Yoon, S.O.; Nakano, I.; et al. Piperlongumine treatment inactivates peroxiredoxin 4, exacerbates endoplasmic reticulum stress, and preferentially kills high-grade glioma cells. Neuro Oncol. 2014, 16, 1354–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezerra, D.P.; Pessoa, C.; De Moraes, M.O.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. 2013, 48, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Burci, L.M.; Pereira, I.T.; Silva, L.M.D.; Rodrigues, R.V.; Facundo, V.A.; Militao, J.S.L.T.; Santos, A.R.S.; Marques, M.C.A.; Baggio, C.H.; Werner, M.F.D.P. Antiulcer and gastric antisecretary effects of dichlomethane fraction and piplartine obtained from fruits of Piper tuberculatum Jacq. In rats. J. Ethnopharmacol. 2013, 148, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Son, D.J.; Kim, S.Y.; Han, S.S.; Kim, C.W.; Kumar, S.; Park, B.S.; Lee, S.E.; Yun, Y.P.; Jo, H.; Park, Y.H. Piperlongumine inhibits atherosclerotic plaque formation and vascular smooth muscle cell proliferation by suppressing PDGF receptor signaling. Biochem. Biophys. Res. Commun. 2012, 427, 349–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, J.; Li, J.; Zhang, Y.; Huang, W.; Zuo, J.; Liu, H.; Xie, D.; Zhu, P. Design, Synthesis and Pharmacological Evaluation of Novel Piperlongumine derivatives as Potential Antiplatelet Aggregation Candidate. Chem. Biol. Drug Des. 2016, 87, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.H.; Chen, X.X.; Wang, H. Piperlongumine induces apoptosis and synergizes with cisplatin or paclitaxel in human ovarian cancer cells. Oxid. Med. Cell. Longev. 2014, 2014, 906804. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, D.P.; Militao, G.C.; de Castro, F.O.; Pessoa, C.; de Moraes, M.O.; Silveira, E.R.; Lima, M.A.S.; Elmiro, F.J.M.; Costa-Lotufo, L.V. Piplartine induces inhibition of leukemia cell proliferation triggering both apoptosis and necrosis pathways. Toxicol. In Vitro 2007, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.J.; Kümmerle, A.E.; Fraga, C.A. The methylation effect in medicinal chemistry. Chem. Rev. 2011, 111, 5215–5246. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Albers, M.; Fischer, R.; Hebler, G.; Lehmann, T.E.; Okigami, H.; Tajimi, M.; Bacon, K.; Rölle, T. Discovery and evaluation of piperidinyl carboxylic acid derivatives as potent α4β1 integrin antagonists. Bioorg. Med. Chem. Lett. 2001, 11, 3019–3021. [Google Scholar] [CrossRef]
- Hernandes, M.Z.; Cavalcanti, S.M.T.; Moreira, D.R.M.; de Azevedo, J.; Filgueira, W.; Leite, A.C.L. Halogen atoms in the modern medicinal chemistry: Hints for the drug design. Curr. Drug Targets 2010, 11, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G. Fundamentals of Medicinal Chemistry; John Wiley & Sons Ltd.: Chichester, UK, 2009. [Google Scholar]
- Capello, T.M.; Martins, E.G.; De Farias, C.F.; Figueiredo, C.R.; Matsuo, A.L.; Passero, L.F.; Oliveira-Silva, D.; Sartorelli, P.; Lago, J.H. Chemical composition and in vitro cytotoxic and antileishmanial activities of extract and essential oil from leaves of Piper cernuum. Nat. Prod. Commun. 2015, 10, 285–288. [Google Scholar] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell. Res. 2011, 21, 103. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.J.; Homer, N.; O’connor, B.D.; Chen, Z.; Eskin, A.; Lee, H.; Merriman, B.; Nelson, S.F. U87MG decoded: The genomic sequence of a cytogenetically aberrant human cancer cell line. PLoS Genet. 2010, 6, e1000832. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.O.; Park, J.A.; Kim, H.A.; Chang, Y.H.; Hong, Y.J.; Park, I.C.; Lee, J.K. Piperlongumine downregulates the expression of HER family in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 486, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Zhan, H.; Bian, Q.; Gu, J. Piperlongumine inhibits gastric cancer cells via suppression of the JAK1, 2/STAT3 signaling pathway. Mol. Med. Rep. 2016, 13, 4475–4480. [Google Scholar] [CrossRef] [PubMed]
- Golovine, K.; Makhov, P.; Naito, S.; Raiyani, H.; Tomaszewski, J.; Mehrazin, R.; Tulin, A.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Piperlongumine and its analogs down-regulate expression of c-Met in renal cell carcinoma. Cancer Biol. Ther. 2015, 16, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meegan, M.J.; Nathwani, S.; Twamley, B.; Zisterer, D.M.; O’Boyle, N.M. Piperlongumine (piplartine) and analogues: Antiproliferative microtubule-destabilising agents. Eur. J. Med. Chem. 2017, 125, 453–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, E.; Kim, Y.; Kim, Y.; Cho, H.; Yu, S.; Kim, K.; Chang, J.; Ahn, S. Piplartine induces caspase-mediated apoptosis in PC-3 human prostate cancer cells. Oncol. Rep. 2008, 20, 785–792. [Google Scholar] [PubMed]
- Bezerra, D.P.; Ferreira, P.M.; Machado, C.M.; de Aquino, N.C.; Silveira, E.R.; Chammas, R.; Pessoa, C. Antitumour efficacy of Piper tuberculatum and piplartine based on the hollow fiber assay. Planta Med. 2015, 81, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Voce, D.J.; Schmitt, A.M.; Uppal, A.; McNerney, M.E.; Bernal, G.M.; Cahill, K.E.; Wahlstrom, J.S.; Nassiri, A.; Yu, X.; Crawley, C.D.; et al. Nfkb1 is a haploinsufficient DNA damage-specific tumor suppressor. Oncogene 2015, 34, 2807–2813. [Google Scholar] [CrossRef] [PubMed]
- Nagai, S.; Washiyama, K.; Kurimoto, M.; Takaku, A.; Endo, S.; Kumanishi, T. Aberrant nuclear factor-κB activity and its participation in the growth of human malignant astrocytoma. J. Neurosurg. Pediatr. 2002, 96, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, B.; Han, Y.; Lu, T.; Vogelbaum, M.A. Aberrant constitutive activation of nuclear factor κB in glioblastoma multiforme drives invasive phenotype. J. Neurooncol. 2007, 85, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Nadiminty, N.; Lou, W.; Sun, M.; Chen, J.; Yue, J.; Kung, H.J.; Evans, C.P.; Zhou, Q.; Gao, A.C. Aberrant activation of the androgen receptor by NF-kappaB2/p52 in prostate câncer cells. Cancer Res. 2010, 70, 3309–3319. [Google Scholar] [CrossRef] [PubMed]
- Ruan, D.; Li, X.; Li, A.; Liu, B.; Xu, F. Paclitaxel inhibits growth and proliferation of glioblastoma through MMP-9-meidated p38/JNK signaling pathway. Biomed. Res. 2017, 28, 7348–7353. [Google Scholar]
- Tseng, S.H.; Bobola, M.S.; Berger, M.S.; Silber, J.R. Characterization of paclitaxel (Taxol®) sensitivity in human glioma- and medulloblastoma-derived cell lines. Neuro Oncol. 1999, 1, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Deng, L.; Brandt, S.L.; Goodwin, C.B.; Ma, P.; Yang, Z.; Mali, R.S.; Liu, Z.; Kapur, R.; Serezani, C.H.; et al. Role of p85α in neutrophil extra- and intracelular reactive oxygen species generation. Oncotarget 2016, 7, 23096–23105. [Google Scholar] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.K.; Edwards, J.; Joshi, A.D.; Siu, I.M.; Riggins, G.J. A survey of glioblastoma genomic amplifications and deletions. J. Neurooncol. 2010, 96, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Zhang, L.J.; Chen, Z.; Liu, L.B. The PTEN inhibitor bpV (pic) promotes neuroprotection against amyloid β-peptide (25–35)-induced oxidative stress and neurotoxicity. Neurol. Res. 2017, 39, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Yuan, G.; Liu, X.; Ren, R.; Li, J.; Zhang, W.; Wu, J.; Xu, X.; Fu, L.; Li, Y.; et al. PTEN deficiency reprogrammes human neural stem cells towards a glioblastoma stem cell-like phenotype. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pollack, I.F.; Hamilton, R.L.; Finkelstein, S.D.; Campbell, J.W.; Martinez, A.J.; Sherwin, R.N.; Bozik, M.E.; Gollin, S.M. The relationship between TP53 mutations and overexpression of p53 and prognosis in malignant gliomas of childhood. Cancer Res. 1997, 57, 304–309. [Google Scholar] [PubMed]
- Robinson, J.P.; VanBrocklin, M.W.; Guilbeault, A.R.; Signorelli, D.L.; Brandner, S.; Holmen, S.L. Activated BRAF induces gliomas in mice when combined with Ink4a/Arf loss or Akt activation. Oncogene 2010, 29, 335. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.Y.; Lee, H.S.; Kim, E.K.; Ha, J.M.; Kim, Y.W.; Bae, S. Reactive oxygen species and PI3K/Akt signaling in cancer. Free Radic. Biol. Med. 2014, 75, S34–S35. [Google Scholar] [CrossRef] [PubMed]
- Okoh, V.O.; Felty, Q.; Parkash, J.; Poppiti, R.; Roy, D. Reactive oxygen species via redox signaling to PI3K/AKT pathway contribute to the malignant growth of 4-hydroxy estradiol-transformed mammary epithelial cells. PLoS ONE 2013, 8, e54206. [Google Scholar] [CrossRef] [PubMed]
- Haas-Kogan, D.; Shalev, N.; Wong, M.; Mills, G.; Yount, G.; Stokoe, D. Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr. Biol. 1998, 8, 1195–1998. [Google Scholar] [CrossRef]
- Holland, E.C.; Celestino, J.; Dai, C.; Schaefer, L.; Sawaya, R.E.; Fuller, G.N. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat. Genet. 2000, 25, 55. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.H.; Hu, S.L.; Shen, G.D.; Shen, G. Tumor suppressor genes and their underlying interactions in paclitaxel resistance in cancer therapy. Cancer Cell Int. 2016, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Błasiak, J.; Gloc, E.; Warszawski, M. A comparison of the in vitro genotoxicity of anticancer drugs idarubicin and mitoxantrone. Acta Biochim. Pol. 2002, 49, 145–155. [Google Scholar] [PubMed]
- Turkez, H.; Tatar, A.; Hacimuftuoglu, A.; Ozdemir, E. Boric acid as a protector against paclitaxel genotoxicity. Acta Biochim. Pol. 2010, 57, 95–97. [Google Scholar] [PubMed]
- Steverding, D.; da Nóbrega, F.R.; Rushworth, S.A.; de Sousa, D.P. Trypanocidal and cysteine protease inhibitory activity of isopentylcaffeate is not linked in Trypanosomabrucei. J. Parasitol. Res. 2016, 115, 4397–4403. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.R.; Russell, M.E.; Surjasasmita, I.B. Synthesis and experimental ionization energies of certain (E)-3-arylpropenoic acids and their methyl esters. J. Chem. Eng. 1988, 33, 534–537. [Google Scholar] [CrossRef]
- Jung, J.C.; Moon, S.; Min, D.; Park, W.K.; Jung, M.; Oh, S. Synthesis and Evaluation of a Series of 3,4,5-Trimethoxycinnamic Acid Derivatives as Potential Antinarcotic Agents. Chem. Biol. Drug Des. 2013, 81, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Arya, P.; Mukherjee, C.; Singh, B.K.; Singh, N.; Parmar, V.S.; Prasad, A.K.; Ghosh, B. Novel aromatic ester from Piper longum and its analogues inhibit expression of cell adhesion molecules on endothelial cells. Biochemistry 2005, 44, 15944–15952. [Google Scholar] [CrossRef] [PubMed]
- Hayat, S.; Choudhary, M.I.; Khan, K.M.; Abbaskhan, A. Two new cinnamic acid esters from marine brown alga Spatoglossum variabile. Chem. Pharm. Bull. 2002, 50, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, B.K.; Arya, P.; Malhotra, S.; Thimmulappa, R.; Prasad, A.K.; Van der Eycken, E.; Olsen, C.E.; Depass, A.L.; Biswal, S.; et al. Novel natural product-based cinnamates and their thio and thiono analogs as potent inhibitors of cell adhesion molecules on human endothelial cells. Eur. J. Med. Chem 2011, 46, 5498–5511. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, N.; Tang, Y.; Li, B.; Liu, L.; Zhang, X.; Fu, H.; Duan, J.A. Biological activity evaluation and structure-activity relationships analysis of ferulic acid and caffeic acid derivatives for anticancer. Bioorg. Med. Chem. Lett. 2012, 22, 6085–6088. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.I.; Tatchell, A.R.; Furniss, B.S.; Hannaford, A.S.; Smith, P.W.G. Textobook of Pratical Organic Chemistry; Longman Scientific & Technical: Essex, UK, 1989. [Google Scholar]
- Itoh, K.; Utsukihara, T.; Funayama, K.; Sakamaki, H.; Kanamori, M.; Takahashi, T.T.; Saitoh, Y.; Matsushita, M.; He, L.; Hashimoto, C.; et al. Reaction of α,β-unsaturated ketones using cerium (IV) sulfate tetrahydrate in acetic acid, applied organometallic chemistry appl. Organomet. Chem. 2007, 21, 1029–1032. [Google Scholar] [CrossRef]
- Magens, S.; Ertelt, M.; Jatsch, A.; Plietker, B. A nucleophilic Fe catalyst for transesterifications under neutral conditions. Org. Lett. 2008, 10, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Rajan, P.; Vedernikova, I.; Cos, P.; Berghe, D.V.; Augustyns, K.; Haemers, A. Synthesis and evaluation of caffeic acid amides as antioxidants. Bioorg. Med. Chem. Lett. 2001, 11, 215–217. [Google Scholar] [CrossRef]
- Nomura, E.; Hosoda, A.; Mori, H.; Taniguchi, H. Rapid base-catalyzed decarboxylation and amide-forming reaction of substituted cinnamic acids via microwave heating. Green Chem. 2005, 7, 863–866. [Google Scholar] [CrossRef]
- Nordstrom, L.U.; Vogt, H.; Madsen, R. Silica gel mediated amide bond formation: An environmentally benign method for liquid-phase synthesis and cytotoxic activities of amides. J. Am. Chem. Soc. 2008, 30, 17672–17673. [Google Scholar]
- Leslie, B.J.; Holaday, C.R.; Nguyen, T.; Hergenrother, P.J. Phenylcinnamides as novel antimicotic agents. J. Med. Chem. 2010, 53, 3964–3972. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.C.; Li, C.C.; Chun, Z.H.; Ngiam, J.S.Y.; Seayad, A.M.; Chen, A. Mesoporous Niobium Oxide Spheres as an Effective Catalyst for the Transamidation of Primary Amides with Amines. Adv. Synth. Catal. 2014, 356, 475–484. [Google Scholar] [CrossRef]
- Yang, X.-D.; Zeng, X.-H.; Zhao, Y.-H.; Wang, X.-Q.; Pan, Z.-Q.; Li, L.; Zhang, H.-B. Silica gel mediated amide bond formation: An environmentally benign method for liquid-phase synthesis and cytotoxic activities of amides. J. Comb. Chem. 2010, 12, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Cacciatore, I.; Fornasari, E.; Marinelli, L.; Eusepi, P.; Ciulla, M.; Ozdemir, O.; Tatar, A.; Turkez, H.; Di Stefano, A. Memantine-derived drugs as potential antitumor agents for the treatment of glioblastoma. Eur. J. Pharm. Sci. 2017, 109, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Togar, B.; Turkez, H.; Hacimuftuoglu, A.; Tatar, A.; Geyikoglu, F. Guaiazulene biochemical activity and cytotoxic and genotoxic effects on rat neuron and N2a neuroblastom cells. J. Intercult. Ethnopharmacol. 2015, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Turkez, H.; Togar, B.; Di Stefano, A.; Taspınar, N.; Sozio, P. Protective effects of cyclosativene on H2O2-induced injury in cultured rat primary cerebral cortex cells. Cytotechnology 2015, 67, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.J.; O’Riordan, M.L. Human peripheral blood lymphocytes for the analysis of chromosome aberrations in mutagen tests. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 1975, 31, 135–148. [Google Scholar] [CrossRef]
- Abdel-Wahab, B.A.; Metwally, M.E. Ginkgo biloba enhances the anticonvulsant and neuroprotective effects of sodium valproate against kainic acid-induced seizures in mice. J. Pharmacol. Toxicol. 2011, 6, 679–690. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds not are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Esters | Amides | ||
|---|---|---|---|
| Compound | IC50 (μg/mL) | Compound | IC50 (μg/mL) |
| 2 | 85.129 ± 0.78 | 14 | 13.196 ± 0.32 |
| 3 | 100.030 ± 1.12 | 15 | 33.128 ± 0.41 |
| 4 | 78.393 ± 0.69 | 16 | 22.741 ± 0.34 |
| 5 | 57.871 ± 0.71 | 17 | 22.654 ± 0.39 |
| 6 | 31.666 ± 0.48 | 18 | 43.396 ± 0.48 |
| 7 | 34.926 ± 0.54 | 19 | 36.072 ± 0.44 |
| 8 | 50.077 ± 0.62 | ||
| 9 | 81.433 ± 0.55 | ||
| 10 | 2.579 ± 0.35 | ||
| 11 | 26.701 ± 0.47 | ||
| 12 | 24.786 ± 0.37 | ||
| 13 | 30.336 ± 0.35 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da Nóbrega, F.R.; Ozdemir, O.; Nascimento Sousa, S.C.S.; Barboza, J.N.; Turkez, H.; De Sousa, D.P. Piplartine Analogues and Cytotoxic Evaluation against Glioblastoma. Molecules 2018, 23, 1382. https://doi.org/10.3390/molecules23061382
Da Nóbrega FR, Ozdemir O, Nascimento Sousa SCS, Barboza JN, Turkez H, De Sousa DP. Piplartine Analogues and Cytotoxic Evaluation against Glioblastoma. Molecules. 2018; 23(6):1382. https://doi.org/10.3390/molecules23061382
Chicago/Turabian StyleDa Nóbrega, Flávio Rogério, Ozlem Ozdemir, Sheila Cristina S. Nascimento Sousa, Joice Nascimento Barboza, Hasan Turkez, and Damião Pergentino De Sousa. 2018. "Piplartine Analogues and Cytotoxic Evaluation against Glioblastoma" Molecules 23, no. 6: 1382. https://doi.org/10.3390/molecules23061382