Novel Sophoridine Derivatives Bearing Phosphoramide Mustard Moiety Exhibit Potent Antitumor Activities In Vitro and In Vivo

Abstract
:
1. Introduction
2. Results
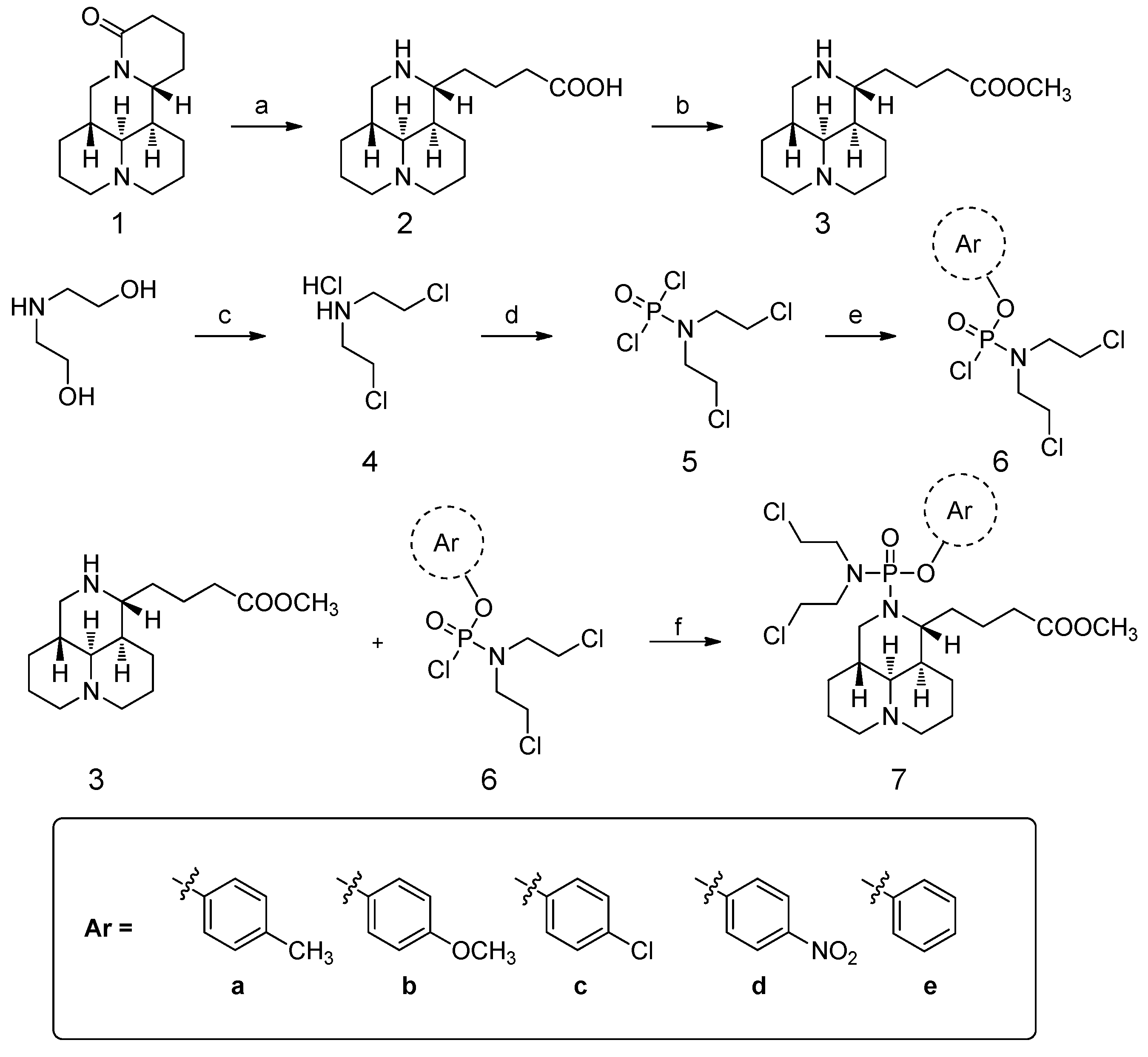
2.1. Chemistry
2.2. In Vitro Activity
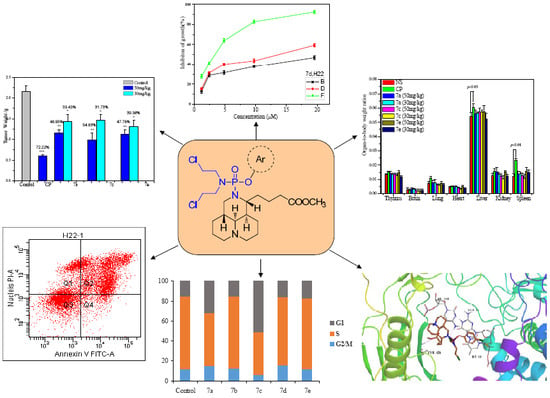
2.3. In Vivo Activity
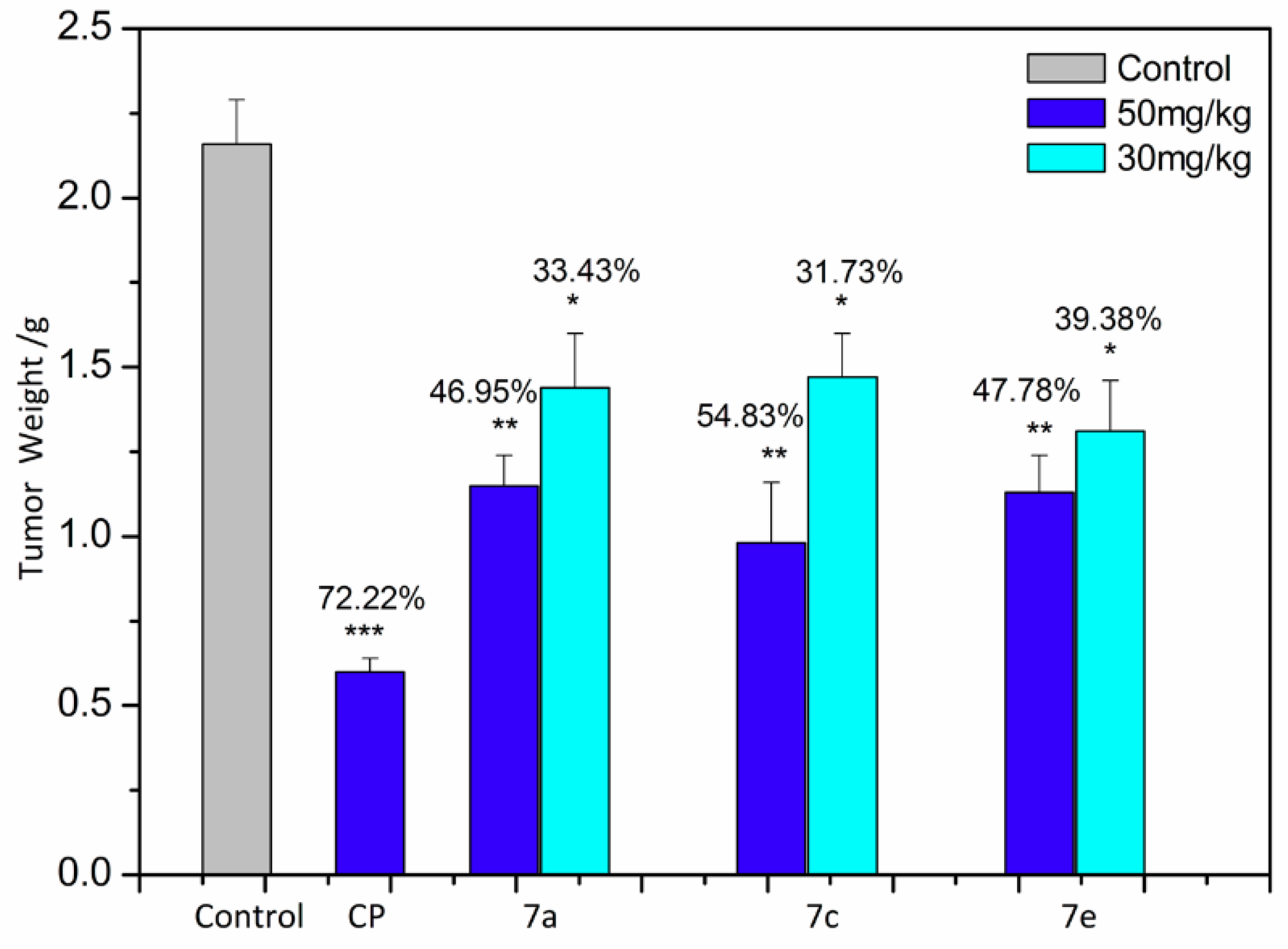
2.4. Apoptosis Analysis
2.5. Cell Cycle Analysis
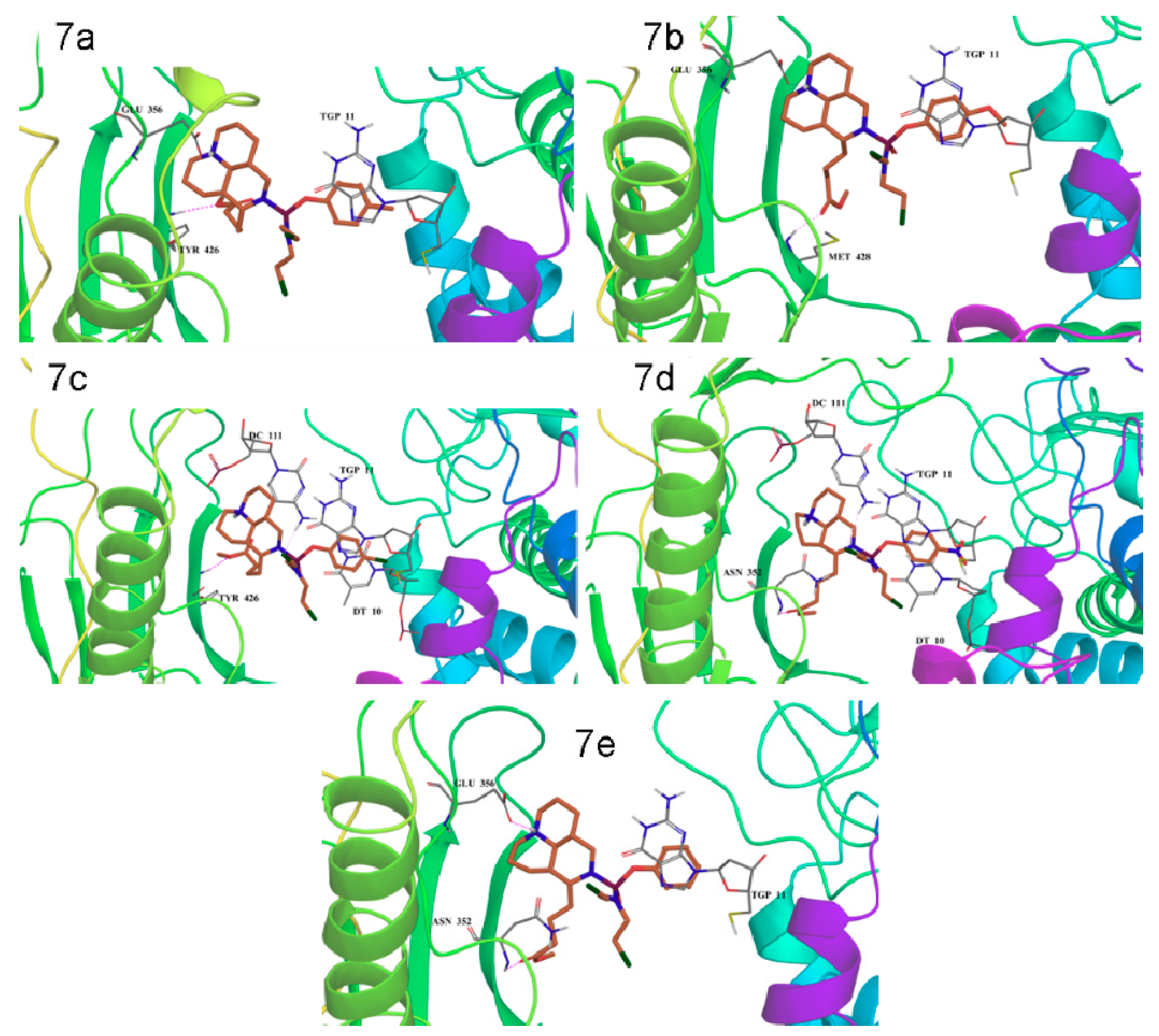
2.6. Molecule Docking
3. Materials and Methods
3.1. Instrumentation
3.2. Materials
3.3. Synthesis of Compounds
3.3.1. Synthesis of 2
3.3.2. Synthesis of 3
3.3.3. Synthesis of 4
3.3.4. Synthesis of 5
3.3.5. Synthesis of 6a–6e
3.3.6. Synthesis of 7a–7e
3.4. Anti-Proliferation Assay
3.5. Measurement of Apoptosis by Annexin V Analysis
3.6. Cell Cycle Analysis
3.7. Molecular Docking
3.8. Antitumor Activity In Vivo
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Newman, D.J.; Gragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Wu, Y.G.; Pan, D.X.; Wu, L.K.; Yu, Y.H.; Zhang, A.H.; Chen, S.L.; Guan, Z.Z.; Yang, X.Y. Sophoridine is a new antitumor medicine with new molecular structure. Chin. J. New Drugs 2006, 8, 654–657. [Google Scholar]
- Zhao, B.; Wang, Q.; Liu, J.; Xiong, W.M.; Li, Y.J.; Xie, X.L.; Xie, C.L. An Anticancer Drug Combination. CN103191107A, 10 July 2013. [Google Scholar]
- Liu, J.; Liu, Y. Influence of erbanxiao solution on inhibiting angiogenesis in stasis toxin stagnation of non-small cell lung cancer. J. Tradit. Chin. Med. 2013, 33, 303–306. [Google Scholar] [CrossRef]
- Zhuang, H.F.; Ren, J.X.; Zhou, Y.H.; Chen, X.Y.; Wu, Y.F. Matrine injection combined with intrapleural cisplatin in treatment of 24 patients with hematologic malignancies complicated by pleural effusion. Chin. J. New Drugs 2012, 21, 1013–1015. [Google Scholar]
- Wang, C.Y.; Bai, X.Y.; Wang, C.H. Traditional Chinese medicine: A treasured natural resource of anticancer drug research and development. Am. J. Chin. Med. 2014, 42, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.Z.; Ma, W.L.; Gao, Y.; Zheng, W.L.; Zhang, B.; Peng, Y.F. Meta-analysis: Therapeutic effect of transcatheter arterial chemoembolization combined with compound kushen injection in hepatocellular carcinoma. Afr. J. Tradit. Complement. Altern. Med. 2012, 9, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.X.; Fan, W.B.; Liu, P.; Tian, J.H. Meta analysis of compound matrine injection combined with cisplatin chemotherapy for advanced gastric cancer. Chin. J. Chin. Mater. Med. 2011, 36, 3198–3202. [Google Scholar]
- Wei, R.; Yang, D.Y.; Jiang, W.Z.; Dai, Y.Y.; Wan, L.Y.; Yang, Z. Efficacy of Yanshu injection (a compound Chinese traditional medicine) combined with concurrent radiochemotherapy in patients with stage III nasopharyngeal carcinoma. Chin. J. Oncol. 2011, 33, 391–394. [Google Scholar]
- Deng, H.; Luo, H.M.; Huang, F.; Li, X.G.; Gao, Q. Inhibition of proliferation and influence of proto-oncogenes expression by matrine in C6 Cell. Chin. Med. Mater. 2004, 27, 416–419. [Google Scholar]
- Zheng, K.B.; Li, C.H.; Shan, X.S.; Liu, H.P.; Fan, W.F.; Wang, Z.S. A study on isolation of chemical constituents from Sophora flavescens Ait and their anti-glioma effects. Afr. J. Tradit. Complement. Altern. Med. 2014, 11, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.G.; Su, G.; Ma, D.Q.; Sun, J.Z.; Fan, Y.Z.; Hao, Y.C. Apoptosis of gastric carcinoma MGC-803 cells induced by sophoridine. Tumour 2003, 23, 197–199. [Google Scholar]
- Ye, G.; Zhu, H.Y.; Li, Z.X.; Ma, C.H.; Fan, M.S.; Sun, Z.L.; Huang, C.G. LC-MS characterization of efficacy substances in serum of experimental animals treated with Sophora flavescens extracts. Biomed. Chromatogr. 2010, 21, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guan, F.; Bai, L.; Zhang, L.; Liu, J.; Pan, X.; Zhang, L. Quinolizidine alkaloids reduced mortality in EV71-infected mice by compensating for the levels of T cells. Bioorg. Med. Chem. Lett. 2015, 25, 3526–3528. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.Z.; Liu, G.M.; Hu, R.J. Inhibitory Effects of sophoridine hydrochlorium on DNA topoisomerase. Lishizhen Med. Mater. Med. Res. 2006, 17, 986–987. [Google Scholar]
- Li, X.; Zhao, W.L.; Jiang, J.D.; Ren, K.H.; Du, N.N.; Li, Y.B.; Wang, Y.X.; Bi, C.W.; Shao, R.G.; Song, D.Q. Synthesis, structure-activity relationship and biological evaluation of anticancer activity for novel N-substituted sophoridinic acid derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 5251–5254. [Google Scholar] [CrossRef] [PubMed]
- Rajski, S.R.; Williams, R.M. DNA Cross-Linking Agents as Antitumor Drugs. Chem. Rev. 1998, 98, 2723–2796. [Google Scholar] [CrossRef] [PubMed]
- Groehler, A.S.; Villalta, P.W.; Campbell, C.; Tretyakova, N.Y. Covalent DNA-Protein Cross-linking by Phosphoramide Mustard and Nornitrogen Mustard in Human Cells. Chem. Res. Toxicol. 2015, 29, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Sartillo-Piscil, F.; Quintero, L.; Cruz-Gregorio, S.; Espinosa-Aguirre, J.; Elinos-Baez, C.M.; Höpfl, H.; Serrano, A. Further evidence on the favorable role of the anomeric effect on the cleavage of HepDirect and cyclophosphamide prodrugs. J. Org. Chem. 2014, 79, 9647–9658. [Google Scholar] [CrossRef] [PubMed]
- Friedman, O.M.; Seligman, A.M. Preparation of N-Phosphorylated Derivatives of Bis-β-chloroethylamine. J. Am. Chem. Soc. 1954, 76, 655–658. [Google Scholar] [CrossRef]
- Hileman, E.O.; Liu, J.; Albitar, M.; Keating, M.J.; Huang, P. Intrinsic oxidative stress in cancer cells: A biochemical basis for therapeutic selectivity. Cancer Chemother. Pharmacol. 2004, 53, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.W.; Zhang, C.X.; Li, Y.H.; Tang, S.; Deng, H.B.; Zhao, W.L.; Wang, Z.; Shao, R.G.; Song, D.Q. Novel N-substituted sophoridinol derivatives as anticancer agents. Eur. J. Med. Chem. 2014, 81, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.W.; Zhang, C.X.; Li, Y.H.; Tang, S.; Wang, S.G.; Shao, R.G.; Fu, H.G.; Su, F.; Song, D.Q. Synthesis and biological evaluation of sophoridinol derivatives as a novel family of potential anticancer agents. ACS Med. Chem. Lett. 2014, 5, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Redinbo, M.R.; Champoux, J.J.; Hol, W.G. Novel insights into catalytic mechanism from a crystal structure of human topoisomerase I in complex with DNA. Biochemistry 2000, 39, 6832–6840. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell Lines (IC50, μM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| S180 | H22 | K562 | MCF-7 | SMMC-7721 | LoVo | L929 | SI * | SI ** | |
| 5 | 12.13 ± 0.55 | 11.70 ± 1.32 | 48.76 ± 2.01 | 111.37 ± 3.95 | 48.76 ± 2.74 | 83.98 ± 2.36 | 52.84 ± 2.80 | 4.36 | 4.52 |
| 7a | 2.89 ± 0.23 | 2.17 ± 0.13 | 27.07 ± 2.51 | 61.20 ± 2.21 | 60.80 ± 2.34 | 62.47 ± 2.32 | 180.66 ± 4.56 | 62.51 | 83.10 |
| 7b | 3.50 ± 0.35 | 2.65 ± 0.25 | 54.50 ± 2.32 | 38.86 ± 2.56 | 65.16 ± 2.67 | 46.17 ± 2.32 | 104.30 ± 3.20 | 29.80 | 39.36 |
| 7c | 2.02 ± 0.17 | 1.01 ± 0.08 | 18.07 ± 1.34 | 31.92 ± 2.45 | 31.73 ± 1.21 | 33.43 ± 2.45 | 162.41 ± 4.12 | 80.40 | 160.80 |
| 7d | 2.01 ± 0.14 | 1.80 ± 0.12 | 20.31 ± 2.05 | 34.62 ± 2.21 | 36.51 ± 2.45 | 32.25 ± 2.21 | 105.32 ± 3.10 | 52.40 | 58.51 |
| 7e | 3.65 ± 0.25 | 1.85 ± 0.16 | 32.73 ± 2.56 | 86.57 ± 2.67 | 77.89 ± 2.78 | 69.95 ± 2.78 | 138.54 ± 3.35 | 37.96 | 74.89 |
| Dox | 29.20 ± 2.07 | 36.51 ± 2.56 | 17.23 ± 1.78 | 0.44 ± 0.08 | 1.22 ± 0.05 | 0.15 ± 0.02 | 0.23 ± 0.08 | - | - |
| 1 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Dai, L.; Zhao, X.; Zhi, S.; Shen, H.; Yang, Z. Novel Sophoridine Derivatives Bearing Phosphoramide Mustard Moiety Exhibit Potent Antitumor Activities In Vitro and In Vivo. Molecules 2018, 23, 1960. https://doi.org/10.3390/molecules23081960
Li D, Dai L, Zhao X, Zhi S, Shen H, Yang Z. Novel Sophoridine Derivatives Bearing Phosphoramide Mustard Moiety Exhibit Potent Antitumor Activities In Vitro and In Vivo. Molecules. 2018; 23(8):1960. https://doi.org/10.3390/molecules23081960
Chicago/Turabian StyleLi, Dongdong, Linlin Dai, Xiumei Zhao, Shuang Zhi, Hongsheng Shen, and Zibo Yang. 2018. "Novel Sophoridine Derivatives Bearing Phosphoramide Mustard Moiety Exhibit Potent Antitumor Activities In Vitro and In Vivo" Molecules 23, no. 8: 1960. https://doi.org/10.3390/molecules23081960