Truly Target-Focused Pharmacophore Modeling: A Novel Tool for Mapping Intermolecular Surfaces


Abstract
:1. Introduction
2. Materials and Methods
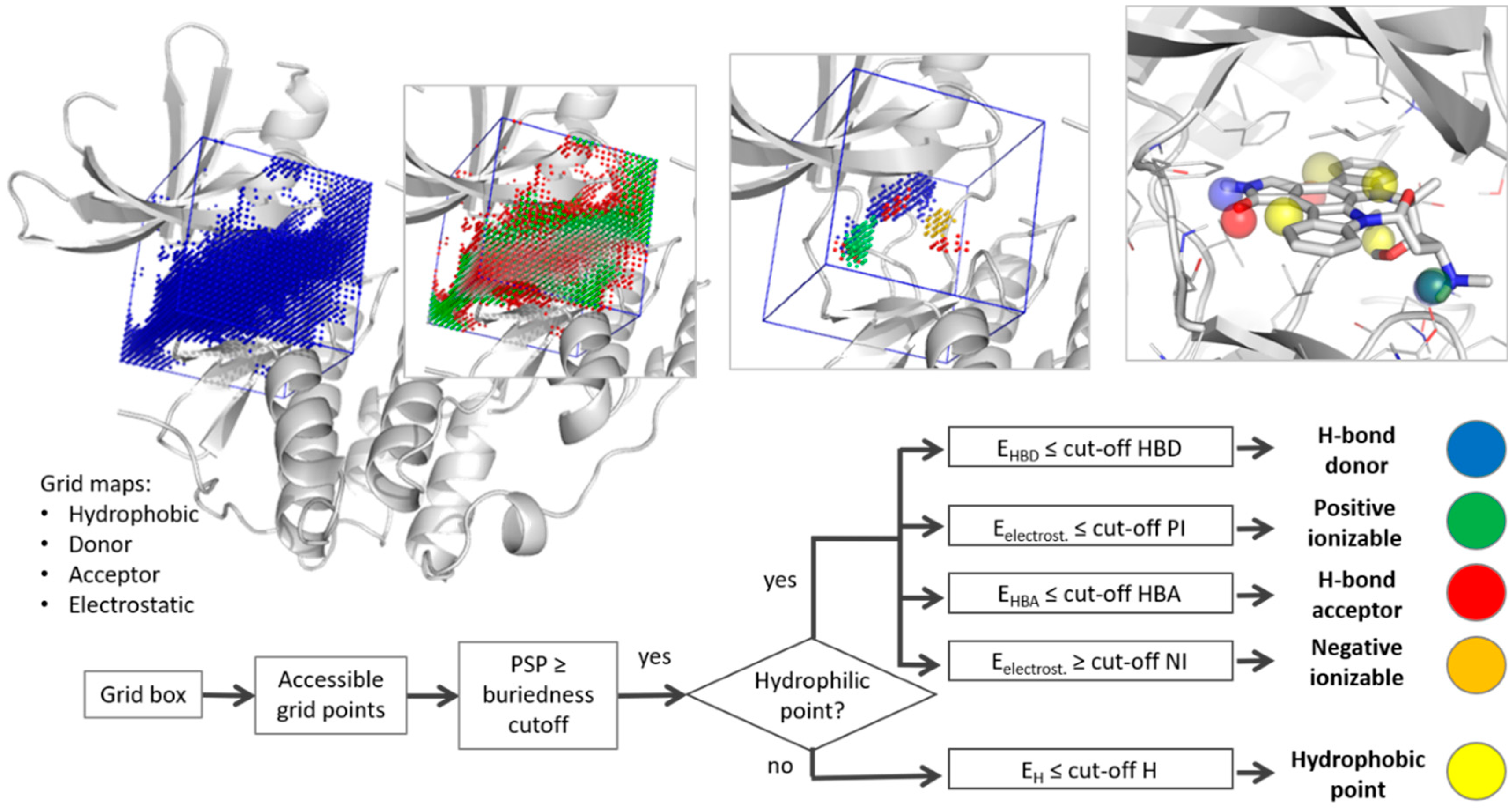
2.1. Pharmacophore Generation
2.2. Evaluation Data Sets
3. Results and Discussion
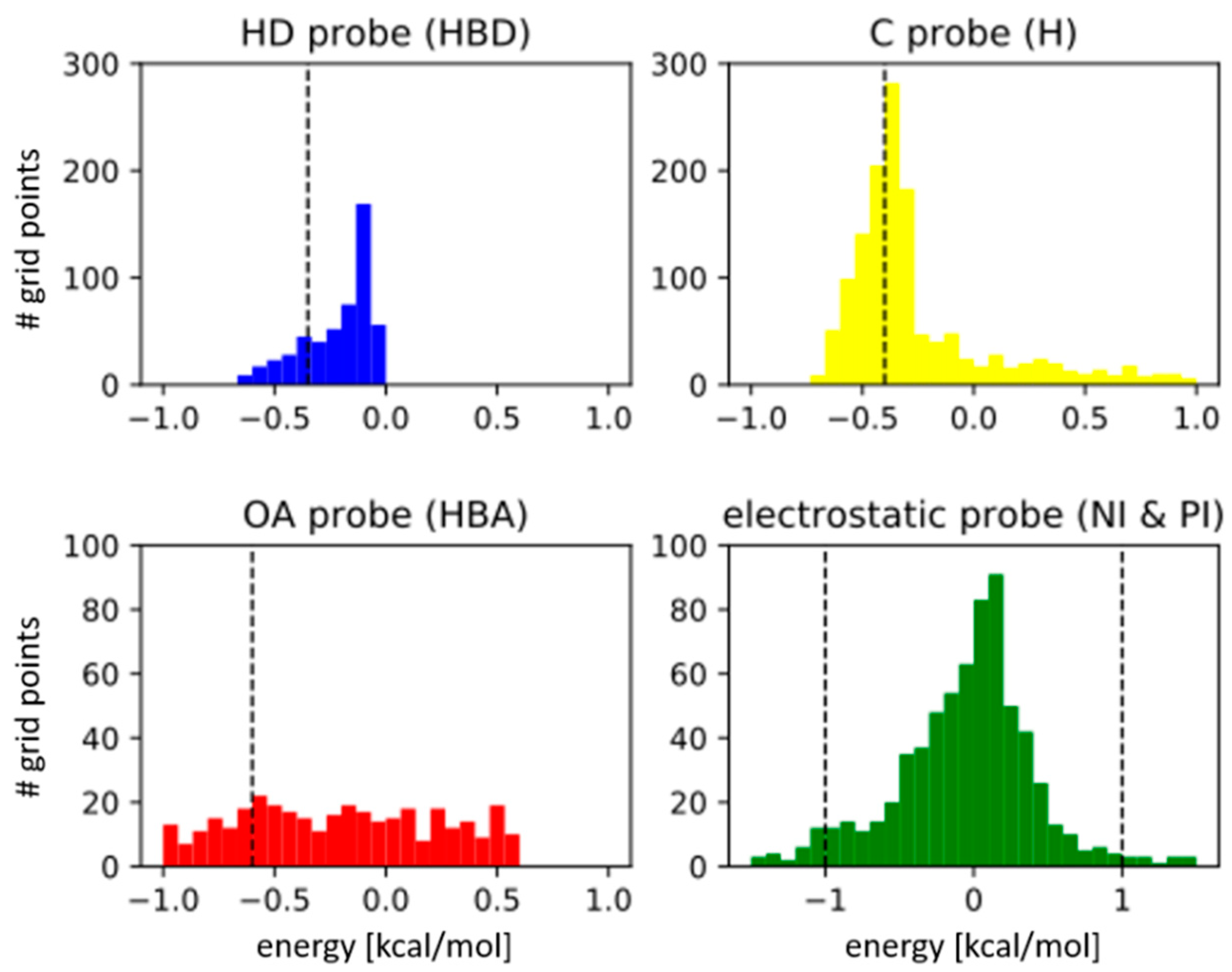
3.1. Parameter Selection
3.2. Evaluation
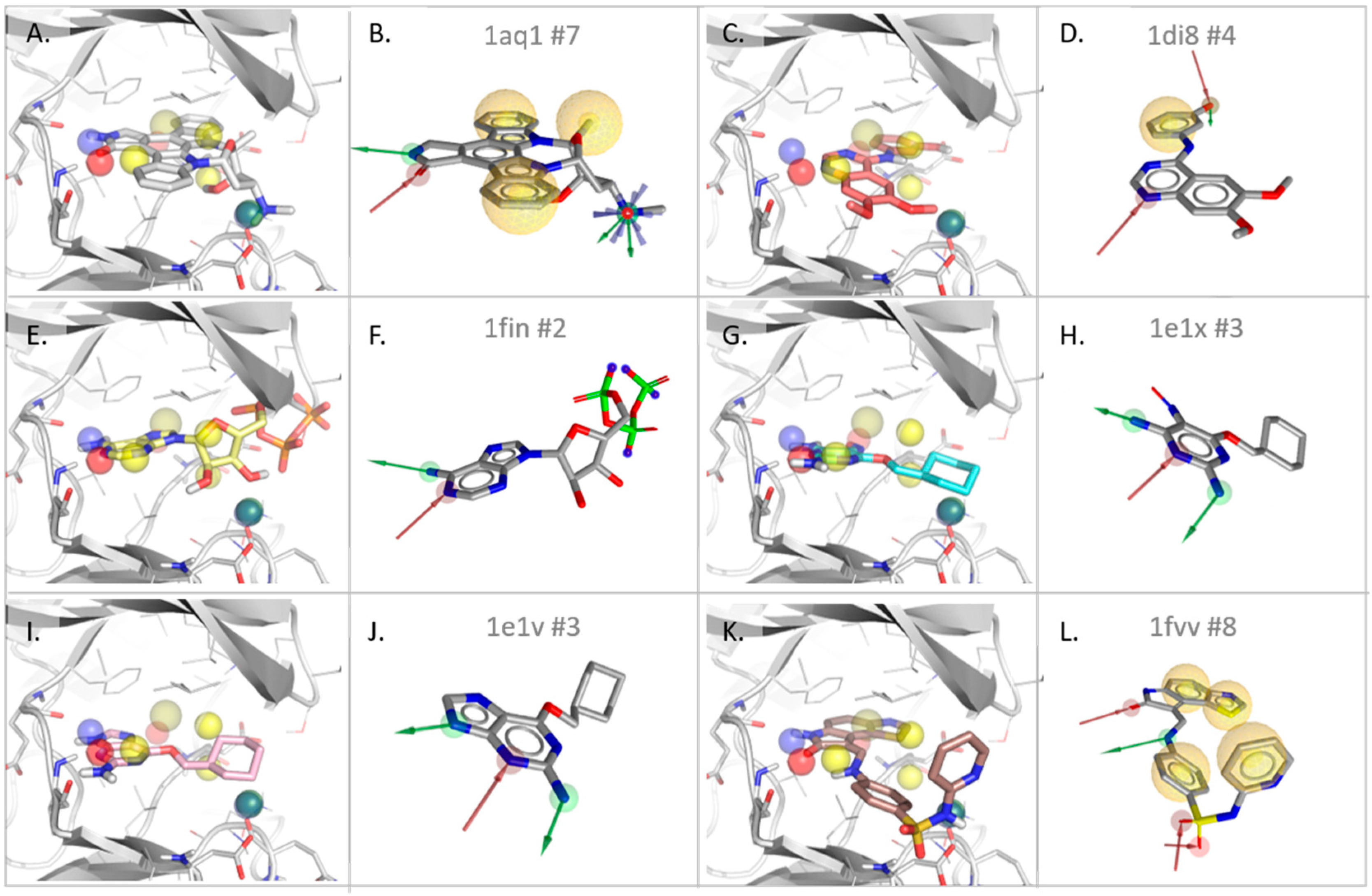
3.2.1. Cyclin-Dependent Kinase
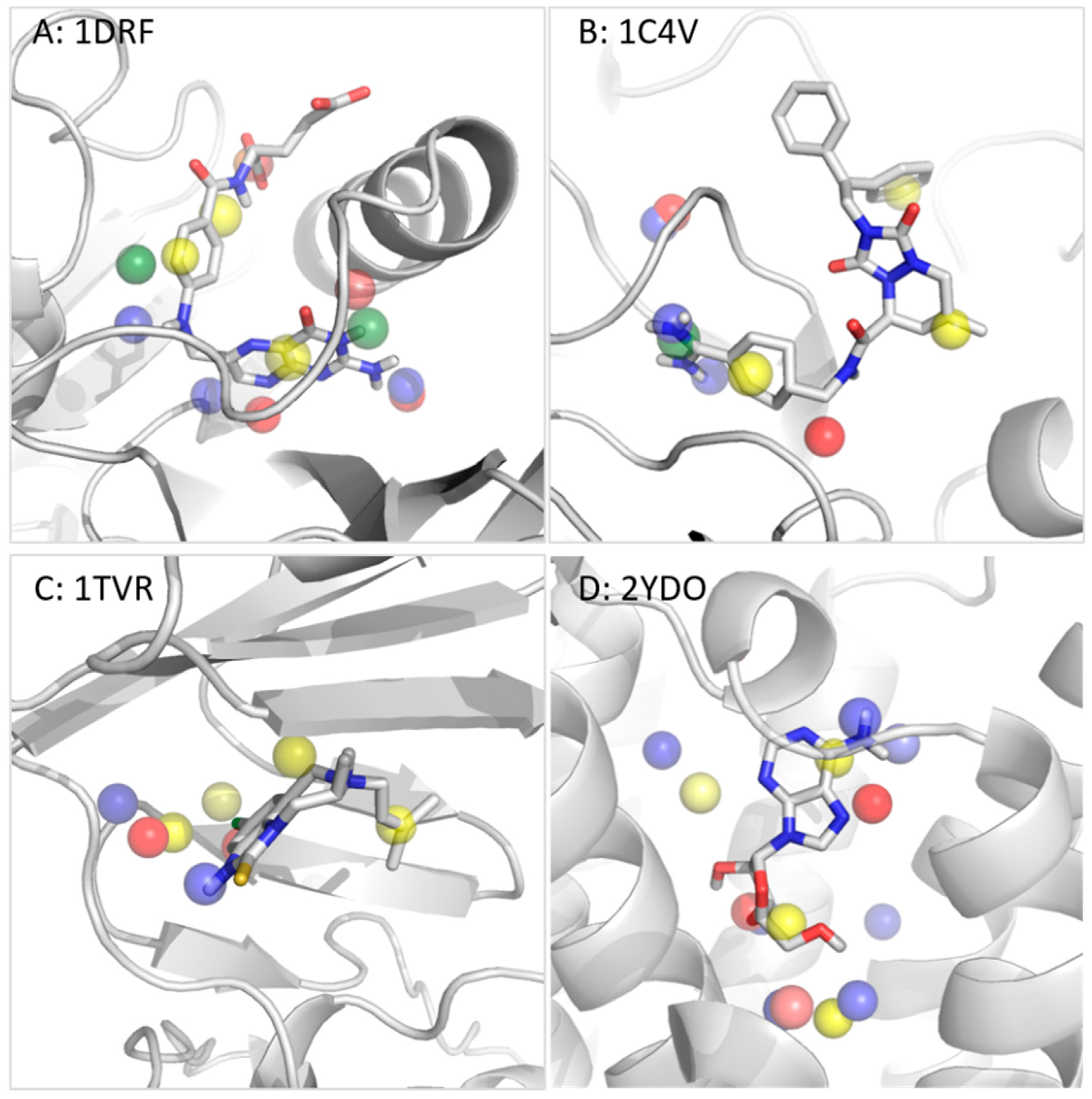
3.2.2. Dihydrofolate Reductase
3.2.3. Thrombin
3.2.4. Reverse Transcriptase
3.2.5. Adenosine A2A Receptor
3.2.6. Sensitivity to Conformational Changes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gund, P. Three-dimensional pharmacophoric pattern searching. In Progress in Molecular and Subcellular Biology; Springer: Berlin, Germany, 1977; pp. 117–143. [Google Scholar]
- Wolber, G.; Seidel, T.; Bendix, F.; Langer, T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov. Today 2008, 13, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Al-Asri, J.; Gyemant, G.; Fazekas, E.; Lehoczki, G.; Melzig, M.F.; Wolber, G.; Mortier, J. α-Amylase Modulation: Discovery of Inhibitors Using a Multi-Pharmacophore Approach for Virtual Screening. ChemMedChem 2016, 21, 2372–2377. [Google Scholar] [CrossRef] [PubMed]
- Al-Asri, J.; Fazekas, E.; Lehoczki, G.; Perdih, A.; Gorick, C.; Melzig, M.F.; Gyemant, G.; Wolber, G.; Mortier, J. From carbohydrates to drug-like fragments: Rational development of novel α-amylase inhibitors. Bioorg. Med. Chem. 2015, 23, 6725–6732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Houri, R.B.; Mortier, J.; Murgueitio, M.S.; Wolber, G.; Christensen, L.P. Identification of PPARgamma Agonists from Natural Sources Using Different In Silico Approaches. Planta Med. 2015, 81, 488–494. [Google Scholar] [PubMed]
- Bock, A.; Bermudez, M.; Krebs, F.; Matera, C.; Chirinda, B.; Sydow, D.; Dallanoce, C.; Holzgrabe, U.; De Amici, M.; Lohse, M.J.; et al. Ligand Binding Ensembles Determine Graded Agonist Efficacies at a G Protein-coupled Receptor. J. Biol. Chem. 2016, 291, 16375–16389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortier, J.; Prevost, J.R.C.; Sydow, D.; Teuchert, S.; Omieczynski, C.; Bermudez, M.; Frederick, R.; Wolber, G. Arginase Structure and Inhibition: Catalytic Site Plasticity Reveals New Modulation Possibilities. Sci. Rep. 2017, 7, 13616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murgueitio, M.S.; Bermudez, M.; Mortier, J.; Wolber, G. In silico virtual screening approaches for anti-viral drug discovery. Drug Discov. Today Technol. 2012, 9, e219–e225. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, G.M.; Heiss, M.; Mangold, M.; Markt, P.; Kirchmair, J.; Wolber, G.; Liedl, K.R. One concept, three implementations of 3D pharmacophore-based virtual screening: Distinct coverage of chemical search space. J. Chem. Inf. Model. 2010, 50, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.P.A.; McGuire, R.; Roumen, L.; de Esch, I.J.P.; de Vlieg, J.; Klomp, J.P.G.; de Graaf, C. From the protein’s perspective: The benefits and challenges of protein structure-based pharmacophore modeling. MedChemComm 2012, 3, 28–38. [Google Scholar] [CrossRef]
- Sanders, M.P.; Verhoeven, S.; de Graaf, C.; Roumen, L.; Vroling, B.; Nabuurs, S.B.; de Vlieg, J.; Klomp, J.P. Snooker: A structure-based pharmacophore generation tool applied to class A GPCRs. J. Chem. Inf. Model. 2011, 51, 2277–2292. [Google Scholar] [CrossRef] [PubMed]
- Klabunde, T.; Giegerich, C.; Evers, A. Sequence-derived three-dimensional pharmacophore models for G-protein-coupled receptors and their application in virtual screening. J. Med. Chem. 2009, 52, 2923–2932. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, N.A.; Malherbe, P.; Lindemann, L.; Ebeling, M.; Hoener, M.C.; Muhlemann, A.; Porter, R.H.; Stahl, M.; Gerber, P.R. An automated system for the analysis of G protein-coupled receptor transmembrane binding pockets: Alignment, receptor-based pharmacophores, and their application. J. Chem. Inf. Model. 2005, 45, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Miranker, A.; Karplus, M. Functionality maps of binding sites: A multiple copy simultaneous search method. Proteins 1991, 11, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Meagher, K.L.; Carlson, H.A. Incorporating protein flexibility in structure-based drug discovery: Using HIV-1 protease as a test case. J. Am. Chem. Soc. 2004, 126, 13276–13281. [Google Scholar] [CrossRef] [PubMed]
- Ghanakota, P.; Carlson, H.A. Driving Structure-Based Drug Discovery through Cosolvent Molecular Dynamics. J. Med. Chem. 2016, 59, 10383–10399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Goto, J.; Kataoka, R.; Hirayama, N. Ph4Dock: Pharmacophore-based protein-ligand docking. J. Med. Chem. 2004, 47, 6804–6811. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lai, L. Pocket v.2: Further developments on receptor-based pharmacophore modeling. J. Chem. Inf. Model. 2006, 46, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Ortuso, F.; Langer, T.; Alcaro, S. GBPM: GRID-based pharmacophore model: Concept and application studies to protein-protein recognition. Bioinformatics 2006, 22, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Tintori, C.; Corradi, V.; Magnani, M.; Manetti, F.; Botta, M. Targets looking for drugs: A multistep computational protocol for the development of structure-based pharmacophores and their applications for hit discovery. J. Chem. Inf. Model. 2008, 48, 2166–2179. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Lill, M.A. Protein pharmacophore selection using hydration-site analysis. J. Chem. Inf. Model. 2012, 52, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): Theory and application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Lill, M.A. Exploring the potential of protein-based pharmacophore models in ligand pose prediction and ranking. J. Chem. Inf. Model. 2013, 53, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.; Mortenson, P.N.; Murray, C.W. Diverse, high-quality test set for the validation of protein-ligand docking performance. J. Med. Chem. 2007, 50, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, L.; Lai, L.; Tang, Y. SCORE: A new empirical method for estimating the binding affinity of a protein-ligand complex. J. Mol. Model. 1998, 4, 379–394. [Google Scholar] [CrossRef]
- Patel, Y.; Gillet, V.J.; Bravi, G.; Leach, A.R. A comparison of the pharmacophore identification programs: Catalyst, DISCO and GASP. J. Comput. Aided Mol. Des. 2002, 16, 653–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, L.; Cross, S.; Baroni, M.; Goracci, L.; Cruciani, G. BioGPS: Navigating biological space to predict polypharmacology, off-targeting, and selectivity. Proteins 2015, 83, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Lill, M.A. PharmDock: A pharmacophore-based docking program. J. Cheminform. 2014, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, O.; Keller, B.G. Density-based cluster algorithms for the identification of core sets. J. Chem. Phys. 2016, 145, 164104. [Google Scholar] [CrossRef] [PubMed]
- Edelsbrunner, H.; Facello, M.; Fu, P.; Liang, J. Measuring proteins and voids in proteins. In Proceedings of the Twenty-Eighth Hawaii International Conference on System Sciences, Wailea, HI, USA, 3–6 January 1995; IEEE: Piscataway, NJ, USA, 1995; pp. 256–264. [Google Scholar]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Rarey, M. Exploiting structural information for drug-target assessment. Future Med. Chem. 2014, 6, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Everitt, B. Cluster Analysis, 5th ed.; Wiley: Chichester, UK, 2011. [Google Scholar]
- Lemke, O.; Keller, B.G. Common Nearest Neighbor Clustering. Available online: https://github.com/BDGSoftware/CNNClustering (accessed on 21 November 2017).
- Keller, B.; Daura, X.; van Gunsteren, W.F. Comparing geometric and kinetic cluster algorithms for molecular simulation data. J. Chem. Phys. 2010, 132, 074110. [Google Scholar] [CrossRef] [PubMed]
- Lemke, O.; Keller, B. Common Nearest Neighbor Clustering—A Benchmark. Algorithms 2018, 11, 19. [Google Scholar] [CrossRef]
- Cross, S.; Baroni, M.; Goracci, L.; Cruciani, G. GRID-based three-dimensional pharmacophores I: FLAPpharm, a novel approach for pharmacophore elucidation. J. Chem. Inf. Model. 2012, 52, 2587–2598. [Google Scholar] [CrossRef] [PubMed]
- Bietz, S.; Urbaczek, S.; Schulz, B.; Rarey, M. Protoss: A holistic approach to predict tautomers and protonation states in protein-ligand complexes. J. Cheminform. 2014, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Hendlich, M.; Rippmann, F.; Barnickel, G. LIGSITE: Automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graph. Model. 1997, 15, 359–363. [Google Scholar] [CrossRef]
- Schrodinger, L.L.C. The PyMOL Molecular Graphics System, Version 1.8; DeLano Scientific: San Carlos, CA, USA, 2015. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Egner, U.; Hillig, R.C. A structural biology view of target drugability. Expert Opin. Drug Discov. 2008, 3, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G. Wirkstoffdesign: Entwurf und Wirkung von Arzneistoffen; Springer: Berlin, Germany, 2009. [Google Scholar]
- Mortier, J.; Masereel, B.; Remouchamps, C.; Ganeff, C.; Piette, J.; Frederick, R. NF-kappaB inducing kinase (NIK) inhibitors: Identification of new scaffolds using virtual screening. Bioorg. Med. Chem. Lett. 2010, 20, 4515–4520. [Google Scholar] [CrossRef] [PubMed]
- Mortier, J.; Frederick, R.; Ganeff, C.; Remouchamps, C.; Talaga, P.; Pochet, L.; Wouters, J.; Piette, J.; Dejardin, E.; Masereel, B. Pyrazolo[4,3-c]isoquinolines as potential inhibitors of NF-kappaB activation. Biochem. Pharmacol. 2010, 79, 1462–1472. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, A.J.; Volkamer, A. Chapter Six—Kinase-Centric Computational Drug Development. In Annual Reports in Medicinal Chemistry; Goodnow, R.A., Ed.; Academic Press: Cambridge, MA, USA, 2017; Volume 50, pp. 197–236. [Google Scholar]
- Bermudez, M.; Mortier, J.; Rakers, C.; Sydow, D.; Wolber, G. More than a look into a crystal ball: Protein structure elucidation guided by molecular dynamics simulations. Drug Discov. Today 2016, 21, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Christin, R.; Marcel, B.; Keller, B.G.; Jérémie, M.; Gerhard, W. Computational close up on protein–protein interactions: How to unravel the invisible using molecular dynamics simulations? Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 345–359. [Google Scholar]
- Mortier, J.; Rakers, C.; Bermudez, M.; Murgueitio, M.S.; Riniker, S.; Wolber, G. The impact of molecular dynamics on drug design: Applications for the characterization of ligand-macromolecule complexes. Drug Discov. Today 2015, 20, 686–702. [Google Scholar] [CrossRef] [PubMed]
- Tautermann, C.S. GPCR structures in drug design, emerging opportunities with new structures. Bioorg. Med. Chem. Lett. 2014, 24, 4073–4079. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Gao, Z.-G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Cavity Definition | Approach | Clustering Method | Evaluation | Year | Refs. |
|---|---|---|---|---|---|---|
| Ph4Dock | cavity detection (Delaunay triangulation/α spheres) | electrostatic interactions (MMFF94 [26]) of charged dummy atoms | single-linkage | CCDC/Astex valida-tion set [27] d | 2004 | [18] |
| Pocket V2 | grid box around ligand (or user-defined pocket residues) | grid (Score) [28] | unclear clustering method a | CDK2, HIV1-PR, ER, 17b-HSD | 2006 | [19] |
| FLAP + BioGPS | grid box around ligand or FLAPsite detection | grid (GRID software) [17] | region-based energy minima | Patel set [29], DUD [30] d | 2007 | [23,31] |
| Tintori et al. | grid box around binding site | grid (GRID software) [17] | no clustering b (GRID minima + interpolation) | TrxR (MTB), HIV1 IN, HIV-1 RT dimer | 2008 | [21] |
| Hydro-Pharm | grid box around ligand (3 Å) | grid (ChemScore [25]) + MD-based hydration site feature reduction c | k-means | HIV1-PR, DHFR, FXa | 2012 | [22] |
| PharmDock | grid box around bound ligand (3 Å) | grid (ChemScore [25]) | k-means | PDB bind, DUD [30] d | 2014 | [24,32] |
| T2F-Pharm | grid box around ligand or user-defined center (& cavity point reduction) | grid (AutoDock) [33] | CNN [34] | Patel set [29] + A2A receptor | 2018 | This paper |
| Group | Reference Structure (Ligand) | Water | Others Structures |
|---|---|---|---|
| Cyclin-dependent kinase 2 (CDK2) | 1AQ1 (STU) | - | 1E1X, 1FVV, 1DI8, 1E1V, 1FIN |
| Dihydrofolate reductase (DHFR) | 1DRF (FOL) | - | 1BOZ, 1DLR, 2DHF, 1OHK, 1HFP |
| Thrombin | 1C4V (IH2) | HOH 404, 408, 410 and 477 | 1D4P, 1D6W, 1D9I, 1DWD, 1TOM, 1FPC |
| HIV-reverse transcriptase (RT) | 1TVR (TB9) | - | 1DTT, 1EP4, 1FK9, 1RT1, 1RT3, 1VRU, 1RT5, 1KLM, 1BMQ |
| A2A receptor | 2DYO (ADN) | - | 2YDV, 3EML |
| Grid box | Center | Ligand CoM * or center coordinates |
| Size of the edge of the cubic box | 16 Å | |
| Distance between two grid points | 0.6 Å | |
| Cavity | Occupancy | 0.6 kcal/mol |
| Buriedness (PSP) | 4 | |
| Feature type | Hydrophilic radius | 3 Å |
| Type specific energy cut-off ** | Hydrophobic (H) | −0.4 kcal/mol (−0.6) |
| H-bond donor (HBD) | −0.35 kcal/mol (−0.3) | |
| H-bond acceptor (HBA) | −0.6 kcal/mol (−0.5) | |
| Negative/Positive ionizable (NI/PI) | ±1.0 kcal/mol | |
| Clustering | Neighbor distance cut-off | 1.21 Å |
| Number of common neighbors | 6 (12, 16) *** | |
| Min. number of points per cluster | 15 |
| Type | Dist ** | Freq *** | 1AQ1 | 1DI8 | 1FIN | 1E1V | 1E1X | 1FVV |
|---|---|---|---|---|---|---|---|---|
| #match * | - | - | 4/7 | 2/4 | 2/2 | 2/3 | 2/3 | 3/8 |
| rmsd **** | 0.94 | 1.59 | 0.50 | 0.69 | 0.96 | 1.18 | ||
| HBD | 0.56 | 4 | X | X | X | X | ||
| HBA | 0.45 | 6 | X | X | X | X | X | X |
| HBD | 1.00 | 1 | X | |||||
| PI | 1.17 | 1 | X | |||||
| H | 1.39 | 2 | X (2.1 Å) | X | 2 * X | |||
| H | X (2.7 Å) | Slightly shifted front pocket H feature | ||||||
| H | Surrounding of Leu134 and Ala144 | |||||||
| H | Not detected in SB models | |||||||
| HBA | HBA towards back pocket water channel (ASP 145, backbone NH) | |||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mortier, J.; Dhakal, P.; Volkamer, A. Truly Target-Focused Pharmacophore Modeling: A Novel Tool for Mapping Intermolecular Surfaces. Molecules 2018, 23, 1959. https://doi.org/10.3390/molecules23081959
Mortier J, Dhakal P, Volkamer A. Truly Target-Focused Pharmacophore Modeling: A Novel Tool for Mapping Intermolecular Surfaces. Molecules. 2018; 23(8):1959. https://doi.org/10.3390/molecules23081959
Chicago/Turabian StyleMortier, Jérémie, Pratik Dhakal, and Andrea Volkamer. 2018. "Truly Target-Focused Pharmacophore Modeling: A Novel Tool for Mapping Intermolecular Surfaces" Molecules 23, no. 8: 1959. https://doi.org/10.3390/molecules23081959
APA StyleMortier, J., Dhakal, P., & Volkamer, A. (2018). Truly Target-Focused Pharmacophore Modeling: A Novel Tool for Mapping Intermolecular Surfaces. Molecules, 23(8), 1959. https://doi.org/10.3390/molecules23081959