Identifying Phage Virion Proteins by Using Two-Step Feature Selection Methods

Abstract
:1. Introduction
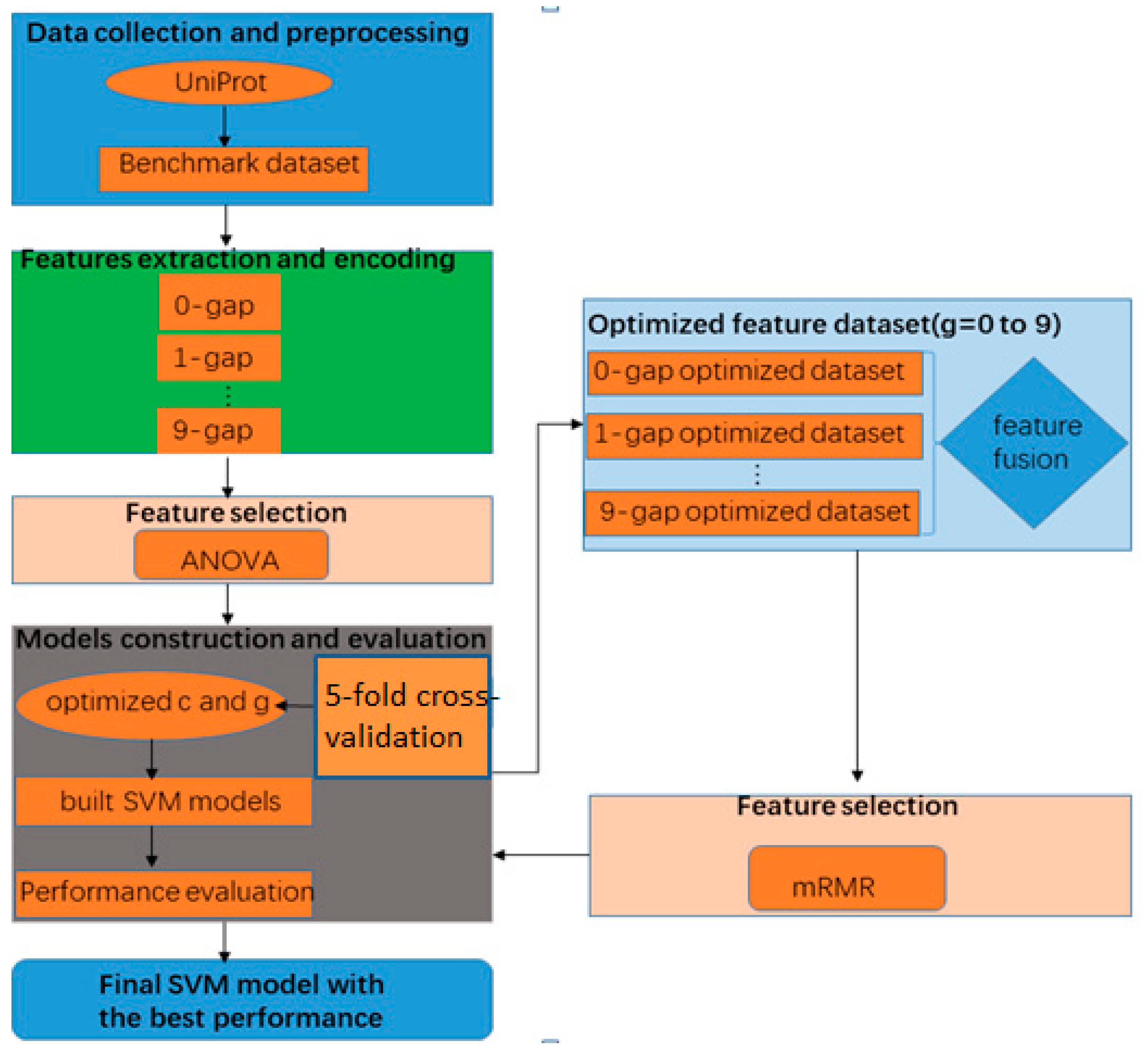
2. Materials and Methods
2.1. Benchmark Dataset
2.2. The g-Gap Dipeptide Composition
2.3. The Analysis of Variance (ANOVA)
2.4. Minimal-Redundancy-Maximal-Relevance (mRMR)
- Max-Relevance:
- Min-Redundancy:where represents the i-th feature attribute, c represents the category variable, S represents the feature subset, and represents the size of the feature subset. Furthermore, if the two metrics are considered equally important, we can abbreviate it by using the formula below.
2.5. Support Vector Machine (SVM)
2.6. Performance Evaluation
3. Result and Discussion
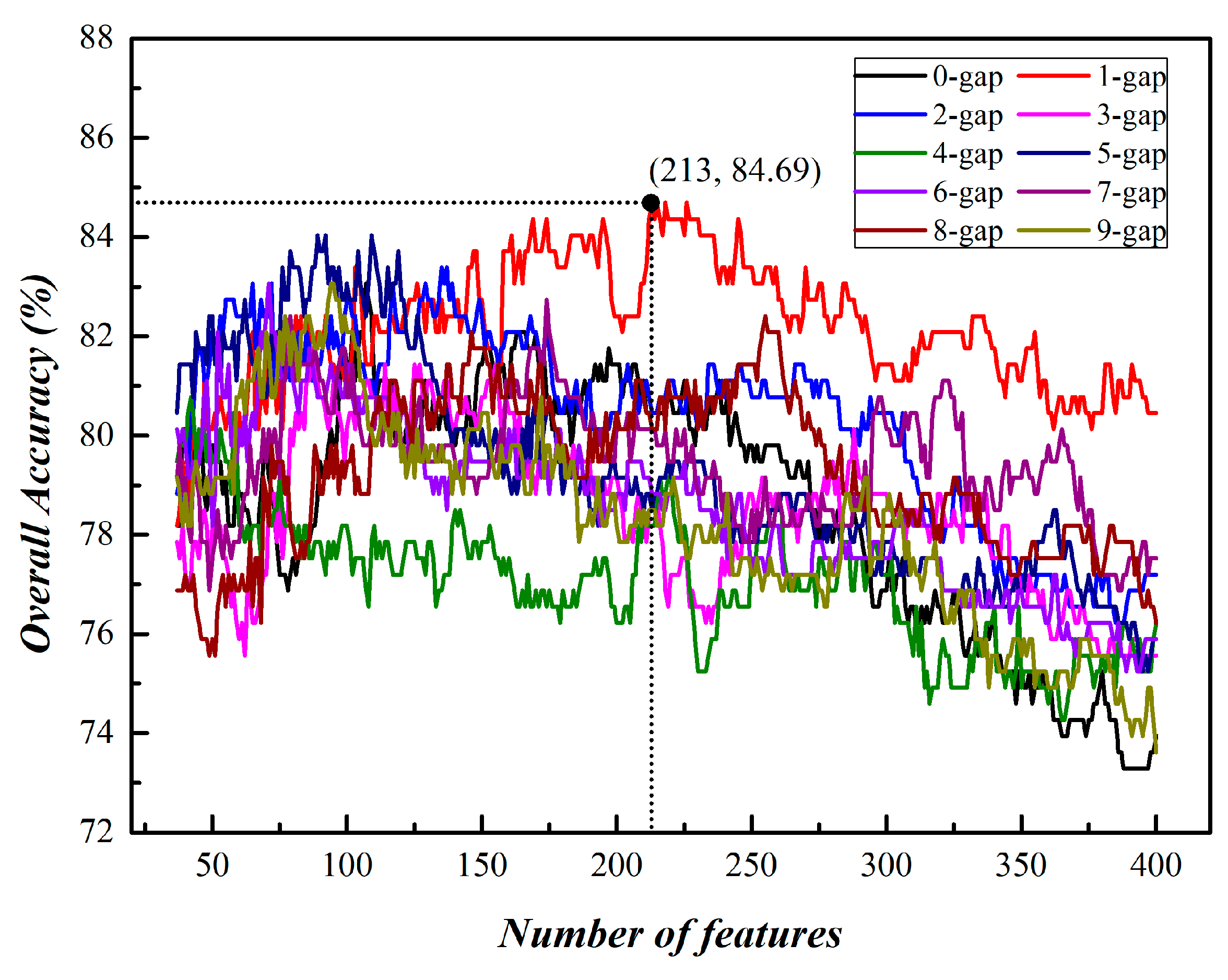
3.1. Prediction of Phage Virion Proteins Based on Single Kind of g-Gap Dipeptides
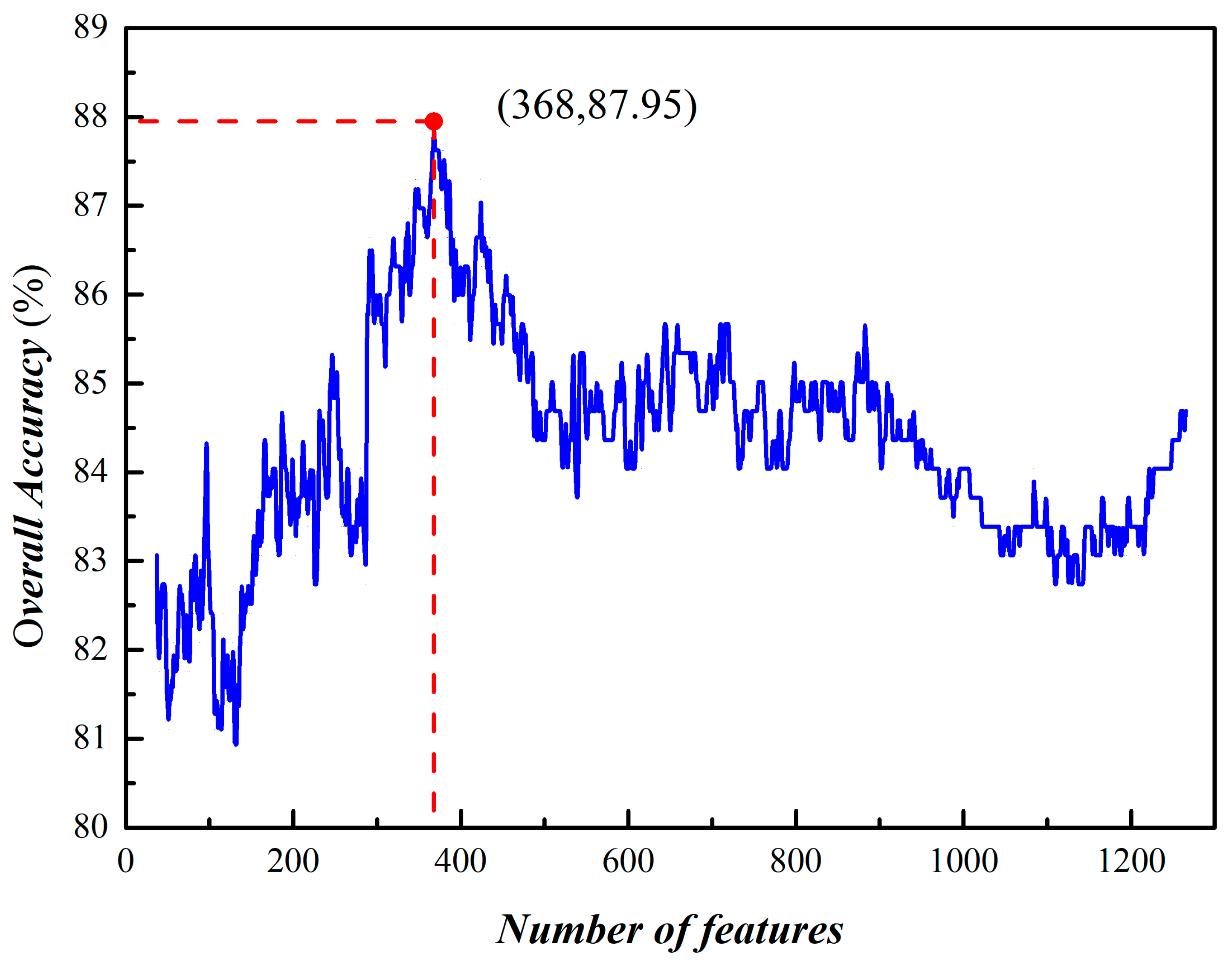
3.2. Prediction of Phage Virion Proteins Based on Fusing Features
3.3. Comparision with Other Published Methods
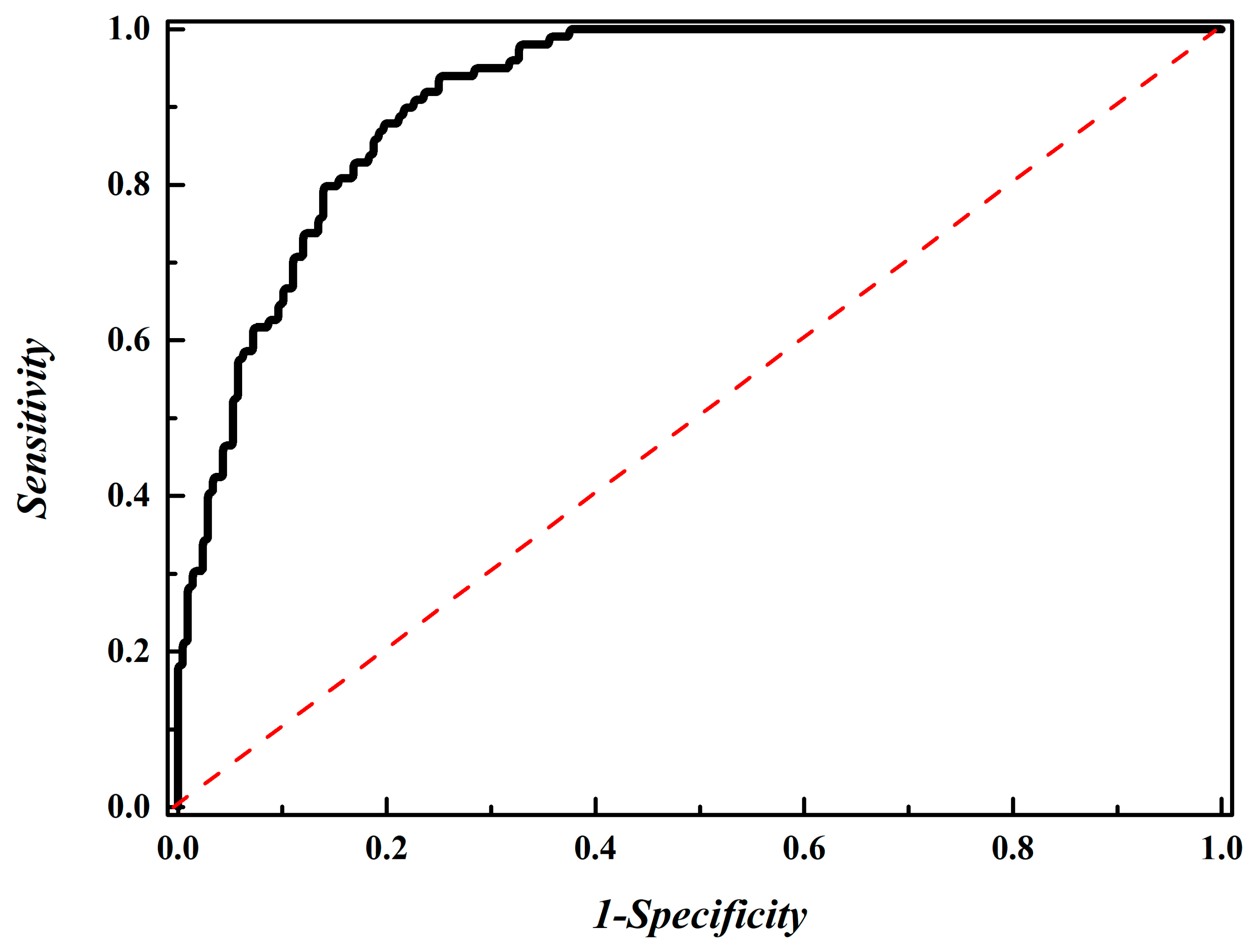
3.4. Performance Evaluation Using an Independent Dataset
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stella, E.J.; Franceschelli, J.J.; Tasselli, S.E.; Morbidoni, H.R. Analysis of novel mycobacteriophages indicates the existence of different strategies for phage inheritance in mycobacteria. PLoS ONE 2013, 8, e56384. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W. Structure and assembly of the virion. Intervirology 1996, 39, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Ceyssens, P.J.; Robben, J. Phage proteomics: Applications of mass spectrometry. Methods Mol. Biol. 2009, 502, 239–251. [Google Scholar] [PubMed]
- Feng, P.M.; Ding, H.; Chen, W.; Lin, H. Naive Bayes classifier with feature selection to identify phage virion proteins. Comput. Math. Method Med. 2013, 2013, 530696. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Feng, P.M.; Chen, W.; Lin, H. Identification of bacteriophage virion proteins by the ANOVA feature selection and analysis. Mol. Biosyst. 2014, 10, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, C.; Gao, R.; Yang, R. An Ensemble Method to Distinguish Bacteriophage Virion from Non-Virion Proteins Based on Protein Sequence Characteristics. Int. J. Mol. Sci. 2015, 16, 21734–21758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manavalan, B.; Shin, T.H.; Lee, G. PVP-SVM: Sequence-Based Prediction of Phage Virion Proteins Using a Support Vector Machine. Front. Microbiol. 2018, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Gao, H.; Lin, H.; Liu, Z.; Tang, L.; Li, S. Identification of Bacteriophage Virion Proteins Using Multinomial Naive Bayes with g-Gap Feature Tree. Int. J. Mol. Sci. 2018, 19, 1779. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013, 41, D43–D47. [Google Scholar]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Chen, W.; Lin, H. Identification of immunoglobulins using Chou’s pseudo amino acid composition with feature selection technique. Mol. Biosyst. 2016, 12, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Yang, W.; Tang, H.; Feng, P.M.; Huang, J.; Chen, W.; Lin, H. PHYPred: A tool for identifying bacteriophage enzymes and hydrolases. Virol. Sin. 2016, 31, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, D.N.; Karakasidis, T.E.; Megaritis, A.C.; Nieto, J.J.; Torres, A. An extension of fuzzy topological approach for comparison of genetic sequences. J. Intell. Fuzzy Syst. 2015, 29, 2259–2269. [Google Scholar] [CrossRef]
- Chou, K.C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Qiu, W.R.; Liu, G.Q.; Guo, F.B.; Chen, W.; Chou, K.C.; Lin, H. iRSpot-Pse6NC: Identifying recombination spots in Saccharomyces cerevisiae by incorporating hexamer composition into general PseKNC. Int. J. Biol. Sci. 2018, 14, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.D.; Huang, Y.; Zhang, Z.Y.; Zhao, Y.W.; Wang, D.; Chen, W.; Chou, K.C.; Lin, H. iLoc-lncRNA: Predict the subcellular location of lncRNAs by incorporating octamer composition into general PseKNC. Bioinformatics 2018. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Wan, S.; Ju, Y.; Tang, J.; Zeng, X. Pretata: Predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 2016, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Wan, S.; Yang, Z.; Teschendorff, A.E.; Zou, Q. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 2018, 34, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tang, H.; Chen, X.X.; Zhang, C.J.; Zhu, P.P.; Ding, H.; Chen, W.; Lin, H. Identification of Secretory Proteins in Mycobacterium tuberculosis Using Pseudo Amino Acid Composition. BioMed Res. Int. 2016, 2016, 5413903. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Tang, H.; Li, W.C.; Wu, H.; Chen, W.; Ding, H.; Lin, H. Identification of Bacterial Cell Wall Lyases via Pseudo Amino Acid Composition. BioMed Res. Int. 2016, 2016, 1654623. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zhao, Y.W.; Zou, P.; Zhang, C.M.; Chen, R.; Huang, P.; Lin, H. HBPred: A tool to identify growth hormone-binding proteins. Int. J. Biol. Sci. 2018, 14, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Peng, H. Minimum redundancy feature selection from microarray gene expression data. J. Bioinf. Comput. Biol. 2005, 3, 185–205. [Google Scholar] [CrossRef]
- Zou, Q.; Zeng, J.; Cao, L.; Ji, R. A novel features ranking metric with application to scalable visual and bioinformatics data classification. Neurocomputing 2016, 173, 346–354. [Google Scholar] [CrossRef]
- Manavalan, B.; Shin, T.H.; Lee, G. DHSpred: Support-vector-machine-based human DNase I hypersensitive sites prediction using the optimal features selected by random forest. Oncotarget 2018, 9, 1944–1956. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Shin, T.H.; Choi, S.; Kim, M.O.; Lee, G. MLACP: Machine-learning-based prediction of anticancer peptides. Oncotarget 2017, 8, 77121–77136. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Lee, J. SVMQA: Support-vector-machine-based protein single-model quality assessment. Bioinformatics 2017, 33, 2496–2503. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chen, W.; Chou, K.C. iDNA6mA-PseKNC: Identifying DNA N(6)-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics 2018. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liang, Z.Y.; Tang, H.; Chen, W. Identifying sigma70 promoters with novel pseudo nucleotide composition. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yang, H.; Feng, P.; Ding, H.; Lin, H. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 2017, 33, 3518–3523. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Cao, R.Z.; Wang, W.; Liu, T.S.; Wang, L.M.; He, C.M. A two-step discriminated method to identify thermophilic proteins. Int. J. Biomath. 2017, 10, 1750050. [Google Scholar] [CrossRef]
- Cao, R.; Wang, Z.; Wang, Y.; Cheng, J. SMOQ: A tool for predicting the absolute residue-specific quality of a single protein model with support vector machines. BMC Bioinform. 2014, 15, 120. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Deng, E.Z.; Yuan, L.F.; Liu, L.; Lin, H.; Chen, W.; Chou, K.C. iCTX-type: A sequence-based predictor for identifying the types of conotoxins in targeting ion channels. BioMed Res. Int. 2014, 2014, 286419. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Deng, E.Z.; Ding, H.; Chen, W.; Chou, K.C. iPro54-PseKNC: A sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 2014, 42, 12961–12972. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.H.; Deng, E.Z.; Xu, L.Q.; Ding, H.; Lin, H.; Chen, W.; Chou, K.C. iNuc-PseKNC: A sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics 2014, 30, 1522–1529. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lv, H.; Ding, H.; Chen, W.; Lin, H. iRNA-2OM: A sequence-based predictor for identifying 2’-O-methylation sites in Homo sapiens. J. Comput. Biol. 2018. [Google Scholar] [CrossRef]
- Zhao, Y.W.; Su, Z.D.; Yang, W.; Lin, H.; Chen, W.; Tang, H. IonchanPred 2.0: A Tool to Predict Ion Channels and Their Types. Int. J. Mol. Sci. 2017, 18, 1838. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ju, Y.; Zou, Q. Protein Folds Prediction with Hierarchical Structured SVM. Curr. Proteom. 2016, 13, 79–85. [Google Scholar] [CrossRef]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. AIPpred: Sequence-Based Prediction of Anti-inflammatory Peptides Using Random Forest. Front. Pharmacol. 2018, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Subramaniyam, S.; Shin, T.H.; Kim, M.O.; Lee, G. Machine-learning-based prediction of cell-penetrating peptides and their uptake efficiency with improved accuracy. J. Proteome Res. 2018, 17, 2715–2726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.J.; Tang, H.; Li, W.C.; Lin, H.; Chen, W.; Chou, K.C. iOri-Human: Identify human origin of replication by incorporating dinucleotide physicochemical properties into pseudo nucleotide composition. Oncotarget 2016, 7, 69783–69793. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.M.; Lin, H.; Chou, K.C. iSS-PseDNC: Identifying splicing sites using pseudo dinucleotide composition. BioMed Res. Int. 2014, 2014, 623149. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.M.; Deng, E.Z.; Lin, H.; Chou, K.C. iTIS-PseTNC: A sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Anal. Biochem. 2014, 462, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.M.; Chen, W.; Lin, H.; Chou, K.C. iHSP-PseRAAAC: Identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 2013, 442, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.Y.; Chen, X.X.; Chen, W.; Tang, H.; Lin, H. Sequence-based predictive modeling to identify cancerlectins. Oncotarget 2017, 8, 28169–28175. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.M.; Lin, H.; Chen, W. Identification of antioxidants from sequence information using naive Bayes. Comput. Math. Method. Med. 2013, 2013, 567529. [Google Scholar] [CrossRef] [PubMed]
- Li, B.Q.; Zhang, Y.H.; Jin, M.L.; Huang, T.; Cai, Y.D. Prediction of Protein-Peptide Interactions with a Nearest Neighbor Algorithm. Curr. Bioinform. 2018, 13, 14–24. [Google Scholar] [CrossRef]
- Naseem, I.; Khan, S.; Togneri, R.; Bennamoun, M. ECMSRC: A Sparse Learning Approach for the Prediction of Extracellular Matrix Proteins. Curr. Bioinform. 2017, 12, 361–368. [Google Scholar] [CrossRef]
- Lin, Y.Q.; Min, X.P.; Li, L.L.; Yu, H.; Ge, S.X.; Zhang, J.; Xia, N.S. Using a Machine-Learning Approach to Predict Discontinuous Antibody-Specific B-Cell Epitopes. Curr. Bioinform. 2017, 12, 406–415. [Google Scholar] [CrossRef]
- Kang, J.; Fang, Y.; Yao, P.; Li, N.; Tang, Q.; Huang, J. NeuroPP: A Tool for the Prediction of Neuropeptide Precursors Based on Optimal Sequence Composition. Interdiscip. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Kang, J.; Jiang, L.; He, B.; Lin, H.; Huang, J. PSBinder: A Web Service for Predicting Polystyrene Surface-Binding Peptides. BioMed Res. Int. 2017, 2017, 5761517. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.P.; Li, W.C.; Zhong, Z.J.; Deng, E.Z.; Ding, H.; Chen, W.; Lin, H. Predicting the subcellular localization of mycobacterial proteins by incorporating the optimal tripeptides into the general form of pseudo amino acid composition. Mol. Biosyst. 2015, 11, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Li, W.C.; Deng, E.Z.; Ding, H.; Chen, W.; Lin, H. iORI-PseKNC: A predictor for identifying origin of replication with pseudo k-tuple nucleotide composition. Chemom. Intell. Lab. Syst. 2015, 141, 100–106. [Google Scholar] [CrossRef]
- Dao, F.Y.; Yang, H.; Su, Z.D.; Yang, W.; Wu, Y.; Hui, D.; Chen, W.; Tang, H.; Lin, H. Recent Advances in Conotoxin Classification by Using Machine Learning Methods. Molecules 2017, 22, 1057. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xing, P.; Zou, Q. Detecting N(6)-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Sci. Rep. 2017, 7, 40242. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Peng, M.M.; Liao, B.; Huang, G.H.; Li, W.B.; Xie, D.F. The Advances and Challenges of Deep Learning Application in Biological Big Data Processing. Curr. Bioinform. 2018, 13, 352–359. [Google Scholar] [CrossRef]
- Patel, S.; Tripathi, R.; Kumari, V.; Varadwaj, P. DeepInteract: Deep Neural Network Based Protein-Protein Interaction Prediction Tool. Curr. Bioinform. 2017, 12, 551–557. [Google Scholar] [CrossRef]
- Long, H.X.; Wang, M.; Fu, H.Y. Deep Convolutional Neural Networks for Predicting Hydroxyproline in Proteins. Curr. Bioinform. 2017, 12, 233–238. [Google Scholar] [CrossRef]
- Cao, R.Z.; Adhikari, B.; Bhattacharya, D.; Sun, M.; Hou, J.; Cheng, J.L. QAcon: Single model quality assessment using protein structural and contact information with machine learning techniques. Bioinformatics 2017, 33, 586–588. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Freitas, C.; Chan, L.; Sun, M.; Jiang, H.; Chen, Z. ProLanGO: Protein Function Prediction Using Neural Machine Translation Based on a Recurrent Neural Network. Molecules 2017, 22, 1732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Tan, P.; Wang, L.; Jin, N.; Li, Y.; Zhang, L.; Yang, H.; Hu, Z.; Zhang, L.; Hu, C.; et al. RNALocate: A resource for RNA subcellular localizations. Nucleic Acids Res. 2017, 45, D135–D138. [Google Scholar] [PubMed]
- Liang, Z.Y.; Lai, H.Y.; Yang, H.; Zhang, C.J.; Yang, H.; Wei, H.H.; Chen, X.X.; Zhao, Y.W.; Su, Z.D.; Li, W.C.; et al. Pro54DB: A database for experimentally verified sigma-54 promoters. Bioinformatics 2017, 33, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Zhang, L.; Huang, Y.; Yi, Y.; Tan, P.; Zhao, Y.; Hu, Y.; Xu, L.; Li, E.; Wang, D. MNDR v2.0: An updated resource of ncRNA-disease associations in mammals. Nucleic Acids Res. 2018, 46, D371–D374. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Zhao, Y.; Li, C.; Zhang, L.; Huang, H.; Li, Y.; Liu, L.; Hou, P.; Cui, T.; Tan, P.; et al. RAID v2.0: An updated resource of RNA-associated interactions across organisms. Nucleic Acids Res. 2017, 45, D115–D118. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| g | Number of Feature | Acc (%) |
|---|---|---|
| 0 | 107 | 83.06 |
| 1 | 213 | 84.69 |
| 2 | 135 | 83.39 |
| 3 | 87 | 81.76 |
| 4 | 42 | 80.78 |
| 5 | 89 | 84.04 |
| 6 | 70 | 82.41 |
| 7 | 174 | 82.73 |
| 8 | 255 | 82.41 |
| 9 | 94 | 83.06 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, J.-X.; Dao, F.-Y.; Lv, H.; Feng, P.-M.; Ding, H. Identifying Phage Virion Proteins by Using Two-Step Feature Selection Methods. Molecules 2018, 23, 2000. https://doi.org/10.3390/molecules23082000
Tan J-X, Dao F-Y, Lv H, Feng P-M, Ding H. Identifying Phage Virion Proteins by Using Two-Step Feature Selection Methods. Molecules. 2018; 23(8):2000. https://doi.org/10.3390/molecules23082000
Chicago/Turabian StyleTan, Jiu-Xin, Fu-Ying Dao, Hao Lv, Peng-Mian Feng, and Hui Ding. 2018. "Identifying Phage Virion Proteins by Using Two-Step Feature Selection Methods" Molecules 23, no. 8: 2000. https://doi.org/10.3390/molecules23082000