
Targeting Dynamical Binding Processes in the Design of Non-Antibiotic Anti-Adhesives by Molecular Simulation—The Example of FimH


Abstract
:1. Introduction
2. The Molecular Binding Mechanism of Small Mannosidic Compounds to the FimH Binding Site
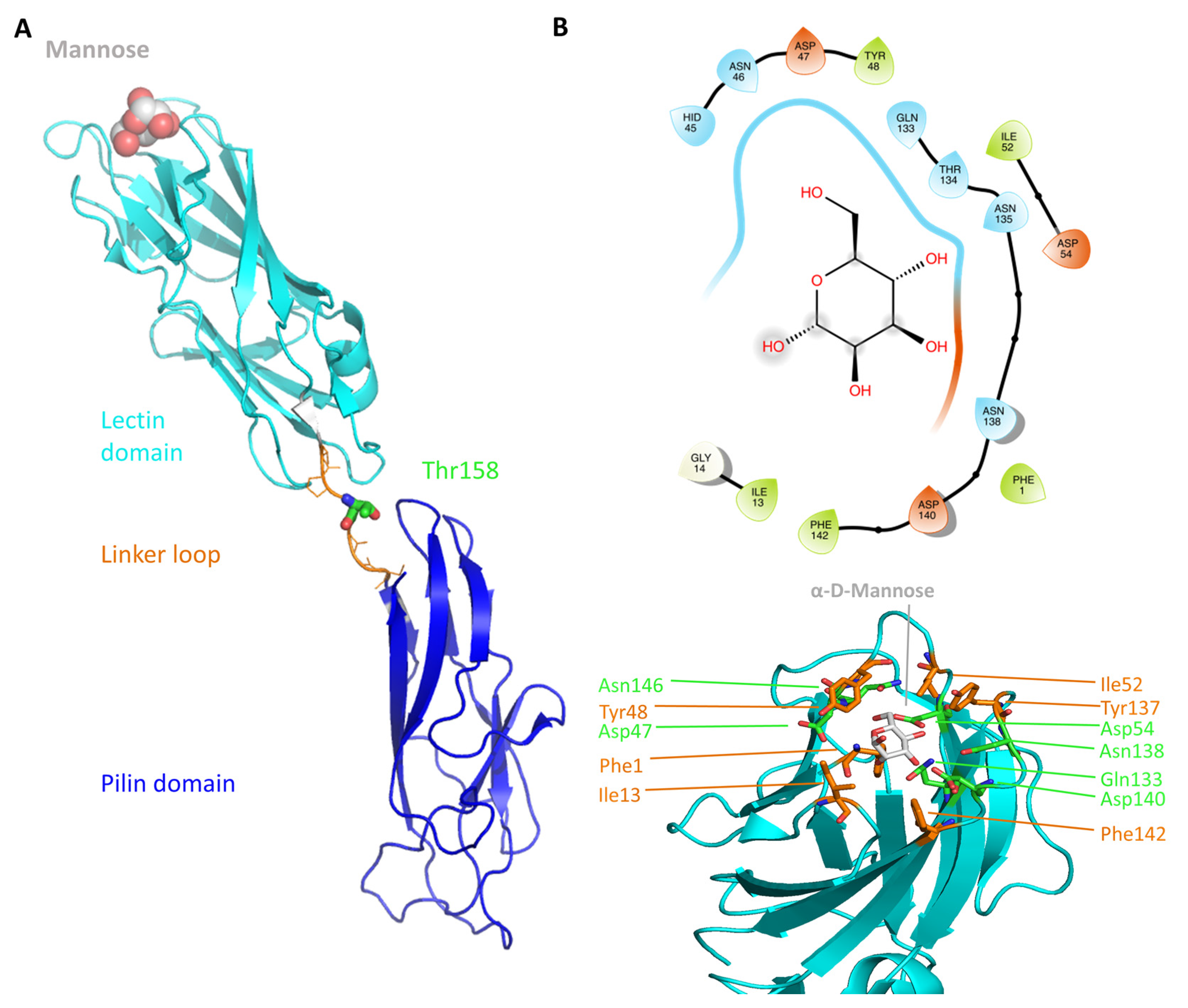
2.1. The FimH Mannose-Binding Site
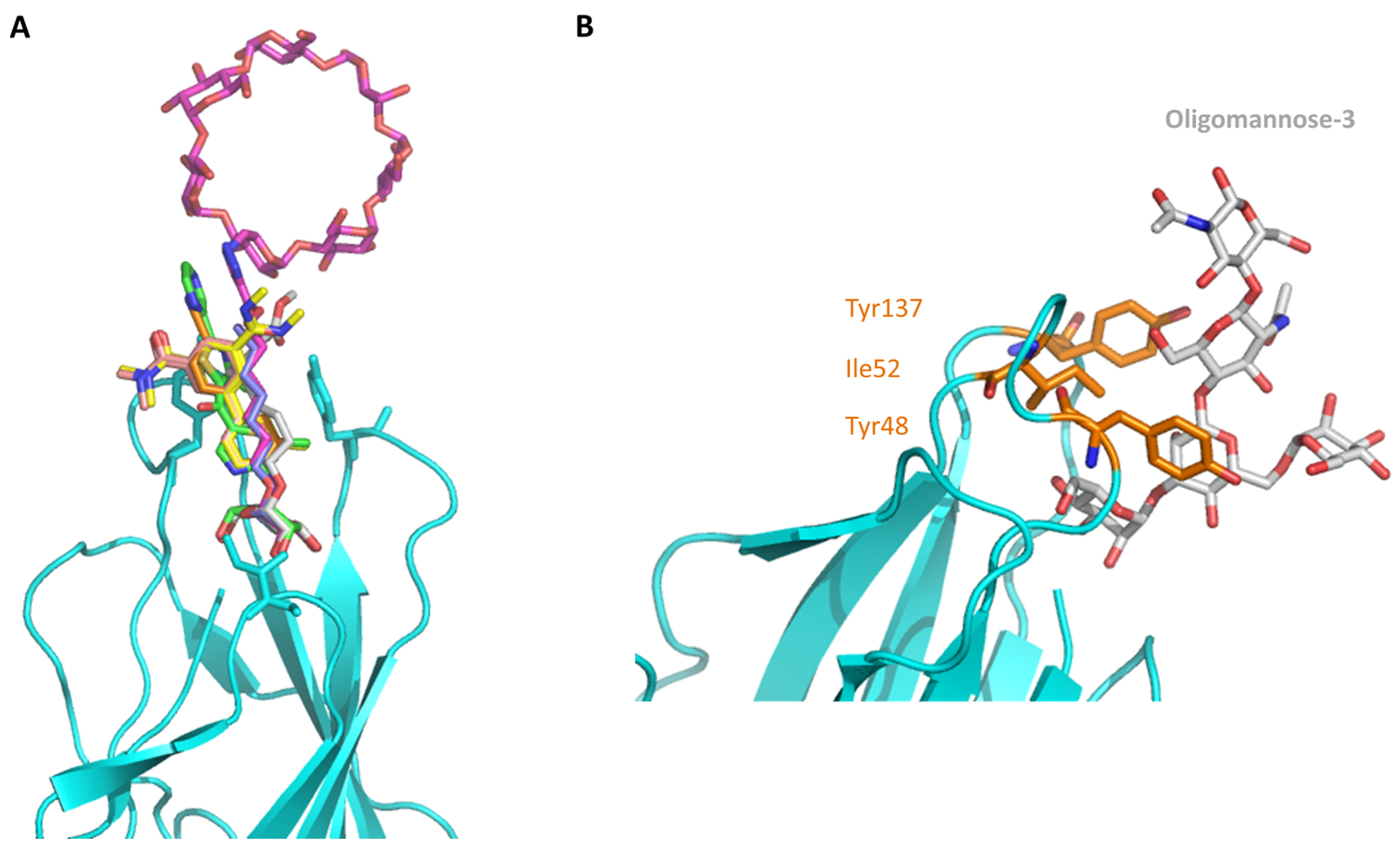
2.2. The Tyrosine Gate and Its Impact on Mannoside Binding
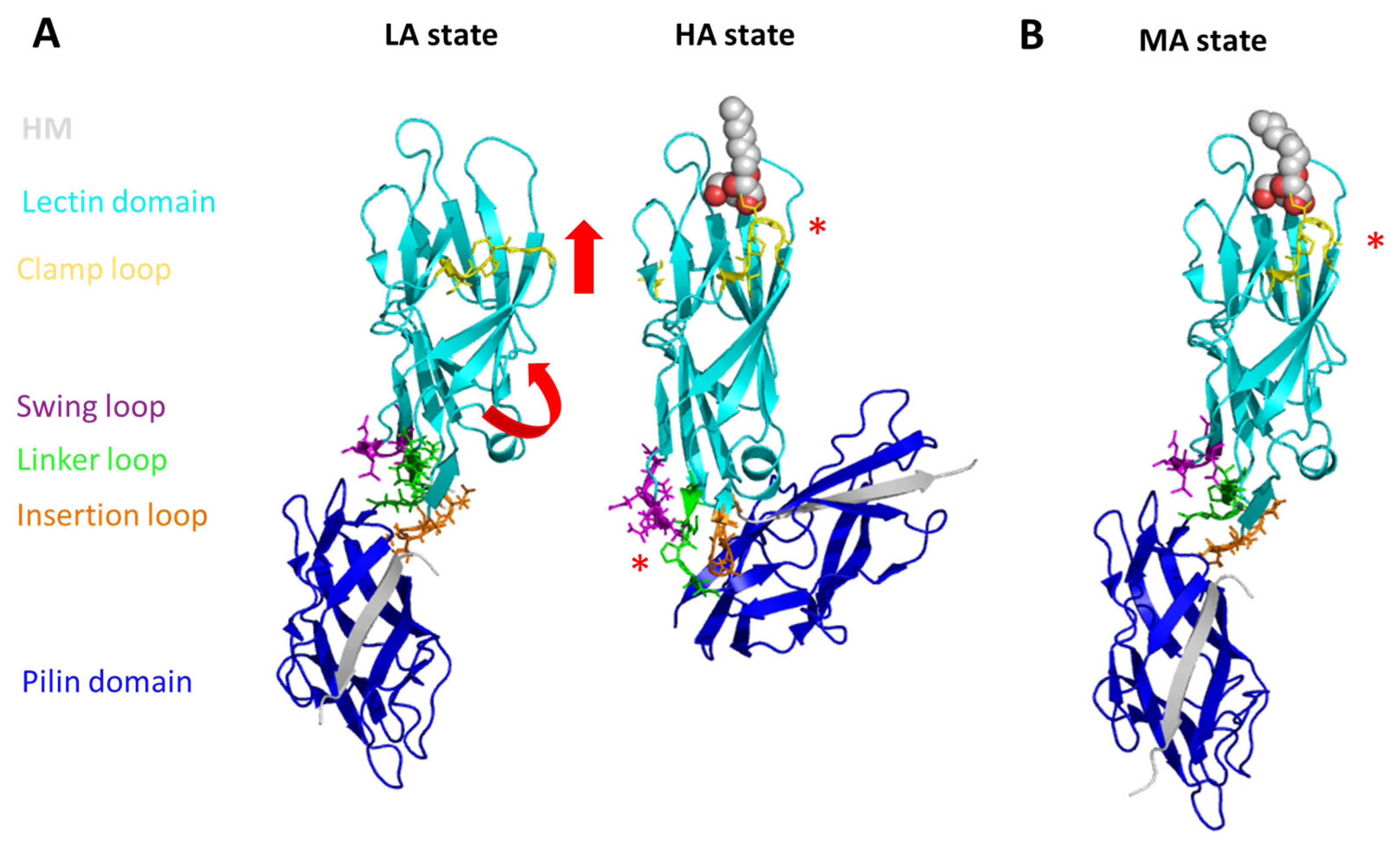
2.3. The Conformational States of FimH
3. Rational Drug Design of FimH Inhibitors
3.1. Monovalent FimH Inhibitors Targeting the Mannose-Binding Pocket of the HA FimH State
3.2. Multivalent FimH Inhibitors Targeting the Mannose-Binding Pocket
3.3. Alternative Binding Positions for Inhibitors
4. Molecular Simulation as a Tool to Study FimH Function and Inhibition
5. Conclusions and Outlook
Funding
Conflicts of Interest
References
- Leimbach, A.; Hacker, J.; Dobrindt, U. E. E. coli as an All-Rounder: The thin line between commensalism and pathogenicity. Curr. Top. Microbiol. Immunol. 2013, 358, 3–32. [Google Scholar] [PubMed]
- Foxman, B. Epidemiology of Urinary tract infections: Incidence, morbidity, and economic costs. Dis. Mon. 2003, 49, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Ronald, A.R.; Nicolle, L.E.; Stamm, E.; Krieger, J.; Warren, J.; Schaeffer, A.; Naber, K.G.; Hooton, T.M.; Johnson, J.; Chambers, S.; et al. Urinary tract infection in adults: Research priorities and strategies. Int. J. Antimicrob. Agents 2001, 17, 343–348. [Google Scholar] [CrossRef]
- Foxman, B. Urinary tract infection syndromes. Infect. Dis. Clin. N. Am. 2014, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.V.; Master, R.N.; Bordon, J. Trimethoprim-sulfamethoxazole may no longer be acceptable for the treatment of acute uncomplicated cystitis in the United States. Clin. Infect. Dis. 2011, 53, 316–317. [Google Scholar] [CrossRef] [PubMed]
- Karlowsky, J.A.; Hoban, D.J.; DeCorby, M.R.; Laing, N.M.; Zhanel, G.G. Fluoroquinolone-resistant urinary isolates of Escherichia coli from outpatients are frequently multidrug resistant: Results from the north american urinary tract infection collaborative alliance-quinolone resistance study. Antimicrob. Agents Chemother. 2006, 50, 2251–2254. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Hisanaga, T.L.; Laing, N.M.; DeCorby, M.R.; Nichol, K.A.; Palatnick, L.P.; Johnson, J.; Noreddin, A.; Harding, G.K.M.; Nicolle, L.E.; et al. Antibiotic resistance in outpatient urinary isolates: Final results from the North American Urinary Tract Infection Collaborative Alliance (NAUTICA). Int. J. Antimicrob. Agents 2005, 26, 380–388. [Google Scholar] [CrossRef] [PubMed]
- McGann, P.; Snesrud, E.; Maybank, R.; Corey, B.; Ong, A.C.; Clifford, R.; Hinkle, M.; Whitman, T.; Lesho, E.; Schaecher, K.E. Escherichia coli Harboring mcr-1 and bla CTX-M on a novel incf plasmid: First report of mcr-1 in the United States. Antimicrob. Agents Chemother. 2016, 60, 4420–4421. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T. Who will develop new antibacterial agents? Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130430. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Fresh approaches to anti-infective therapies. Sci. Transl. Med. 2012, 4, 140sr2. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Lindhorst, T.K. The bacterial lectin fimh, a target for drug discovery–carbohydrate inhibitors of type 1 fimbriae-mediated bacterial adhesion. Eur. J. Org. Chem. 2011, 2011, 3583–3609. [Google Scholar] [CrossRef]
- Pieters, R.J. Intervention with bacterial adhesion by multivalent carbohydrates. Med. Res. Rev. 2007, 27, 796–816. [Google Scholar] [CrossRef] [PubMed]
- Langermann, S. Prevention of mucosal Escherichia coli infection by fimh-adhesin-based systemic vaccination. Science 1997, 276, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-S.; Bouckaert, J.; Hung, D.; Pinkner, J.; Widberg, C.; DeFusco, A.; Auguste, C.G.; Strouse, R.; Langermann, S.; Waksman, G.; et al. Structural basis of tropism of escherichia coli to the bladder during urinary tract infection. Mol. Microbiol. 2002, 44, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Eto, D.S.; Jones, T.A.; Sundsbak, J.L.; Mulvey, M.A. Integrin-mediated host cell invasion by type 1–piliated uropathogenic Escherichia coli. PLoS Pathog. 2007, 3, e100. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.A.; Hooton, T.M.; Stamm, W.E.; Humphrey, P.A.; Hultgren, S.J. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med. 2007, 4, e329. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.J. Type 1 Pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 2000, 19, 2803–2812. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Mo, W.J.; Sebbel, P.; Min, G.; Neubert, T.A.; Glockshuber, R.; Wu, X.R.; Sun, T.T.; Kong, X.P. Uroplakin Ia is the urothelial receptor for uropathogenic Escherichia coli: Evidence from in vitro FimH binding. J. Cell Sci. 2001, 114, 4095–4103. [Google Scholar] [PubMed]
- Baumgart, M.; Dogan, B.; Rishniw, M.; Weitzman, G.; Bosworth, B.; Yantiss, R.; Orsi, R.H.; Wiedmann, M.; McDonough, P.; Kim, S.G.; et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of clostridiales in crohn’s disease involving the ileum. ISME J. 2007, 1, 403–418. [Google Scholar] [CrossRef] [PubMed]
- DeFilippis, E.M.; Longman, R.; Harbus, M.; Dannenberg, K.; Scherl, E.J. Crohn’s Disease: Evolution, epigenetics, and the emerging role of microbiome-targeted therapies. Curr. Gastroenterol. Rep. 2016, 18, 13. [Google Scholar] [CrossRef] [PubMed]
- Sivignon, A.; Bouckaert, J.; Bernard, J.; Gouin, S.G.; Barnich, N. The potential of fimh as a novel therapeutic target for the treatment of crohn’s disease. Expert Opin. Ther. Targets 2017, 21, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Barnich, N.; Carvalho, F.A.; Glasser, A.-L.; Darcha, C.; Jantscheff, P.; Allez, M.; Peeters, H.; Bommelaer, G.; Desreumaux, P.; Colombel, J.-F.; et al. CEACAM6 Acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in crohn disease. J. Clin. Investig. 2007, 117, 1566–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez Dorta, D.; Sivignon, A.; Chalopin, T.; Dumych, T.I.; Roos, G.; Bilyy, R.O.; Deniaud, D.; Krammer, E.-M.; De Ruyck, J.; Lensink, M.F.; et al. The Antiadhesive strategy in crohn’s disease: Orally active mannosides to decolonize pathogenic Escherichia coli from the gut. ChemBioChem 2016, 17, 936–952. [Google Scholar] [CrossRef] [PubMed]
- Mydock-McGrane, L.K.; Hannan, T.J.; Janetka, J.W. Rational design strategies for fimh antagonists: New drugs on the horizon for urinary tract infection and crohn’s sisease. Expert Opin. Drug Discov. 2017, 12, 711–731. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, C.N.; Klein, R.D.; Schreiber, H.L.; Janetka, J.W.; Hultgren, S.J. Precision antimicrobial therapeutics: The path of least resistance? NPJ Biofilms Microbiomes 2018, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, J.; Berglund, J.; Schembri, M.; De Genst, E.; Cools, L.; Wuhrer, M.; Hung, C.-S.; Pinkner, J.; Slättegård, R.; Zavialov, A.; et al. Receptor binding studies disclose a novel class of high-affinity inhibitors of the escherichia coli fimh adhesin. Mol. Microbiol. 2004, 55, 441–455. [Google Scholar] [CrossRef] [PubMed]
- De Ruyck, J.; Roos, G.; Krammer, E.-M.; Prévost, M.; Lensink, M.F.; Bouckaert, J. Molecular mechanisms of drug action: X-ray crystallography at the basis of structure-based and ligand-based drug design. In Biophysical Techniques in Drug Discovery; The Royal Society of Chemstry: London, UK, 2017; Chapter 4; pp. 67–86. ISBN 9781788010016. [Google Scholar]
- Chen, S.L.; Hung, C.S.; Pinkner, J.S.; Walker, J.N.; Cusumano, C.K.; Li, Z.; Bouckaert, J.; Gordon, J.I.; Hultgren, S.J. Positive selection identifies an in vivo role for fimh during urinary tract infection in addition to mannose binding. Proc. Natl. Acad. Sci. USA 2009, 106, 22439–22444. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger LLC: New York, NY, USA, 2018.
- Sager, C.P.; Fiege, B.; Zihlmann, P.; Vannam, R.; Rabbani, S.; Jakob, R.P.; Preston, R.C.; Zalewski, A.; Maier, T.; Peczuh, M.W.; et al. The Price of flexibility—A case study on septanoses as pyranose mimetics. Chem. Sci. 2018, 9, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Roos, G.; Wellens, A.; Touaibia, M.; Yamakawa, N.; Geerlings, P.; Roy, R.; Wyns, L.; Bouckaert, J. Validation of reactivity descriptors to assess the aromatic stacking within the tyrosine gate of FimH. ACS Med. Chem. Lett. 2013, 4, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Alvarez Dorta, D.; Chalopin, T.; Sivignon, A.; de Ruyck, J.; Dumych, T.I.; Bilyy, R.O.; Deniaud, D.; Barnich, N.; Bouckaert, J.; Gouin, S.G. Physiochemical tuning of potent Escherichia coli anti-adhesives by microencapsulation and methylene homologation. ChemMedChem 2017, 12, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, C.; Han, Z.; Kalas, V.; Klein, R.; Pinkner, J.S.; Ford, B.; Binkley, J.; Cusumano, C.K.; Cusumano, Z.; Mydock-McGrane, L.; et al. Antivirulence isoquinolone mannosides: Optimization of the biaryl aglycone for fimh lectin binding affinity and efficacy in the treatment of chronic UTI. Chem. Med. Chem. 2016, 11, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Wellens, A.; Lahmann, M.; Touaibia, M.; Vaucher, J.; Oscarson, S.; Roy, R.; Remaut, H.; Bouckaert, J. The tyrosine gate as a potential entropic lever in the receptor-binding site of the bacterial adhesin FimH. Biochemistry 2012, 51, 4790–4799. [Google Scholar] [CrossRef] [PubMed]
- Kleeb, S.; Pang, L.; Mayer, K.; Eris, D.; Sigl, A.; Preston, R.C.; Zihlmann, P.; Sharpe, T.; Jakob, R.P.; Abgottspon, D.; et al. FimH antagonists: Bioisosteres to improve the in vitro and in vivo pk/pd profile. J. Med. Chem. 2015, 58, 2221–2239. [Google Scholar] [CrossRef] [PubMed]
- Mydock-McGrane, L.; Cusumano, Z.; Han, Z.; Binkley, J.; Kostakioti, M.; Hannan, T.; Pinkner, J.S.; Klein, R.; Kalas, V.; Crowley, J.; et al. Antivirulence C-mannosides as antibiotic-sparing, oral therapeutics for urinary tract infections. J. Med. Chem. 2016, 59, 9390–9408. [Google Scholar] [CrossRef] [PubMed]
- Wellens, A.; Garofalo, C.; Nguyen, H.; Van Gerven, N.; Slättegård, R.; Hernalsteens, J.-P.; Wyns, L.; Oscarson, S.; De Greve, H.; Hultgren, S.; et al. Intervening with urinary tract infections using anti-adhesives based on the crystal structure of the Fimh–oligomannose-3 complex. PLoS ONE 2008, 3, e2040. [Google Scholar] [CrossRef]
- Rabbani, S.; Krammer, E.-M.; Roos, G.; Zalewski, A.; Preston, R.; Eid, S.; Zihlmann, P.; Prévost, M.; Lensink, M.F.; Thompson, A.; et al. Mutation of Tyr137 of the Universal Escherichia coli Fimbrial Adhesin FimH relaxes the tyrosine gate prior to mannose binding. IUCrJ 2017, 4, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Touaibia, M.; Krammer, E.-M.; Shiao, T.; Yamakawa, N.; Wang, Q.; Glinschert, A.; Papadopoulos, A.; Mousavifar, L.; Maes, E.; Oscarson, S.; et al. Sites for dynamic protein-carbohydrate interactions of O- and C-linked mannosides on the E. coli FimH Adhesin. Molecules 2017, 22, 1101. [Google Scholar] [CrossRef] [PubMed]
- de Ruyck, J.; Lensink, M.F.; Bouckaert, J. Structures of C-mannosylated anti-adhesives bound to the type 1 fimbrial fimh adhesin. IUCrJ 2016, 3, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Pinkner, J.S.; Ford, B.; Obermann, R.; Nolan, W.; Wildman, S.A.; Hobbs, D.; Ellenberger, T.; Cusumano, C.K.; Hultgren, S.J.; et al. Structure-based drug design and optimization of mannoside bacterial fimh antagonists. J. Med. Chem. 2010, 53, 4779–4792. [Google Scholar] [CrossRef] [PubMed]
- Gouin, S.G.; Roos, G.; Bouckaert, J. Discovery and Application of FimH Antagonists. In Carbohydrates as Drugs; Topics in Medicinal Chemistry; Seeberger, P., Rademacher, C., Eds.; Springer: Berlin, Germany, 2014; Volume 12, pp. 123–168. [Google Scholar]
- Le Trong, I.; Aprikian, P.; Kidd, B.A.; Thomas, W.E.; Sokurenko, E.V.; Stenkamp, R.E. Donor strand exchange and conformational changes during E. coli fimbrial formation. J. Struct. Biol. 2010, 172, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Tomašić, T.; Rabbani, S.; Gobec, M.; Raščan, I.M.; Podlipnik, Č.; Ernst, B.; Anderluh, M. Branched α-d-mannopyranosides: A new class of potent fimh antagonists. Med. Chem. Commun. 2014, 5, 1247–1253. [Google Scholar] [CrossRef]
- Feenstra, T.; Thøgersen, M.S.; Wieser, E.; Peschel, A.; Ball, M.J.; Brandes, R.; Satchell, S.C.; Stockner, T.; Aarestrup, F.M.; Rees, A.J.; et al. Adhesion of Escherichia coli under flow conditions reveals potential novel effects of Fimh mutations. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Szunerits, S.; Zagorodko, O.; Cogez, V.; Dumych, T.; Chalopin, T.; Alvarez Dorta, D.; Sivignon, A.; Barnich, N.; Harduin-Lepers, A.; Larroulet, I.; et al. Differentiation of Crohn’s disease-associated isolates from other pathogenic Escherichia coli by Fimbrial adhesion under shear force. Biology (Basel) 2016, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Sokurenko, E.V.; Chesnokova, V.; Dykhuizen, D.E.; Ofek, I.; Wu, X.-R.; Krogfelt, K.A.; Struve, C.; Schembri, M.A.; Hasty, D.L. Pathogenic adaptation of Escherichia coli by natural variation of the fimh adhesin. Proc. Natl. Acad. Sci. USA 1998, 95, 8922–8926. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.E.; Trintchina, E.; Forero, M.; Vogel, V.; Sokurenko, E. V Bacterial adhesion to target cells enhanced by shear force. Cell 2002, 109, 913–923. [Google Scholar] [CrossRef]
- Vanwetswinkel, S.; Volkov, A.N.; Sterckx, Y.G.J.; Garcia-Pino, A.; Buts, L.; Vranken, W.F.; Bouckaert, J.; Roy, R.; Wyns, L.; van Nuland, N.A.J. Study of the structural and dynamic effects in the FimH adhesin upon α-d-Heptyl Mannose Binding. J. Med. Chem. 2014, 57, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Aprikian, P.; Tchesnokova, V.; Kidd, B.; Yakovenko, O.; Yarov-Yarovoy, V.; Trinchina, E.; Vogel, V.; Thomas, W.; Sokurenko, E. Interdomain interaction in the FimH adhesin of Escherichia coli regulates the affinity to mannose. J. Biol. Chem. 2007, 282, 23437–23446. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, L.M.; Thomas, W.E.; Sokurenko, E.V.; Vogel, V. Elevated shear stress protects Escherichia coli cells adhering to surfaces via catch bonds from detachment by soluble inhibitors. Appl. Environ. Microbiol. 2006, 72, 3005–3010. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.N.; Ding, A.M.; Nilsson, L.M.; Kusuma, K.; Tchesnokova, V.; Vogel, V.; Sokurenko, E.V.; Thomas, W.E. Weak rolling adhesion enhances bacterial surface colonization. J. Bacteriol. 2007, 189, 1794–1802. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.G.; Dodson, K.W.; Hooton, T.M.; Hultgren, S.J. Intracellular Bacterial communities of uropathogenic Escherichia coli in urinary tract pathogenesis. Trends Microbiol. 2004, 12, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.E.; Nilsson, L.M.; Forero, M.; Sokurenko, E.V.; Vogel, V. Shear-dependent ‘Stick-and-Eoll’ adhesion of type 1 Fimbriated Escherichia coli. Mol. Microbiol. 2004, 53, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Aprikian, P.; Interlandi, G.; Kidd, B.A.; Le Trong, I.; Tchesnokova, V.; Yakovenko, O.; Whitfield, M.J.; Bullitt, E.; Stenkamp, R.E.; Thomas, W.E.; et al. The bacterial fimbrial tip acts as a mechanical force sensor. PLoS Biol. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, L.M.; Thomas, W.E.; Sokurenko, E.V.; Vogel, V. Beyond Induced-fit receptor-ligand interactions: Structural changes that can significantly extend bond lifetimes. Structure 2008, 16, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Le Trong, I.; Aprikian, P.; Kidd, B.A.; Forero-Shelton, M.; Tchesnokova, V.; Rajagopal, P.; Rodriguez, V.; Interlandi, G.; Klevit, R.; Vogel, V.; et al. Structural basis for mechanical force regulation of the adhesin fimh via finger trap-like β sheet twisting. Cell 2010, 141, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Sterpone, F.; Doutreligne, S.; Tran, T.T.; Melchionna, S.; Baaden, M.; Nguyen, P.H.; Derreumaux, P. Multi-Scale Simulations of biological systems using the opep coarse-grained model. Biochem. Biophys. Res. Commun. 2018, 498, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Sauer, M.M.; Jakob, R.P.; Eras, J.; Baday, S.; Eriş, D.; Navarra, G.; Bernèche, S.; Ernst, B.; Maier, T.; Glockshuber, R. Catch-bond mechanism of the bacterial adhesin FimH. Nat. Commun. 2016, 7, 10738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, D. X-ray Structure of the fimc-fimh chaperone-adhesin complex from uropathogenic Escherichia coli. Science 1999, 285, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Sokurenko, E.V.; Vogel, V.; Thomas, W.E. Catch-Bond mechanism of force-enhanced adhesion: Counterintuitive, elusive, but… widespread? Cell Host Microbe 2008, 4, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, M.; Ghose, T.; Thomas, W. Shear-stabilized rolling behavior of E. coli examined with simulations. Biophys. J. 2010, 99, 2470–2478. [Google Scholar] [CrossRef] [PubMed]
- Rohl, C.A.; Strauss, C.E.M.; Misura, K.M.S.; Baker, D. Protein Structure Prediction Using Rosetta. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2004; Volume 383, pp. 66–93. ISBN 9780121827885. [Google Scholar]
- Rodriguez, V.B.; Kidd, B.A.; Interlandi, G.; Tchesnokova, V.; Sokurenko, E.V.; Thomas, W.E. Allosteric coupling in the bacterial adhesive protein FimH. J. Biol. Chem. 2013, 288, 24128–24139. [Google Scholar] [CrossRef] [PubMed]
- Interlandi, G.; Thomas, W.E. Mechanism of allosteric propagation across a β-sheet structure investigated by molecular dynamics simulations. Proteins Struct. Funct. Bioinf. 2016, 84, 990–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchesnokova, V.; Aprikian, P.; Kisiela, D.; Gowey, S.; Korotkova, N.; Thomas, W.; Sokurenko, E. Type 1 fimbrial adhesin Fimh elicits an immune response that enhances cell adhesion of Escherichia coli. Infect. Immun. 2011, 79, 3895–3904. [Google Scholar] [CrossRef] [PubMed]
- Kisiela, D.I.; Avagyan, H.; Friend, D.; Jalan, A.; Gupta, S.; Interlandi, G.; Liu, Y.; Tchesnokova, V.; Rodriguez, V.B.; Sumida, J.P.; et al. Inhibition and reversal of microbial attachment by an antibody with parasteric activity against the FimH adhesin of uropathogenic E. coli. PLoS Pathog. 2015, 11, e1004857. [Google Scholar] [CrossRef] [PubMed]
- Kisiela, D.I.; Rodriguez, V.B.; Tchesnokova, V.; Avagyan, H.; Aprikian, P.; Liu, Y.; Wu, X.-R.; Thomas, W.E.; Sokurenko, E.V. Conformational inactivation induces immunogenicity of the receptor-binding pocket of a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2013, 110, 19089–19094. [Google Scholar] [CrossRef] [PubMed]
- Singaravelu, M.; Selvan, A.; Anishetty, S. Molecular dynamics simulations of lectin somain of FimH and immunoinformatics for the design of potential vaccine candidates. Comput. Biol. Chem. 2014, 52, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Hahn, E.; Wild, P.; Hermanns, U.; Sebbel, P.; Glockshuber, R.; Häner, M.; Taschner, N.; Burkhard, P.; Aebi, U.; Müller, S.A. Exploring the 3D molecular architecture of Escherichia coli type 1 Pili. J. Mol. Biol. 2002, 323, 845–857. [Google Scholar] [CrossRef]
- Gossert, A.D.; Bettendorff, P.; Puorger, C.; Vetsch, M.; Herrmann, T.; Glockshuber, R.; Wüthrich, K. NMR structure of the Escherichia coli type 1 pilus subunit fimf and its interactions with other pilus subunits. J. Mol. Biol. 2008, 375, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Forero, M.; Yakovenko, O.; Sokurenko, E.V.; Thomas, W.E.; Vogel, V. Uncoiling mechanics of Escherichia coli type i fimbriae are optimized for catch bonds. PLoS Biol. 2006, 4, e298. [Google Scholar] [CrossRef] [PubMed]
- Yakovenko, O.; Sharma, S.; Forero, M.; Tchesnokova, V.; Aprikian, P.; Kidd, B.; Mach, A.; Vogel, V.; Sokurenko, E.; Thomas, W.E. FimH forms catch bonds that are enhanced by mechanical force due to allosteric regulation. J. Biol. Chem. 2008, 283, 11596–11605. [Google Scholar] [CrossRef] [PubMed]
- Rakshit, S.; Sivasankar, S. Biomechanics of cell adhesion: How force regulates the lifetime of adhesive bonds at the single molecule level. Phys. Chem. Chem. Phys. 2014, 16, 2211. [Google Scholar] [CrossRef] [PubMed]
- Mydock-McGrane, L.K.; Cusumano, Z.T.; Janetka, J.W. Mannose-derived FimH antagonists: A promising anti-virulence therapeutic strategy for urinary tract infections and Crohn’s disease. Expert Opin. Ther. Pat. 2016, 26, 175–197. [Google Scholar] [CrossRef] [PubMed]
- Fiege, B.; Rabbani, S.; Preston, R.C.; Jakob, R.P.; Zihlmann, P.; Schwardt, O.; Jiang, X.; Maier, T.; Ernst, B. The tyrosine gate of the bacterial lectin FimH: A conformational analysis by NMR spectroscopy and X-ray crystallography. Chem. Bio. Chem. 2015, 16, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Brument, S.; Sivignon, A.; Dumych, T.I.; Moreau, N.; Roos, G.; Guérardel, Y.; Chalopin, T.; Deniaud, D.; Bilyy, R.O.; Darfeuille-Michaud, A.; et al. Thiazolylaminomannosides As potent antiadhesives of Type 1 piliated escherichia coli isolated from Crohn’s disease patients. J. Med. Chem. 2013, 56, 5395–5406. [Google Scholar] [CrossRef] [PubMed]
- Chalopin, T.; Brissonnet, Y.; Sivignon, A.; Deniaud, D.; Cremet, L.; Barnich, N.; Bouckaert, J.; Gouin, S.G. Inhibition profiles of mono- and polyvalent FimH antagonists against 10 different Escherichia coli strains. Org. Biomol. Chem. 2015, 13, 11369–11375. [Google Scholar] [CrossRef] [PubMed]
- Chalopin, T.; Alvarez Dorta, D.; Sivignon, A.; Caudan, M.; Dumych, T.I.; Bilyy, R.O.; Deniaud, D.; Barnich, N.; Bouckaert, J.; Gouin, S.G. Second Generation of thiazolylmannosides, FimH antagonists for E. coli-induced Crohn’s disease. Org. Biomol. Chem. 2016, 14, 3913–3925. [Google Scholar] [CrossRef] [PubMed]
- Sperling, O.; Fuchs, A.; Lindhorst, T.K. Evaluation of the carbohydrate recognition domain of the bacterial adhesin FimH: Design, synthesis and binding properties of mannoside ligands. Org. Biomol. Chem. 2006, 4, 3913. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Kleeb, S.; Lemme, K.; Rabbani, S.; Scharenberg, M.; Zalewski, A.; Schädler, F.; Schwardt, O.; Ernst, B. FimH antagonists: Structure-activity and structure-property relationships for biphenyl α-d-mannopyranosides. ChemMedChem 2012, 7, 1404–1422. [Google Scholar] [CrossRef] [PubMed]
- Grabosch, C.; Hartmann, M.; Schmidt-Lassen, J.; Lindhorst, T.K. Squaric Acid Monoamide Mannosides as Ligands for the Bacterial Lectin FimH: Covalent Inhibition or Not? ChemBioChem 2011, 12, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Eid, S.; Zalewski, A.; Smieško, M.; Ernst, B.; Vedani, A. A Molecular-Modeling Toolbox Aimed at Bridging the gap between medicinal chemistry and computational sciences. Int. J. Mol. Sci. 2013, 14, 684–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivignon, A.; Yan, X.; Alvarez Dorta, D.; Bonnet, R.; Bouckaert, J.; Fleury, E.; Bernard, J.; Gouin, S.G.; Darfeuille-Michaud, A.; Barnich, N. Development of heptylmannoside-based glycoconjugate antiadhesive compounds against adherent-invasive Escherichia coli bacteria associated with Crohn’s disease. MBio 2015, 6, e01298-15. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, J.; Li, Z.; Xavier, C.; Almant, M.; Caveliers, V.; Lahoutte, T.; Weeks, S.D.; Kovensky, J.; Gouin, S.G. Heptyl α-d-Mannosides grafted on a β-Cyclodextrin core to interfere with Escherichia coli adhesion: An in vivo multivalent effect. Chem. Eur. J. 2013, 19, 7847–7855. [Google Scholar] [CrossRef] [PubMed]
- Dumych, T.; Yamakawa, N.; Sivignon, A.; Garenaux, E.; Robakiewicz, S.; Coddeville, B.; Bongiovanni, A.; Bray, F.; Barnich, N.; Szunerits, S.; et al. Oligomannose-rich membranes of dying intestinal epithelial cells promote host colonization by adherent-invasive E. coli. Front. Microbiol. 2018, 9, 742. [Google Scholar] [CrossRef] [PubMed]
- Lonardi, E.; Moonens, K.; Buts, L.; de Boer, A.; Olsson, J.; Weiss, M.; Fabre, E.; Guérardel, Y.; Deelder, A.; Oscarson, S.; et al. Structural sampling of glycan interaction profiles reveals mucosal receptors for fimbrial adhesins of enterotoxigenic Escherichia coli. Biology 2013, 2, 894–917. [Google Scholar] [CrossRef] [PubMed]
- Almant, M.; Moreau, V.; Kovensky, J.; Bouckaert, J.; Gouin, S.G. Clustering of Escherichia coli Type-1 fimbrial adhesins by using multimeric heptyl α-d-mannoside probes with a carbohydrate core. Chem. Eur. J. 2011, 17, 10029–10038. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Bronowska, A.K.; Řezáč, J.; Přenosil, O.; Konvalinka, J.; Hobza, P. A Reliable Docking/scoring scheme based on the semiempirical quantum mechanical pm6-dh2 method accurately covering dispersion and H-bonding: HIV-1 protease with 22 ligands. J. Phys. Chem. B 2010, 114, 12666–12678. [Google Scholar] [CrossRef] [PubMed]
- Lepšík, M.; Řezáč, J.; Kolář, M.; Pecina, A.; Hobza, P.; Fanfrlík, J. The Semiempirical quantum mechanical scoring function for in silico drug design. Chempluschem 2013, 78, 921–931. [Google Scholar] [CrossRef]
- Savar, N.S.; Jahanian-Najafabadi, A.; Mahdavi, M.; Shokrgozar, M.A.; Jafari, A.; Bouzari, S. In silico and in vivo studies of Truncated Forms of Flagellin (FliC) of enteroaggregative Escherichia coli fused to FimH from uropathogenic Escherichia coli as a Vaccine candidate against urinary tract infections. J. Biotechnol. 2014, 175, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Habibi, M.; Reza, M.; Karam, A.; Bouzari, S. In silico design of fusion protein of fimh from uropathogenic Escherichia coli and mrph from proteus mirabilis against urinary tract infections. Adv. Biomed. Res. 2015, 4, 217. [Google Scholar] [CrossRef] [PubMed]
- Luna-Pineda, V.M.; Reyes-Grajeda, J.P.; Cruz-Córdova, A.; Saldaña-Ahuactzi, Z.; Ochoa, S.A.; Maldonado-Bernal, C.; Cázares-Domínguez, V.; Moreno-Fierros, L.; Arellano-Galindo, J.; Hernández-Castro, R.; et al. Dimeric and Trimeric fusion proteins generated with fimbrial adhesins of uropathogenic Escherichia coli. Front. Cell. Infect. Microbiol. 2016, 6, 135. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | Important Due to | Exp. Evidence | Insight from Molecular Simulation |
|---|---|---|---|
| Ile13 | Located in the clamp loop (changes conformation due to shear force) Possibly involved in alternative binding position | Ile13 forms van der Waals interactions with the C1–C2 bond of mannose [42] Crystal structures of the HA and LA state highlight the movement of the clamp loop [43] | The aglycon moiety of the C117 and of biantennary mannosides orients towards Ile13 [39,44]. |
| Glu50 | Part of a possible new binding site for anti-adhesives | EDTA binding site [38] Implied in the shear-force dependent conformational change [45] Less adhesion of the E50A mutant under shear [45] | |
| Ile52 | Belongs to the tyrosine gate | Attributed to the tyrosine gate on the basis of crystal structures [42] | Mediates coupled motion of Tyr48 and Tyr137 [38] |
| Thr53 | Part of a possible new binding site for anti-adhesives | EDTA binding site [38] Implied in the shear-force dependent conformational change [45] Less adhesion of the T53A mutant under shear [45] | |
| Asn136 | Part of a possible new binding site for anti-adhesives | EDTA binding site [38] | |
| Tyr137 | Belongs to the tyrosine gate Binding of the aglycon part in the mannose-binding moiety | Y137A mutation significantly reduces FimH affinity towards f HM [38] | The flexibility of the bound HM is increased in the Y137A mutant; The apo mutant already is in a quasi-bound configuration [38] |
| Thr158 | Implicated in the shear-force dependent conformational change | Natural variation leads to bacteria with different stress responses [22,46,47] | A force was applied to this residue in the sMD simulation [48] |
| (A) Different O-Linked Mannosidic Compounds | ||||||
 | ||||||
| Compound Type | Example (s) (R=) | Measure (Technique) | Value [nM] | Ref. | PDB Code | Ref. |
| Mannose | H | KD (ITC) | 1672.2 | [34] | 1KEF | [14] |
| KD (SPR) | 2300.0 | [24] | ||||
| EC90 (HAI) | >1 mM | [24] | ||||
| Alkyl mannosides |  | KD (SPR) | 5.0 | [26] | 4BUQ 4LOV 4XOE 4XOB | [31] [49] [59] [59] |
| KD (ITC) | 28.9 | [38] | ||||
| KD (ITC) | 7.3 | [34] | ||||
| KD (FDA) | 28.3 | [35] | ||||
| EC90 (HAI) | 1500.0 | [24] | ||||
| EC90 (HAI) | 6300.0 | [39] | ||||
| IC50 (ELLSA) | 160.0 | [24] | ||||
 | KD (ITC) | 23.6 | [34] | |||
| Aryl mannosides |  | KD (ITC) | 18.3 | [34] | ||
 | IC50 (Bioassay) | 1730.0 | [82] | |||
| Biaryl mannosides |  | KD (ITC) | 17.7 | [38] | 5FWR | [38] |
| KD (FPA) | 15.1 | [35] | ||||
 | KD (ITC) | 3.5 | [81] | |||
| (B) Mannose Ring Modifications | ||||||
 | ||||||
| Ring modification | Example(s) (R=) | Measure (Technique) | Value [nM] | Ref. | PDB Code | Ref. |
| N-linked compounds X = N |  | IC50 (ELLSA) | 70.0 | [32] | 5MTS | [32] |
 | IC50 (ELLSA) | 205.0 | [32] | 3LZ2 | [77] | |
| C-linked compounds X = C |  | EC90 (HAI) | 3.1 | [36] | ||
 | IC50 (ELLSA) | 194.0 | [32] | |||
| S-linked compounds X = S |  | IC50 (ELLSA) | 146.0 | [23] | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krammer, E.-M.; De Ruyck, J.; Roos, G.; Bouckaert, J.; Lensink, M.F. Targeting Dynamical Binding Processes in the Design of Non-Antibiotic Anti-Adhesives by Molecular Simulation—The Example of FimH. Molecules 2018, 23, 1641. https://doi.org/10.3390/molecules23071641
Krammer E-M, De Ruyck J, Roos G, Bouckaert J, Lensink MF. Targeting Dynamical Binding Processes in the Design of Non-Antibiotic Anti-Adhesives by Molecular Simulation—The Example of FimH. Molecules. 2018; 23(7):1641. https://doi.org/10.3390/molecules23071641
Chicago/Turabian StyleKrammer, Eva-Maria, Jerome De Ruyck, Goedele Roos, Julie Bouckaert, and Marc F. Lensink. 2018. "Targeting Dynamical Binding Processes in the Design of Non-Antibiotic Anti-Adhesives by Molecular Simulation—The Example of FimH" Molecules 23, no. 7: 1641. https://doi.org/10.3390/molecules23071641