
New Octadecanoid Enantiomers from the Whole Plants of Plantago depressa

 ,
,
Abstract
:
1. Introduction
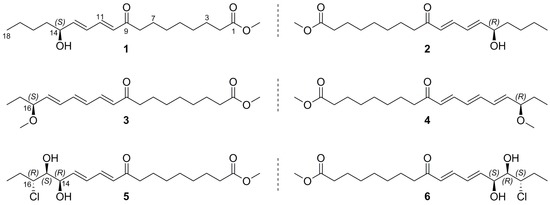
2. Results
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Antimicrobial Assay
3.5. Anti-Acetylcholinesterase Assay
3.6. Anti-Inflammatory Assay
3.7. ECD Calculations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Flora of China Editorial Committee of Chinese Academy of Sciences. The Flora of China; Science Press: Beijing, China, 2002; pp. 318–332. [Google Scholar]
- National Pharmacopoeia Committee. Pharmacopoeia of The People's Republic of China; Part 1; China Press of Traditional Chinese Medicine: Beijing, China, 2015; p. 69. [Google Scholar]
- Olennikov, D.N.; Tankhaeva, L.M.; Stolbikova, A.V.; Petrov, E.V. Phenylpropanoids and polysaccharides from Plantago depressa and P. media growing in Buryatia. Chem. Nat. Compd. 2011, 47, 165–169. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Sun, Y.-C.; Chen, G. A New Phenylethanoid glucoside from Plantago depressa Willd. Nat. Prod. Res. 2013, 27, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Nishibe, S.; Sasahara, M.; Jiao, Y.; Yuan, C.-L.; Tanaka, T. Phenylethanoid glycosides from Plantago depressa. Phytochemistry 1993, 32, 975–977. [Google Scholar] [CrossRef]
- Yan, P.-F.; Liu, G.-Y.; Zhao, S.-M.; Song, L.-L.; Tan, L.-N.; Jin, Y.-R.; Li, X.-W. Studies on chemical constituents of Plantago depressa Willd. Chin. Pharm. J. 2009, 44, 19–21. [Google Scholar]
- Zhang, Z.; Li, F.; Yuan, C.; Zheng, T.; Liu, Y. Studies on the chemical constituents of Plantago depressa var. montata. Chin. J. Med. Chem. 1996, 6, 196–197. [Google Scholar]
- Zheng, X.-M.; Meng, F.-W.; Geng, F.; Qi, M.; Luo, C.; Yang, L.; Wang, Z.-T. Plantadeprate A, a tricyclic monoterpene zwitterionic guanidium, and related derivatives from the seeds of Plantago depressa. J. Nat. Prod. 2015, 78, 2822–2826. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.-H.; Liang, J.-Y.; Chen, R.; Wang, Q.-Z. Chemical constituents and hepatoprotective activity of Plantago depressa var. montata Kitag. Chin. J. Nat. Med. 2006, 4, 435–439. [Google Scholar]
- Smith, M.A.; Zhang, H.-X.; Purves, R.W. Identification and distribution of oxygenated fatty acids in Plantago seed lipids. J. Am. Oil Chem. Soc. 2014, 91, 1313–1322. [Google Scholar] [CrossRef]
- Amakura, Y.; Kondo, K.; Akiyama, H.; Ito, H.; Hatano, T.; Yoshida, T.; Maitani, T. Conjugated ketonic fatty acids from Pleurocybella porrigens. Chem. Pharm. Bull. 2006, 54, 1213–1215. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Ishibashi, M.; Takata, T.; Takano, F.; Ohta, T. Cytotoxic fatty acid from Pleurocybella porrigens. Chem. Pharm. Bull. 2007, 55, 1748–1749. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Iwabuchi, J.; Yokota, Y.; Uda, H.; Nakanishi, K. A chiroptical method for determining the absolute configuration of allylic alcohols. J. Am. Chem. Soc. 1981, 103, 5590–5591. [Google Scholar] [CrossRef]
- Bernart, M.W.; Whatley, G.G.; Gerwick, W.H. Unprecedented oxylipins from the marine green alga Acrosiphonia coalita. J. Nat. Prod. 1993, 56, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Murakami, T.; Shimada, H.; Yoshizumi, S.; Saka, M.; Yamahara, J.; Matsuda, H. Medicinal foodstuffs. Xiv. on the bioactive constituents of moroheiya. (2): New fatty acids, corchorifatty acids A, B, C, D, E, and F, from the leaves of Corchorus olitorius L. (Tiliaceae): Structures and inhibitory effect on No production in mouse periton. Chem. Pharm. Bull. 1998, 46, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, R.; Liu, Y.-X.; Paul, V.J.; Luesch, H. Cultivated sea lettuce is a multiorgan protector from oxidative and inflammatory stress by enhancing the endogenous antioxidant defense system. Cancer Prev. Res. 2013, 6, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, C.; Rogers, H.J.; Thomas, B.; Feussner, I.; Griffiths, G. Characterization of a novel lipoxygenase-independent senescence mechanism in Alstroemeria peruviana floral tissue. Plant Physiol. 2002, 130, 273–283. [Google Scholar]
- Liu, H.-Y.; Liang, J.-U.; Chen, R.; Juan, Y. Study on chemical constituents of (Hemsl.). Hara Herb. Chem. Ind. For. Prod. 2008, 28, 8–12. [Google Scholar]
- Marwah, R.G.; Fatope, M.O.; Deadman, M.L.; Al-Maqbali, Y.M.; Husband, J. Musanahol: A new aureonitol-related metabolite from a Chaetomium sp. Tetrahedron 2007, 63, 8174–8180. [Google Scholar] [CrossRef]
- Ogihara, T.; Amano, N.; Mitsui, Y.; Fujino, K.; Ohta, H.; Takahashi, K.; Matsuura, H. Determination of the absolute configuration of a monoglyceride antibolting compound and isolation of related compounds from radish leaves (Raphanus sativus). J. Nat. Prod. 2017, 80, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Katayama, M.; Marumo, S. (-)-R-Glycerol monolinolate, a minor sporogenic substance of Sclerotinia fructicola. Agric. Biol. Chem. 1978, 42, 1431–1433. [Google Scholar] [CrossRef]
- Kim, K.H.; Moon, E.; Sun, Y.K.; Kang, R.L. Antimelanogenic fatty acid derivatives from the Tuber-barks of Colocasia antiquorum var. esculenta. Bull. Korean Chem. Soc. 2010, 31, 2051–2053. [Google Scholar] [CrossRef]
- Sato, T.; Morita, I.; Shimizu, M.; Seo, S.; Watanabe, M.; Uchida, M.; Ono, M. Platelet Production Promoting Factor and Use Thereof. WO Patent WO 2008/078453 A1, 3 July 2008. [Google Scholar]
- Wang, P.; Yu, J.-H.; Zhu, K.-K.; Wang, Y.; Cheng, Z.-Q.; Jiang, C.-S.; Dai, J.-G.; Wu, J.; Zhang, H. Phenolic bisabolane sesquiterpenoids from a Thai mangrove endophytic fungus, Aspergillus sp. xy02. Fitoterapia 2018, 127, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-C.; Zhang, J.; Rodrigues, M.C.; Ding, D.-J.; Longo, J.P.; Azevedo, R.B.; Muehlmann, L.A.; Jiang, C.-S. Synthesis and evaluation of novel 1,2,3-triazole-based acetylcholinesterase inhibitors with neuroprotective activity. Bioorg. Med. Chem. Lett. 2016, 26, 3881–3885. [Google Scholar] [CrossRef] [PubMed]
- OPLS3. Schrödinger, Inc.: New York, NY, USA, 2013. Available online: https://www.schrodinger.com/opls3 (accessed on 20 June 2018).
- Shivakumar, D.; Harder, E.; Damm, W.; Friesner, R.A.; Sherman, W. Improving the prediction of absolute solvation free energies using the Next Generation OPLS force field. J. Chem. Theory Comput. 2012, 8, 2553–2558. [Google Scholar] [CrossRef] [PubMed]
- MacroModel; Schrödinger, LLC: New York, NY, USA, 2009.
- Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010.
Sample Availability: Samples are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1/2 a | 3/4 a | 5/6 b | 9 b |
|---|---|---|---|---|
| 2 | 2.32, t (7.4) | 2.31, t (7.4) | 2.29, t (7.6) | 2.30, t (7.5) |
| 3 | 1.60, m | 1.61, m | 1.61, m | 1.62, m |
| 4 | 1.34, m | 1.33, m | 1.32, m | 1.32, m |
| 5 | 1.34, m | 1.33, m | 1.32, m | 1.32, m |
| 6 | 1.34, m | 1.33, m | 1.32, m | 1.32, m |
| 7 | 1.60, m | 1.61, m | 1.61, m | 1.61, m |
| 8 | 2.61, t (7.3) | 2.61, t (7.4) | 2.55, t (7.5) | 2.55, t (7.4) |
| 10 | 6.20, d (15.6) | 6.23, d (15.5) | 6.20, d (15.6) | 6.21, d (15.5) |
| 11 | 7.27, dd (15.6, 10.8) | 7.31, dd (15.5, 11.2) | 7.15, dd (15.6, 10.8) | 7.16, dd (15.6, 11.0) |
| 12 | 6.41, dd (15.3, 10.8) | 6.43, dd (15.1, 11.2) | 6.49, dd (15.5, 10.8) | 6.50, dd (15.3, 11.0) |
| 13 | 6.25, dd (15.3, 5.9) | 6.75, dd (15.1, 10.9) | 6.21, dd (15.5, 6.0) | 6.25, dd (15.3, 6.8) |
| 14 | 4.17, m | 6.39, dd (15.2, 10.9) | 4.70, dd (6.0, 2.7) | 4.62, dd (6.8, 5.1) |
| 15 | 1.55, m | 5.81, dd (15.2, 7.8) | 3.62, dd (7.6, 2.7) | 3.86, dd (7.7, 5.1) |
| 16 | 1.34, m | 3.62, m | 3.97, ddd (9.3, 7.6, 2.9) | 3.76, ddd (9.4, 7.7, 2.8) |
| 17 | 1.37, m | 1.61, m | 1.76, m | 1.74, m |
| 2.07, m | 2.08, m | |||
| 18 | 0.92, t (7.4) | 0.90, t (7.4) | 1.08, t (7.3) | 1.07, t (7.2) |
| 1-OMe | 3.65, s | 3.65, s | 3.67, s | 3.67, s |
| 16-OMe | 3.28, s |
| Position | 1/2 a | 3/4 a | 5/6 b | 9 b |
|---|---|---|---|---|
| 1 | 176.0 | 176.0 | 174.5 | 174.5 |
| 2 | 34.8 | 34.8 | 34.2 | 34.2 |
| 3 | 26.0 | 26.0 | 25.0 | 25.0 |
| 4 | 30.0 c | 30.0 d | 29.1 e | 29.1 f |
| 5 | 30.1 c | 30.2 d | 29.2 e | 29.2 f |
| 6 | 30.2 c | 30.2 d | 29.2 e | 29.2 f |
| 7 | 25.5 | 25.5 | 24.3 | 24.3 |
| 8 | 41.0 | 41.1 | 40.8 | 40.8 |
| 9 | 203.7 | 203.5 | 201.0 | 200.9 |
| 10 | 130.3 | 130.5 | 130.6 | 130.9 |
| 11 | 144.3 | 144.5 | 141.1 | 141.0 |
| 12 | 128.8 | 131.9 | 130.3 | 131.5 |
| 13 | 148.5 | 142.3 | 141.7 | 139.4 |
| 14 | 72.6 | 133.2 | 71.0 | 72.5 |
| 15 | 37.8 | 140.2 | 76.5 | 76.6 |
| 16 | 28.7 | 84.5 | 64.6 | 64.7 |
| 17 | 23.7 | 29.2 | 26.8 | 26.6 |
| 18 | 14.4 | 9.9 | 10.7 | 10.6 |
| 1-OMe | 52.0 | 52.0 | 51.7 | 51.6 |
| 16-OMe | 56.7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, X.-Q.; Zhu, K.; Yu, J.-H.; Zhang, Q.; Zhang, Y.; He, F.; Cheng, Z.-Q.; Jiang, C.-S.; Bao, J.; Zhang, H. New Octadecanoid Enantiomers from the Whole Plants of Plantago depressa. Molecules 2018, 23, 1723. https://doi.org/10.3390/molecules23071723
Song X-Q, Zhu K, Yu J-H, Zhang Q, Zhang Y, He F, Cheng Z-Q, Jiang C-S, Bao J, Zhang H. New Octadecanoid Enantiomers from the Whole Plants of Plantago depressa. Molecules. 2018; 23(7):1723. https://doi.org/10.3390/molecules23071723
Chicago/Turabian StyleSong, Xiu-Qing, Kongkai Zhu, Jin-Hai Yu, Qianqian Zhang, Yuying Zhang, Fei He, Zhi-Qiang Cheng, Cheng-Shi Jiang, Jie Bao, and Hua Zhang. 2018. "New Octadecanoid Enantiomers from the Whole Plants of Plantago depressa" Molecules 23, no. 7: 1723. https://doi.org/10.3390/molecules23071723