Transcriptomic Analysis of the Candidate Genes Related to Aroma Formation in Osmanthus fragrans

Abstract
:1. Introduction
2. Results
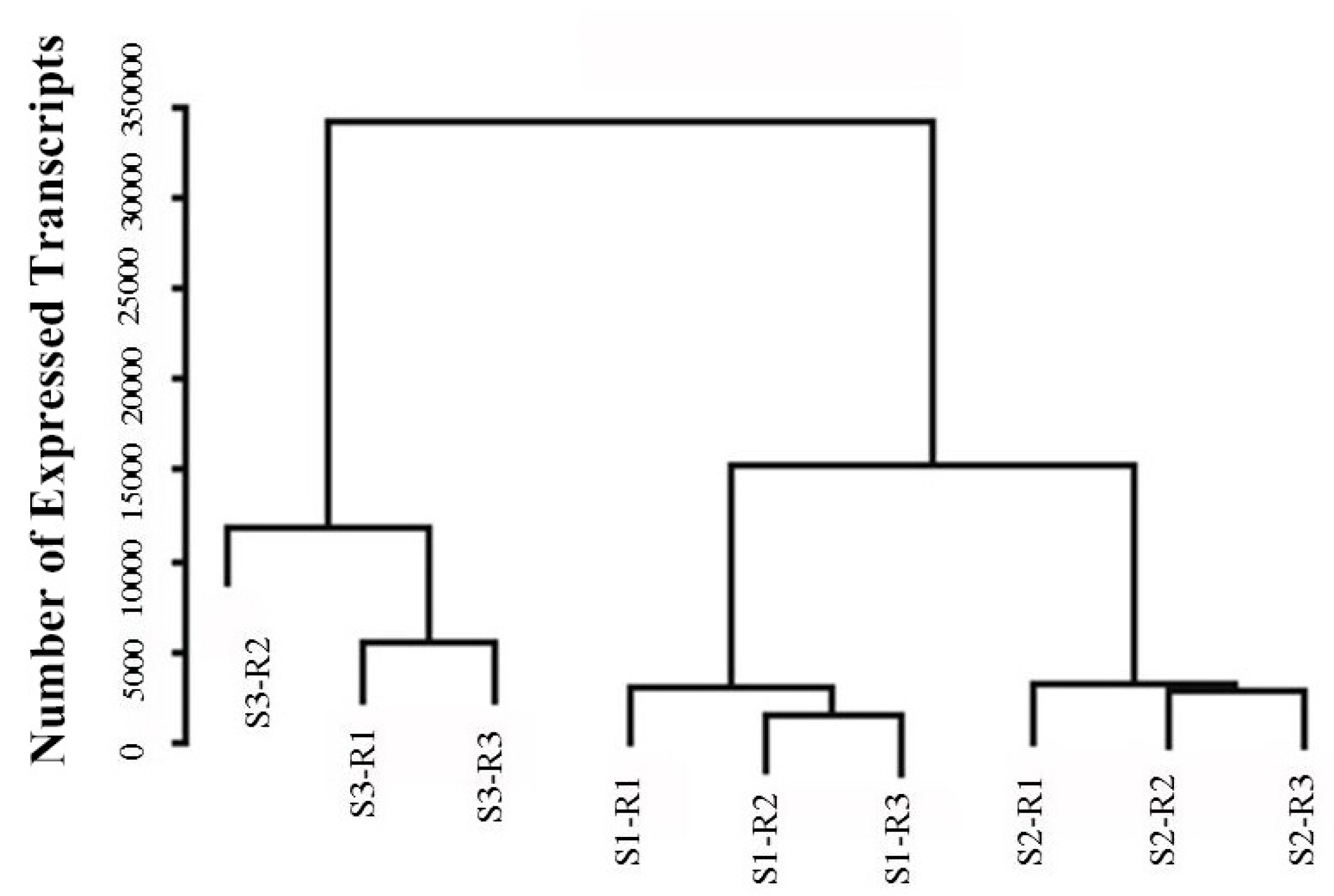
2.1. Sequencing and De Novo Assembly
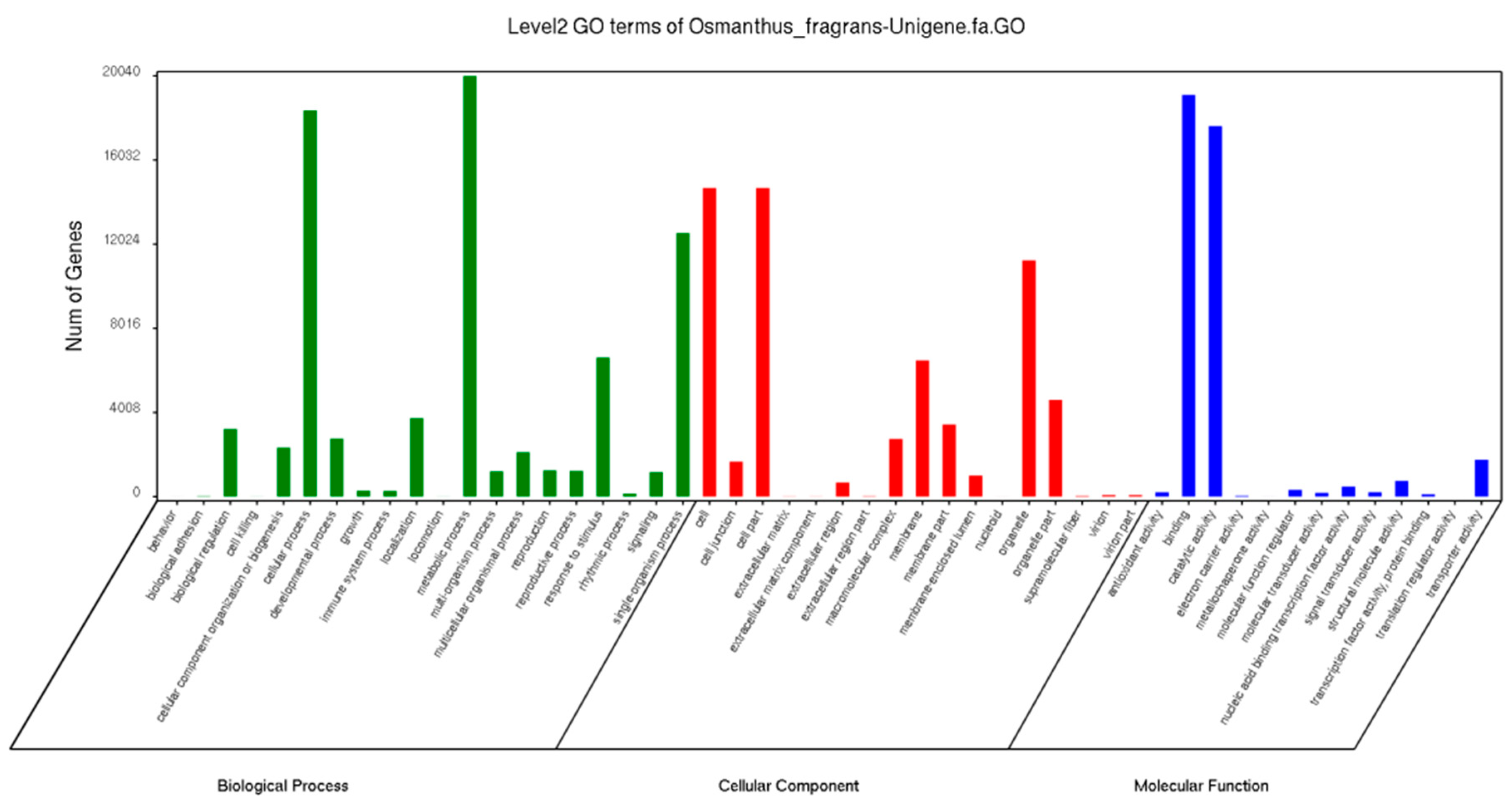
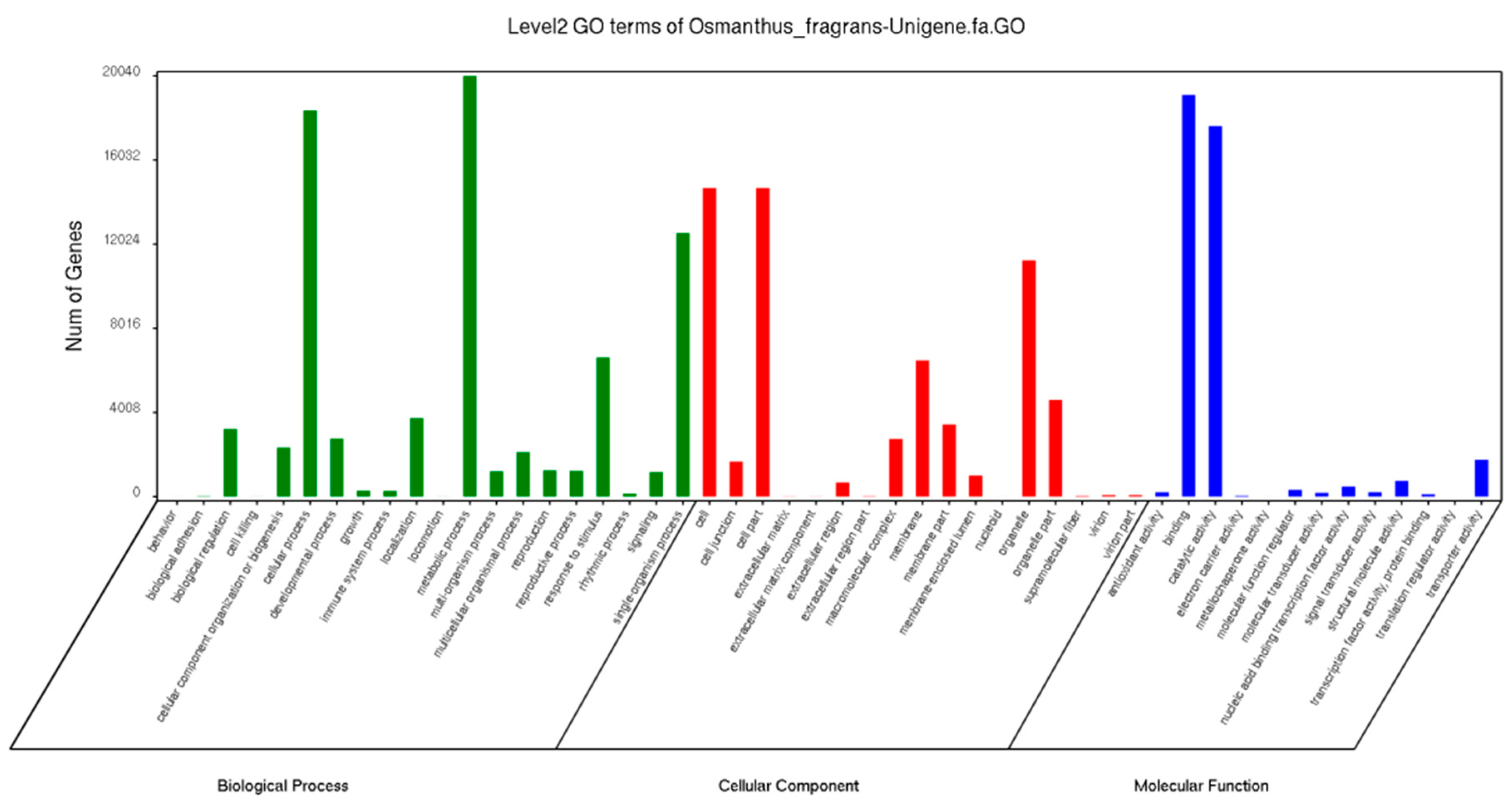
2.2. Annotation and Functional Classification
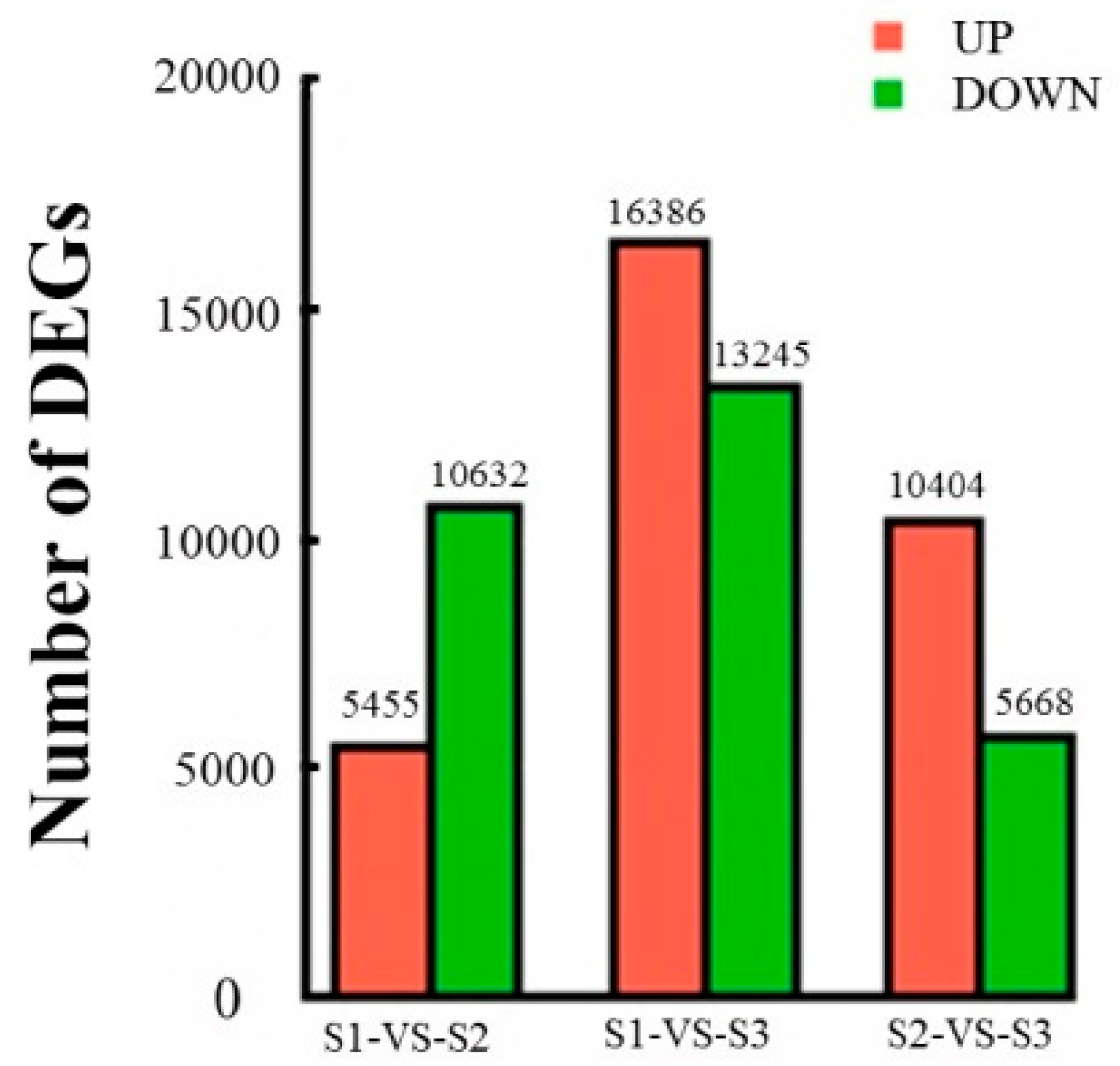
2.3. Analysis of DEGs
2.4. Identification of UnigenesInvolved in Scent Metabolism
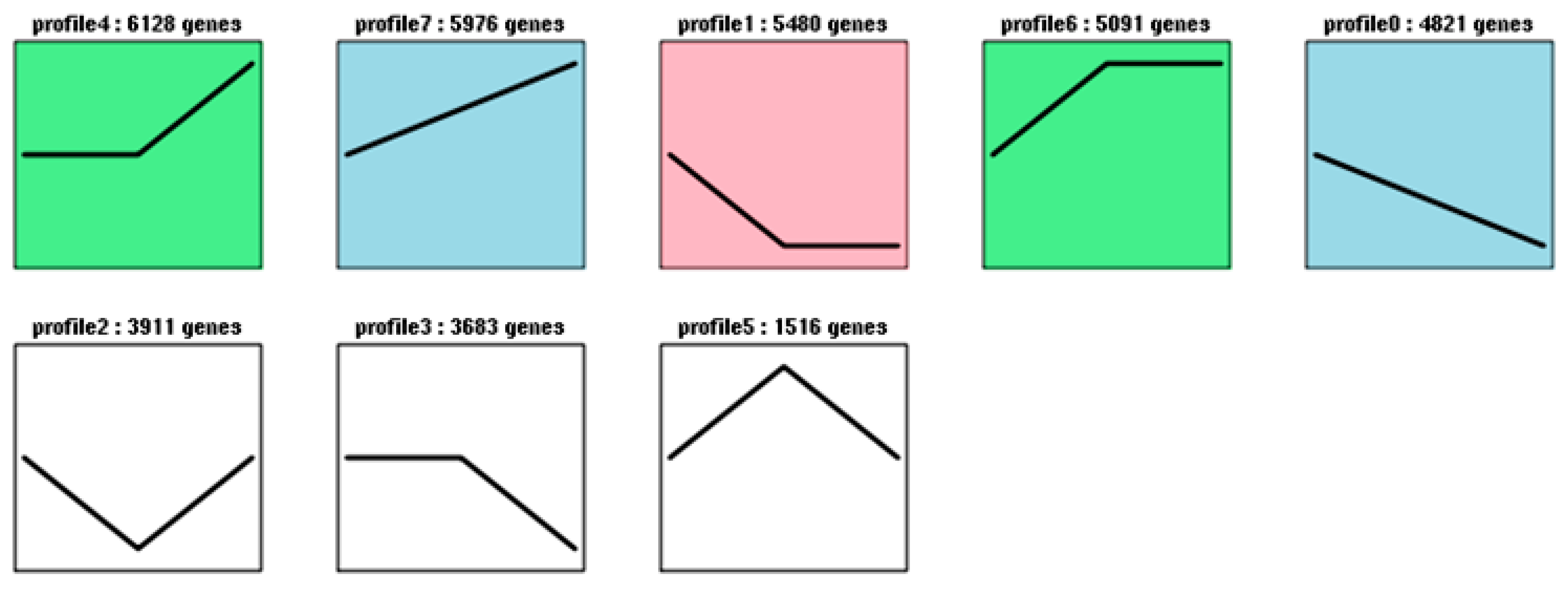
2.5. Analysis of TFs during Blooming
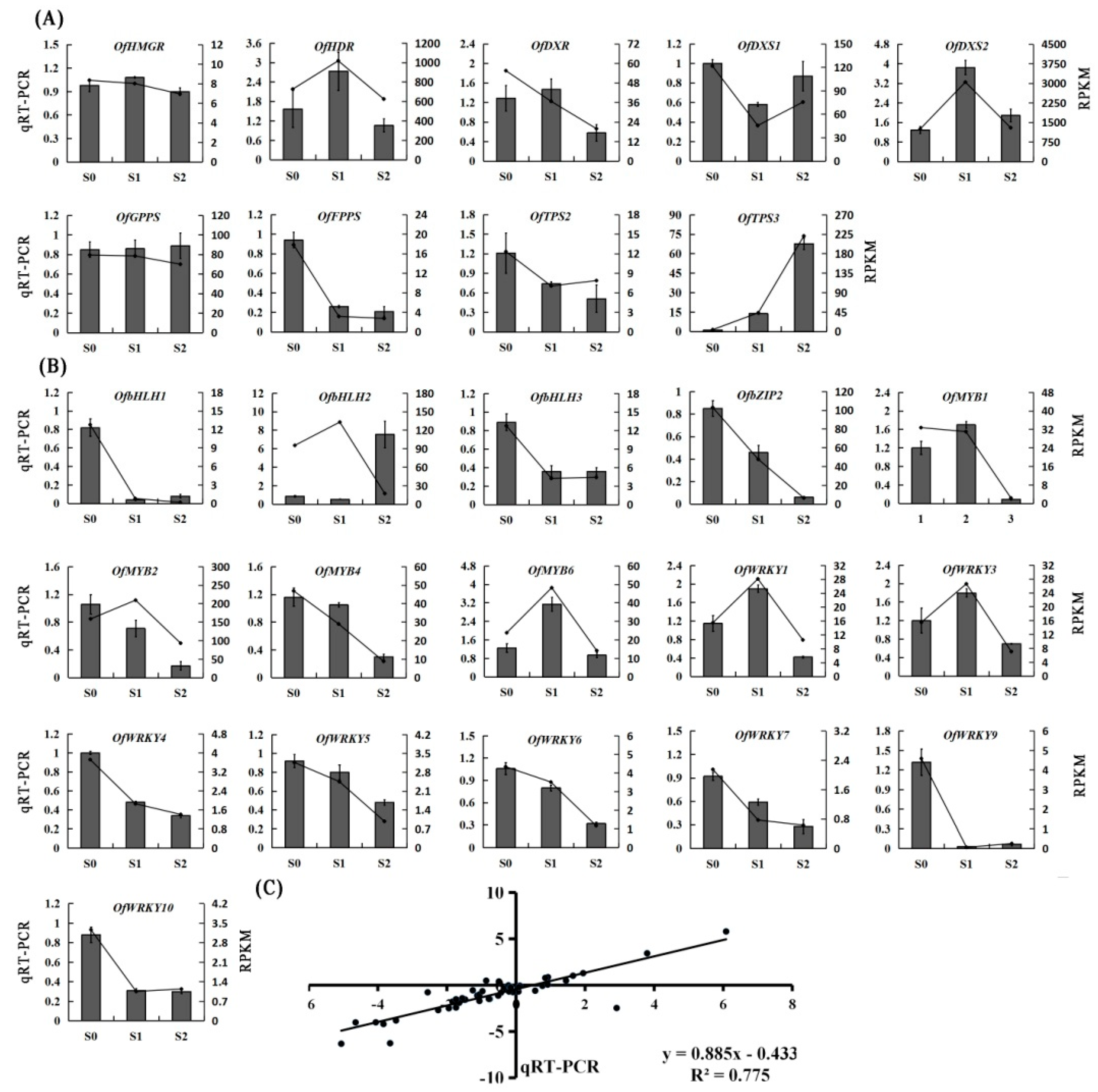
2.6. Validation of RNA-Seq Results by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Plant Materials and RNA Extraction
4.2. Libraries Construction and Illumina Sequencing
4.3. Assembly and Functional Annotation
4.4. Gene Expression Analysis
4.5. qRT-PCR Validation
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Zang, D.K.; Xiang, Q.B.; Liu, Y.L.; Hao, R.M. The studying history and the application to international cultivar registration authority of sweet osmanthus (Osmanthus fragrans Lour.). J. Plant Resour. Environ. 2003, 12, 49–53. [Google Scholar]
- Knudsen, J.T.; Eriksson, R.; Gershenzon, J.; Stahl, B. Diversity and distribution of floral scent. Bot. Rev. 2006, 72, 1–120. [Google Scholar] [CrossRef] [Green Version]
- Gang, D.R. Evolution of flavors and scents. Annu. Rev. Plant Biol. 2005, 56, 301–325. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Wu, B.; Zhang, H.; Wang, C.; Li, J.; Yang, B.; Li, S. Characterization of volatile compounds in flowers from four groups of sweet osmanthus (Osmanthus fragrans) cultivars. Can. J. Plant Sci. 2013, 93, 923–931. [Google Scholar] [CrossRef]
- Cai, X.; Mai, R.Z.; Zou, J.J.; Zhang, H.Y.; Zeng, X.L.; Zheng, R.R.; Wang, C.Y. Analysis of aroma-active compounds in three sweet osmanthus (Osmanthus fragrans) cultivars by GC-olfactometry and GC-MS. J. Zhejiang Univ. Sci. B 2014, 15, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.M.; Li, M.T.; Jin, W.W.; Li, S.; Zhang, S.Q.; Yu, L.J. Variationsin the components of Osmanthus fragrans Lour. essential oil atdifferent stages of flowering. Food Chem. 2009, 114, 233–236. [Google Scholar] [CrossRef]
- Lichtenthaler, H.K. The 1-deoxy-d-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Cordoba, E.; Salmi, M.; Leon, P. Unravelling the regulatory mechanisms that modulate the MEP pathway in higher plants. J. Exp. Bot. 2009, 60, 2933–2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Li, H.; Yang, X.; Gu, C.; Mu, H.; Yue, Y.; Wang, L. Cloning and Expression Analysis of MEP Pathway Enzyme-encoding Genes in Osmanthus fragrans. Genes 2016, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Tholl, D.; Chen, F.; Petri, J.; Gershenzon, J.; Pichersky, E. Two sesquiterpene synthases are responsible for the complex mixture of sesquiterpenes emitted from Arabidopsis flowers. Plant J. 2005, 42, 757–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeder, S.; Hartmann, A.M.; Effmert, U.; Piechulla, B. Regulation of simultaneous synthesis of floral scent terpenoids by the 1,8-cineole synthase of Nicotiana suaveolens. Plant Mol. Biol. 2007, 65, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Nagegowda, D.A.; Gutensohn, M.; Wilkerson, C.G.; Dudareva, N. Two nearly identical terpene synthases catalyze the formation of nerolidol and linalool in snapdragon flowers. Plant J. 2008, 55, 224–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Liu, C.; Zheng, R.; Cai, X.; Luo, J.; Zou, J.; Wang, C. Emission and Accumulation of Monoterpene and the Key Terpene Synthase (TPS) Associated with Monoterpene Biosynthesis in Osmanthus fragrans Lour. Front. Plant Sci. 2015, 6, 1232. [Google Scholar] [CrossRef] [PubMed]
- Susanne, B.; Masaya, K.; Miwako, K.; Yoshiko, K.; Akira, F.; Peter, F.; Naoharu, W. Functional characterization of a carotenoid cleavagedioxygenase1 and its relation to the carotenoid accumulation and volatile emission during the floral development of Osmanthus fragrans Lour. J. Exp. Bot. 2010, 61, 2967–2977. [Google Scholar]
- Xu, Y.H.; Wang, J.W.; Wang, S.; Wang, J.Y.; Chen, X.Y. Characterization of GaWRKY1, a cotton transcription factor that regulates the sesquiterpene synthase gene (+)-delta-cadinene synthase-A. Plant Physiol. 2004, 135, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Skibbe, M.; Qu, N.; Galis, I.; Baldwin, I.T. Induced plant defenses in the natural environment: Nicotiana attenuata WRKY3 and WRKY6 coordinate responses to herbivory. Plant Cell 2008, 20, 1984–2000. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.J.; Xue, X.Y.; Mao, Y.B.; Wang, L.J.; Chen, X.Y. Arabidopsis MYC2 interacts with DELLA proteins in regulating sesquiterpene synthase gene expression. Plant Cell 2012, 24, 2635–2648. [Google Scholar] [CrossRef] [PubMed]
- Bedon, F.; Bomal, C.; Caron, S.; Levasseur, C.; Boyle, B.; Mansfield, S.D.; Schmidt, A.; Gershenzon, J.; Grima-Pettenati, J.; Seguin, A.; et al. Subgroup 4 R2R3-MYBs in conifer trees: Gene family expansion and contribution to the isoprenoid- and flavonoid-oriented responses. J. Exp. Bot. 2010, 61, 3847–3864. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Sun, T.H.; Zhao, L.; Pan, X.W.; Lu, S. The bZIP transcription factor HY5 interacts with the promoter of the monoterpene synthase gene QH6 in modulating its rhythmic expression. Front. Plant Sci. 2015, 6, 304. [Google Scholar] [CrossRef] [PubMed]
- Zvi, M.M.; Shklarman, E.; Masci, T.; Kalev, H.; Debener, T.; Shafir, S.; Ovadis, M.; Vainstein, A. PAP1 transcription factor enhances production of phenylpropanoid and terpenoid scent compounds in rose flowers. New Phytol. 2012, 195, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spyropoulou, E.A.; Haring, M.A.; Schuurink, R.C. RNA sequencing on Solanum lycopersicum trichomes identifies transcription factors that activate terpene synthase promoters. BMC Genom. 2014, 15, 402. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.M.; Zhu, W.J. Osmanthus fragrans; Shanghai Press of Science and Technology: Shanghai, China, 2000. [Google Scholar]
- Huang, B.; Chen, H.; Shao, L. The ethanol extract of Osmanthus fragrans attenuates Porphyromonasg in givalis lipopolysaccharide-stimulated inflammatory effect through the nuclear factor erythroid 2-related factor-mediated antioxidant signalling pathway. Arch. Oral Biol. 2015, 60, 1030–1038. [Google Scholar]
- Zhang, C.; Wang, Y.; Fu, J.; Bao, Z.; Zhao, H. Transcriptomic analysis and carotenogenic gene expression related to petal coloration in Osmanthus fragrans ‘Yanhong Gui’. Trees 2016, 30, 1207–1223. [Google Scholar] [CrossRef]
- Mu, H.N.; Li, H.G.; Wang, L.G.; Yang, X.L.; Sun, Z.T.; Xu, C. Transcriptome sequencing and analysis of sweet osmanthus (Osmanthus fragrans Lour.). Genes Genom. 2014, 36, 777–788. [Google Scholar] [CrossRef]
- Dudareva, N.; Pichersky, E. Biochemical and molecular genetic aspects of floral scents. Plant Physiol. 2000, 122, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Mc Garvey, D.J.; Croteau, R. Terpenoid metabolism. Plant Cell 1995, 7, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Dudareva, N.; Negre, F.; Nagegowda, D.A.; Orlova, I. Plant Volatiles: Recent Advances and Future Perspectives. Crit. Rev. Plant Sci. 2006, 25, 417–440. [Google Scholar] [CrossRef]
- Chen, F.; Tholl, D.; D’Auria, J.C.; Farooq, A.; Pichersky, E.; Gershenzon, J. Biosynthesis and emission of terpenoid volatiles from Arabidopsis flowers. Plant Cell 2003, 15, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Dudareva, N. (E)-beta-Ocimene and Myrcene Synthase Genes of Floral Scent Biosynthesis in Snapdragon: Function and Expression of Three Terpene Synthase Genes of a New Terpene Synthase Subfamily. Plant Cell 2003, 15, 1227–1241. [Google Scholar] [CrossRef] [PubMed]
- Muhlemann, J.K.; Maeda, H.; Chang, C.Y.; Miguel, P.S.; Baxter, I.; Cooper, B.; Perera, A.M.; Nikolau, J.B.; Vitek, O.; Morgan, A.J.; et al. Developmental changesin the metabolic network of snapdragon flowers. PLoS ONE 2012, 7, e40381. [Google Scholar] [CrossRef] [PubMed]
- Dudareva, N.; Cseke, L.; Blanc, V.M.; Pichersky, E. Molecular characterization and cell type-specific expression of linalool synthase gene from Clarkia. Plant Physiol. 1996, 111, 815. [Google Scholar]
- Dudareva, N.; Cseke, L.; Blanc, V.M.; Pichersky, E. Evolution of floral scent inClarkia: Novel patterns of S-linalool synthase gene expression in the C. breweri flower. Plant Cell 1996, 8, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Dudareva, N.; D’Auria, J.C.; Nam, K.H.; Raguso, R.A.; Pichersky, E. Acetyl-Co A: Benzylalcohol acetyltransferase-an enzyme involved in floral scent production in Clarkia breweri. Plant J. 1998, 14, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Tholl, D.; Kish, C.M.; Orlova, I.; Sherman, D.; Gershenzon, J.; Pichersky, E.; Dudareva, N. Formation of monoterpenes in Antirrhinum majus and Clarkia breweri flowers involves heterodimeric geranyl diphosphate synthases. Plant Cell 2004, 16, 977–992. [Google Scholar] [CrossRef] [PubMed]
- Boatright, J.; Negre, F.; Chen, X.; Kish, C.M.; Wood, B.; Peel, G.; Orlova, I.; Gang, D.; Rhodes, D.; Dudareva, N. Understanding in vivo benzenoid metabolism in petunia petal tissue. Plant Physiol. 2004, 135, 1993–2011. [Google Scholar] [CrossRef] [PubMed]
- Spiller, M.; Berger, R.G.; Debener, T. Genetic dissection of scent metabolicprofiles in diploid rose populations. Theor. Appl. Genet. 2010, 120, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Guterman, I. Rose Scent: Genomics Approach to Discovering Novel Floral Fragrance-Related Genes. Plant Cell 2002, 14, 2325–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudareva, N.; Klempien, A.; Muhlemann, J.K.; Kaplan, I. Biosynthesis, function and metabolic engineering of plant volatile organic compounds. New Phytol. 2013, 198, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Yu, R.; Fan, Y. Transcriptome profiling provides new insights into the formation of floral scent in Hedychiumcoronarium. BMC Genom. 2015, 16, 470. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.S.; Croteau, R.B. Metabolic engineering of essential oil yield and composition in mint by altering expression of deoxyxylulose phosphate reductoisomerase and menthofuran synthase. Proc. Natl. Acad. Sci. USA 2001, 98, 8915–8920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carretero-Paulet, L.; Ahumada, I.; Cunillera, N.; Rodriguez-Concepcion, M.; Ferrer, A.; Boronat, A.; Campos, N. Expression and molecular analysis of the Arabidopsis DXR gene encoding 1-deoxy-d-xylulose 5-phosphate reductoisomerase, the first committed enzyme of the 2-C-methyl-d-erythritol 4-phosphate pathway. Plant Physiol. 2002, 129, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Koes, R.; Verweij, W.; Quattrocchio, F. Flavonoids: A colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005, 10, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Schwinn, K.; Venail, J.; Shang, Y.J.; Mackay, S.; Alm, V.; Butelli, E.; Oyama, R.; Bailey, P.; Davies, K.; Martin, C. A small familyof MYB-regulatory genes controls floral pigmentation intensity and patterning in the genus Antirrhinum. Plant Cell 2006, 18, 831–851. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, A.; Galili, G. Metabolic engineering of the plant primary-secondary metabolism interface. Curr. Opin. Biotechnol. 2011, 22, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Feller, A.; Machemer, K.; Braun, E.L.; Grotewold, E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011, 66, 94–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonk, J.C.; Haring, M.A.; van Tunen, A.J.; Schuurink, R.C. ODORANT1 regulates fragrance biosynthesis in petunia flowers. Plant Cell 2005, 17, 1612–1624. [Google Scholar] [CrossRef] [PubMed]
- Spitzer-Rimon, B.; Marhevka, E.; Barkai, O.; Marton, I.; Edelbaum, O.; Masci, T.; Prathapani, N.K.; Schklarman, E.; Ovadis, M.; Vainstein, A. EOBII, a gene encodinga flower-specific regulator of phenylpropanoid volatiles’ biosynthesis in petunia. Plant Cell 2010, 22, 1961–1976. [Google Scholar] [CrossRef] [PubMed]
- Spitzer-Rimon, B.; Farhi, M.; Albo, B.; Cna’ani, A.; Zvi, M.B.; Masci, T.; Edelbaum, O.; Yu, Y.X.; Schklarman, E.; Ovadis, M.; et al. The R2R3-MYB-like regulatory factor EOBI, acting downstream of EOBII, regulates scent production by activating ODO1 and structuralscent-related genes in petunia. Plant Cell 2012, 24, 5089–5105. [Google Scholar] [CrossRef] [PubMed]
- Colquhoun, T.A.; Kim, J.Y.; Wedde, A.E.; Levin, L.A.; Schmitt, K.C.; Schuurink, R.C.; Clark, D.G. PhMYB4 fine-tunes the floral volatile signature of Petunia x hybrida through PhC4H. J. Exp. Bot. 2011, 62, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with orwithout a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time seriesgene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification usingthe KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data usingreal-time quantitative PCR and the 2-ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the petals are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript | Unigene |
|---|---|
| Total assembled bases | 108,311,010 |
| Total number of genes | 136,611 |
| Max length of unigenes (bp) | 16,876 |
| Min length of unigenes (bp) | 201 |
| Average length ofunigenes (bp) | 792 |
| N50 (bp) | 1424 |
| GC percentage (%) | 38.9215 |
| Database | Number of Annotated Unigenes | Percentage of Annotated Unigenes (%) |
|---|---|---|
| Nr | 58,556 | 42.86 |
| Swiss-prot | 47,294 | 34.62 |
| KOG | 39,043 | 28.58 |
| KO (KEGG Orthology) | 23,825 | 17.44 |
| Total | 64,795 | 47.43 |
| KEGG Pathway | All Genes with Pathway Annotation | DGEs Genes with Pathway Annotation | Pathway ID |
|---|---|---|---|
| Phenylpropanoid | 319 | 178 | Ko00940 |
| Monoterpenoid biosynthesis | 17 | 8 | Ko00902 |
| Terpenoid backbone biosynthesis | 184 | 78 | Ko00900 |
| Sesquiterpenoidand triterpenoid biosynthesis | 69 | 45 | Ko00909 |
| Diterpenoid biosynthesis | 45 | 25 | Ko00904 |
| Limonene and pinene degradation | 15 | 10 | Ko00903 |
| Transcription Factors | Profile0 | Profile1 | Profile4 | Profile6 | Profile7 | All Profiles |
|---|---|---|---|---|---|---|
| C2H2 | 3 | 2 | 1 | 0 | 2 | 234 |
| ERF | 56 | 88 | 50 | 21 | 50 | 193 |
| bHLH | 24 | 26 | 13 | 5 | 4 | 168 |
| MYB-related | 4 | 6 | 6 | 3 | 5 | 155 |
| NAC | 101 | 158 | 110 | 47 | 80 | 134 |
| WRKY | 3 | 4 | 41 | 7 | 24 | 112 |
| MYB | 18 | 13 | 24 | 9 | 19 | 96 |
| bZIP | 4 | 3 | 2 | 0 | 0 | 89 |
| GRAS | 2 | 0 | 8 | 11 | 15 | 82 |
| C3H | 0 | 1 | 0 | 0 | 2 | 73 |
| FAR1 | 3 | 6 | 11 | 6 | 10 | 67 |
| Dof | 11 | 3 | 2 | 0 | 1 | 59 |
| G2-Like | 3 | 0 | 1 | 1 | 1 | 56 |
| HD-ZIP | 0 | 1 | 0 | 0 | 0 | 52 |
| Trihelix | 4 | 2 | 2 | 0 | 3 | 50 |
| HSF | 2 | 1 | 0 | 1 | 0 | 43 |
| TCP | 6 | 11 | 2 | 0 | 7 | 41 |
| B3 | 6 | 11 | 10 | 8 | 8 | 40 |
| GATA | 14 | 10 | 0 | 1 | 3 | 34 |
| SBP | 4 | 5 | 0 | 0 | 0 | 32 |
| ARF | 0 | 2 | 2 | 1 | 1 | 29 |
| M-type | 4 | 3 | 11 | 5 | 7 | 22 |
| NF-YC | 0 | 1 | 0 | 0 | 0 | 16 |
| AP2 | 4 | 7 | 5 | 2 | 1 | 14 |
| CPP | 0 | 1 | 0 | 0 | 0 | 12 |
| WOX | 1 | 1 | 0 | 0 | 0 | 11 |
| LSD | 0 | 0 | 1 | 0 | 0 | 9 |
| YABBY | 1 | 1 | 0 | 0 | 0 | 8 |
| BES1 | 2 | 2 | 0 | 0 | 0 | 8 |
| SRS | 1 | 0 | 1 | 2 | 1 | 6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.-L.; Li, H.-Y.; Yue, Y.-Z.; Ding, W.-J.; Xu, C.; Shi, T.-T.; Chen, G.-W.; Wang, L.-G. Transcriptomic Analysis of the Candidate Genes Related to Aroma Formation in Osmanthus fragrans. Molecules 2018, 23, 1604. https://doi.org/10.3390/molecules23071604
Yang X-L, Li H-Y, Yue Y-Z, Ding W-J, Xu C, Shi T-T, Chen G-W, Wang L-G. Transcriptomic Analysis of the Candidate Genes Related to Aroma Formation in Osmanthus fragrans. Molecules. 2018; 23(7):1604. https://doi.org/10.3390/molecules23071604
Chicago/Turabian StyleYang, Xiu-Lian, Hai-Yan Li, Yuan-Zheng Yue, Wen-Jie Ding, Chen Xu, Ting-Ting Shi, Gong-Wei Chen, and Liang-Gui Wang. 2018. "Transcriptomic Analysis of the Candidate Genes Related to Aroma Formation in Osmanthus fragrans" Molecules 23, no. 7: 1604. https://doi.org/10.3390/molecules23071604