Cinnamoyl-Oxaborole Amides: Synthesis and Their in Vitro Biological Activity

 , , , , ,
, , , , ,
Abstract
:
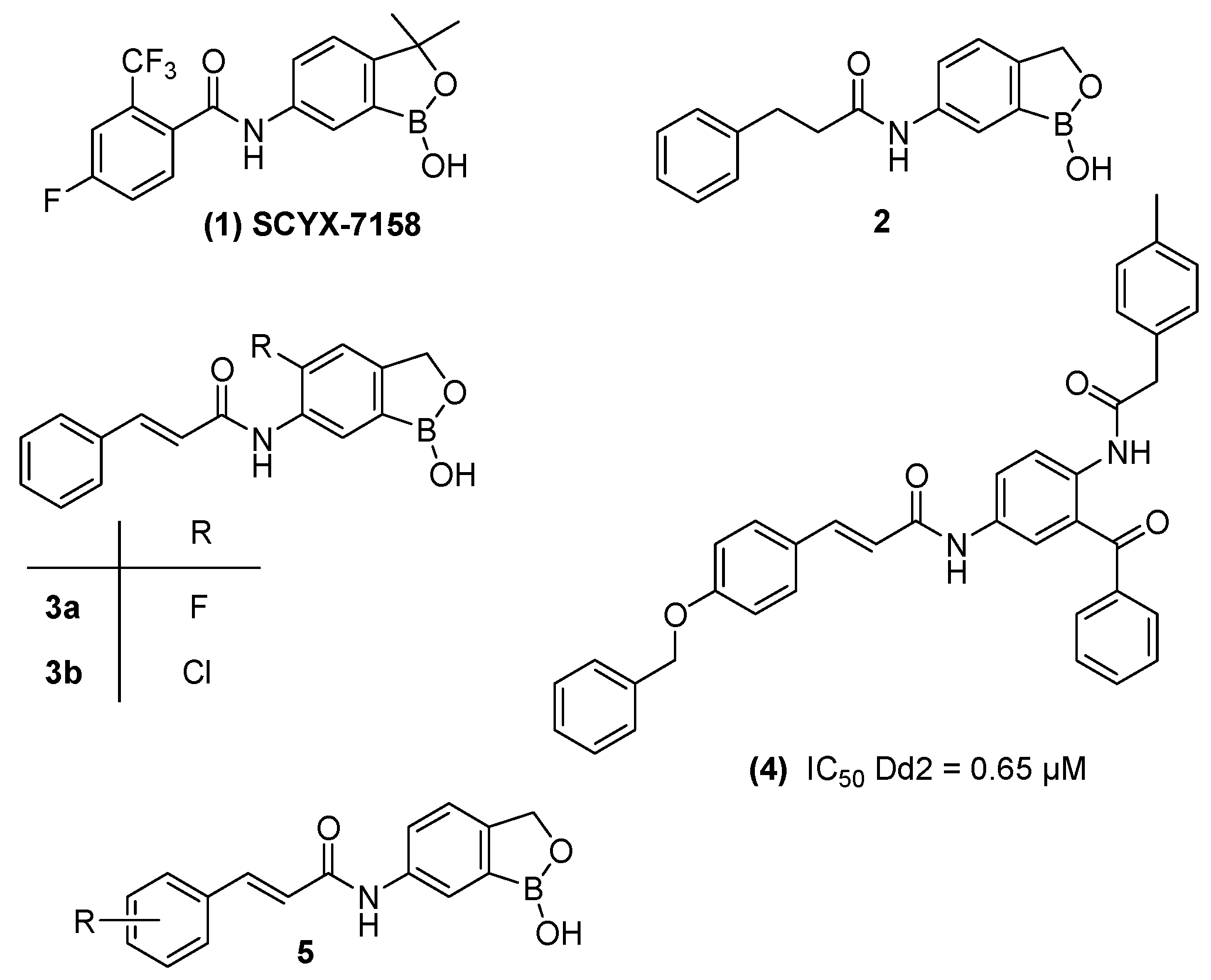
1. Introduction
2. Results and Discussion
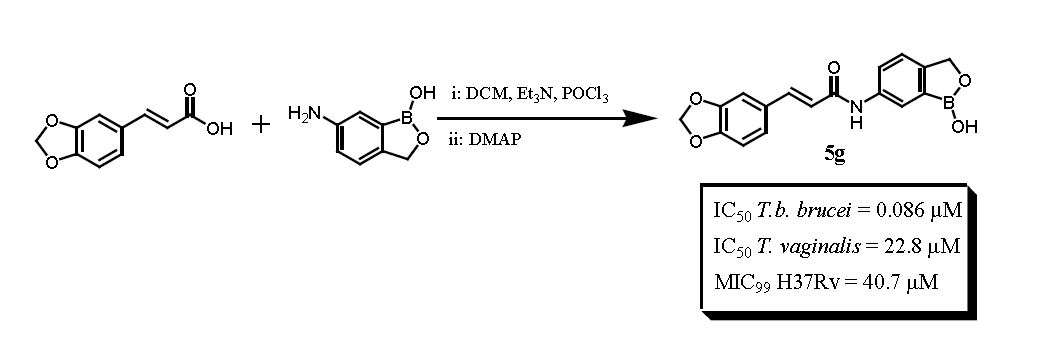
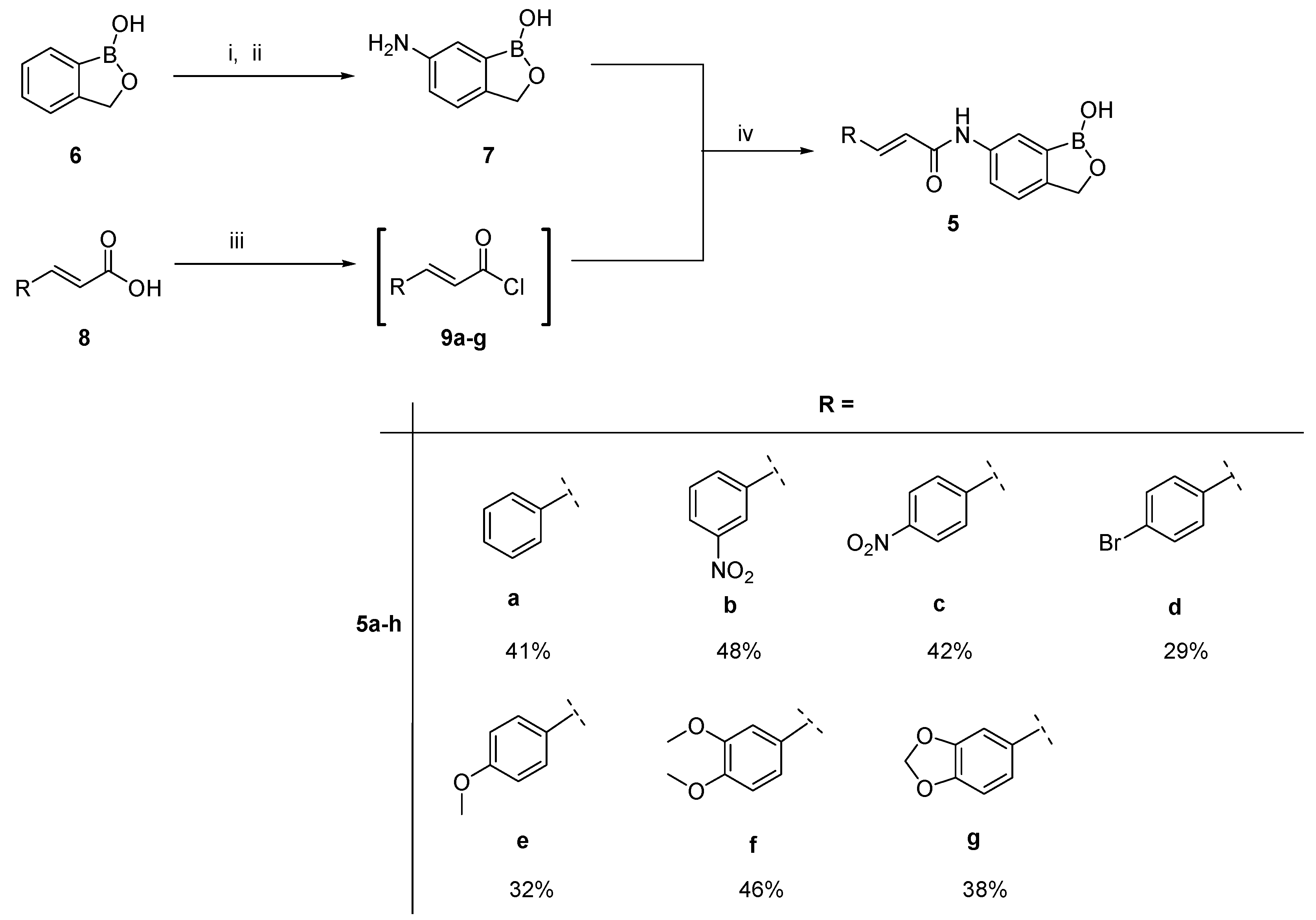
2.1. Chemistry
2.2. Pharmacology
3. Materials and Methods
3.1. General Information
3.2. General Synthetic Procedure for the Cinnamic Acid-Benzoxaborole Hybrids, 5a–g
3.3. In Vitro Antitrypanosomal Assay
3.4. In Vitro Cytotoxicity Assay
3.5. In Vitro Antitrichomonal Assay and Mucosal Normal Flora Susceptibility Assay
3.6. In Vitro Antimycobacterial Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Njogu, P.M.; Guantai, E.M.; Pavadai, E.; Chibale, K. Computer-aided drug discovery approaches against the tropical infectious diseases malaria, tuberculosis, trypanosomiasis, and leishmaniasis. ACS Infect. Dis. 2015, 2, 8–31. [Google Scholar] [CrossRef] [PubMed]
- WHO Global Tubercoulosis Report 2017. Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 30 May 2018).
- Jakobsen, P.H.; Wang, M.W.; Nwaka, S. Innovative partnerships for drug discovery against neglected diseases. PLoS Negl. Trop. Dis. 2011, 5, e1221. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Zhou, X.N. Con-infection of tuberculosis and parasitic diseases in humans: a systematic review. Parasites Vectors 2013, 6, 79. [Google Scholar] [CrossRef] [PubMed]
- Berninger, M.; Schimidt, I.; Ponte-Sucre, A.; Holzgrabe, U. Novel lead compounds in pre-clinical development against African sleeping sickness. Med. Chem. Commun. 2017, 8, 1872–1890. [Google Scholar] [CrossRef]
- Njoroge, M.; Njuguna, N.M.; Mutai, P.; Ongarora, D.S.B.; Smith, P.W.; Chibale, K. Recent approaches to chemical discovery and development against malaria and the neglected tropical diseases human African trypanosomiasis and schistosomiasis. Chem. Rev. 2014, 114, 11138–11163. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Acosta, V.; Ruiz-Pérez, L.; Etxebarria, J.; Reichardt, N.; Navarro, M.; Igarashi, Y.; Liekens, S.; Balzarini, J.; González-Pacanowska, D. Open source drug discovery with the malaria box compound collection for neglected diseases and beyong. PLoS Pathog. 2016, 12, e1005851. [Google Scholar]
- Burrows, J.N.; Elliott, R.L.; Kaneko, T.; Mowbray, C.E.; Waterson, D. The role of modern drug discovery in the fight against neglected and tropical diseases. Med. Chem. Commun. 2014, 5, 688–700. [Google Scholar] [CrossRef]
- Kwofie, K.D.; Tung, N.H.; Suzuki-Ohashi, M.; AmoaBosompem, M.; Adegle, R.; Sakyiamah, M.M.; Ayertey, F.; Owusu, K.B.-A.; Tuffour, I.; Atchoglo, P.; et al. Antitrypanosomal activities and mechanisms of action of novel tetracyclic iridoids from Morinda lucida benth. Antimicrob. Agents Chemother. 2016, 60, 3283–3290. [Google Scholar] [CrossRef] [PubMed]
- WHO: Trypanosomiasis, Human African (Sleeping Sickness). Available online: http://www.who.int/mediacentre/factsheets/fs259/en/ (accessed on 30 June 2018).
- Nwodo, N.J.; Ibezim, A.; Ntie-Kang, F.; Adikwu, M.U.; Mbah, C.J. Anti-trypanosomal activity of Nigerian plants and their constituents. Molecules 2015, 20, 7751–7771. [Google Scholar] [CrossRef] [PubMed]
- CDC. Parasites African Trypanosomiasis (Also known as Sleeping Sickness). Available online: https://www.cdc.gov/parasites/sleepingsickness/ (accessed on 30 June 2018).
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Jannin, J.G. Epidemiology of human African trypanosomiasis. Clin. Epidemiol. 2014, 6, 257–275. [Google Scholar] [PubMed]
- Babokhov, P.; Sanyaolu, A.O.; Oyibo, W.A.; Fagbenro-Beyioku, A.F.; Iriemenam, N.C. A current analysis of chemotherapy strategies for the treatment of human African trypanosomiasis. Pathog. Glob. Health. 2013, 107, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, M.; Bray, M.A.; Cal, M.; Trunz, B.; Torreele, E.; Brun, R. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 2011, 55, 5602–5608. [Google Scholar] [CrossRef] [PubMed]
- Mishina, Y.V.; Krishna, S.; Haynes, R.K.; Meade, J.C. Artemisinins inhibit Trypanosoma cruzi and Trypanosoma bruce rhodesiense in vitro growth. Antimicrob. Agents Chemother. 2007, 51, 1852–1854. [Google Scholar] [CrossRef] [PubMed]
- Willyard, C. Putting sleeping sickness to bed. Nat. Med. 2011, 17, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Balasegaram, M.; Young, H.; Chappuis, F.; Priotto, G.; Raguenaud, M.E.; Checchi, F. Effectiveness of melarsoprol and eflornithine as first-line regimens for gambiense sleeping sickness in nine Médecins Sans Frontières programmes. Trans. R. Soc. Trop. Med. Hyg. 2009, 103, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.Y.; Wang, M.; Li, L.; Wu, H.; He, C.Y.; Yao, S.Q. Design, synthesis and biological evaluation of potent azadipeptide nitrile inhibitors and activity-based probes as promising anti-Trypanosoma brucei agents. Chem. Eur. J. 2012, 18, 6528–6541. [Google Scholar] [CrossRef] [PubMed]
- Giordani, F.; Morrison, L.J.; Rowan, T.G.; De Koning, H.P.; Barrett, M.P. The animal trypanosomiases and their chemotherapy: A review. Parasitology 2016, 143, 1862–1889. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Report 2007: Global Strategy for the Prevention and Control of Sexually Transmitted Infections: 2006–2015: Breaking the Chain of Transmission; WC 142; WHO Press: Geneva, Switzerland, 2007; pp. 1–60. [Google Scholar]
- World Health Organization Department of Reproductive Health and Research (2011) WHO. Available online: http://www.who.int/reproductivehealth/publications/rtis/9789241502450/en/index.html/ (accessed on 30 June 2018).
- Secor, W.E.; Meites, E.; Starr, M.C.; Workowski, K.A. Neglected parasitic infections in the United States: Trichomoniasis. Am. J. Trop. Med. Hyg. 2014, 90, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Kirkcaldy, R.D.; Augostini, P.; Asbel, L.E.; Bernstein, K.T.; Kerani, R.P.; Mettenbrink, C.J.; Pathela, P.; Schwebke, J.R.; Secor, W.E.; Workowski, K.A.; et al. Trichomonas vaginalis antimicrobial drug resistance in six US cities, STD surveillance network, 2009–2010. Emerg. Infect. Dis. 2012, 18, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Workowski, K.A.; Bolan, G.A. Sexually transmitted diseases treatment guidelines. MMWR Recomm. Rep. 2015, 64, 1–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.T.; Tomsho, J.W.; Benkovic, S.J. The unique chemistry of benzoxaboroles: Current and emerging applications in biotechnology and therapeutic treatments. Bioorg. Med. Chem. 2014, 22, 4462–4473. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Tomsho, J.W.; Benkovic, S.J. Boron-containing inhibitors of synthetases. Chem. Soc. Rev. 2011, 40, 4279–4285. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.T.; Plattner, J.J.; Keenan, M. Boron-based drugs as antiprotozoals. Curr. Opin. Infect. Dis. 2011, 24, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk-Woźniak, A.; Borys, K.M.; Sporzyński, A. Recent developments in the chemistry and biological applications of benzoxaboroles. Chem. Rev. 2015, 115, 5224–5247. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk-Woźniak, A.; Cyrański, M.K.; Żubrowska, A.; Sporzyński, A. Benzoxaboroles-Old compounds with new applications. J. Organomet. Chem. 2009, 694, 3533–3541. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, M.Y.; Lin, Y.N.; Zhou, H.C. The synthesis of benzoxaboroles and their applications in medicinal chemistry. Sci. China Chem. 2013, 56, 1372–1381. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Plattner, J.J.; Easome, E.E.; Zhou, Y.; Akama, T.; Bu, W.; White, W.H.; Defauw, J.M.; Winkle, J.R.; Balko, T.W.; et al. Discovery of an orally bioavailable isoxazoline benzoxaborole (AN8030) as a long acting animal ectoparasiticide. Bioorg. Med. Chem. Lett. 2015, 25, 5589–5593. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T.; et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis. 2011, 5, e1151. [Google Scholar] [CrossRef] [PubMed]
- Torssell, K. Bromination of tolylboronic acids according to Wohl-Ziegler. Ark. Kemi. 1957, 10, 507–511. [Google Scholar]
- Brun, R.; Don, R.; Jacobs, R.; Wang, M.; Barrett, M. Devlopment of novel drugs for human African trypanosomiasis. Future Microbiol. 2011, 6, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Drugs for Neglected Diseases Initiative (Dndi). Dndi Announces Successful Completion of SCYX-7158 Phase I Study for Treatment of Sleeping Sicknes. Available online: http://www.news-medical.net/news/20150909/DNDi-announces-successful-completion-of-SCYX-7158-Phase-I-study-for-treatment-of-sleeping-sickness.aspx (accessed on 25 June 2018).
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.; Orr, M.; Wring, S.; Chen, D.; Zhou, H.; Ding, D.; Feng, Y.; Ye, L.; Hernandez, V.S.; Zhang, Y.K.; et al. Boron-Containing Small Molecules as Anti-Protozoal Agents. WO2010045503A1, 15 November 2016. [Google Scholar]
- Stierli, D.; Renold, P.; Rajan, R. Novel microbiocides. WO2014173880A1, 30 October 2014. [Google Scholar]
- De, P.; Bedos-Belval, F.; Vanucci-Bacque, C.; Baltas, M. Cinnamic acid derivatives iin tuberculosis, malaria and cardiovascular diseases-a review. Curr. Org. Chem. 2012, 16, 747–768. [Google Scholar]
- Sharma, P. Cinnamic acid derivatives: A new chapter of various pharmacological activities. J. Chem. Pharm. Res. 2011, 3, 403–423. [Google Scholar]
- Guzman, J.D. Natural cinnamic acids, synthetic derivatives and hybrids with antimicrobial activity. Molecules 2014, 19, 19292–19349. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Wiβner, P.; Dahse, H.M.; Jomaa, H.; Schlitzer, M. Discovery of novel lead structure for antimalarials. Bioorg. Med. Chem. 2001, 9, 785–792. [Google Scholar] [CrossRef]
- Joardar, S.; Bhattacharyya, A.; Das, S. A palladium on carbon catalyzed one-pot synthesis of substituted benzyimidazoles. Synthesis 2014, 46, 3121–3132. [Google Scholar] [CrossRef]
- Joullié, M.M.; Lassen, K.M. Evolution of bond formation. Arkivoc 2010, 189–250. [Google Scholar]
- Montalbetti, C.A.G.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Leggio, A.; Belsito, E.L.; De Luca, G.; Di Gioia, M.L.; Leotta, V.; Romio, E.; Siciliano, C.; Liguori, A. One-pot synthesis of amides from carboxylic acids activated using thionyl chloride. RSC Adv. 2016, 6, 34468–34475. [Google Scholar] [CrossRef]
- Chen, P.J.; Wang, H.Y.; Peng, A.Y. A mild and efficient amide formation reaction mediated by P(OEt)3 and iodine. RSC Adv. 2015, 5, 94328–94331. [Google Scholar] [CrossRef]
- Adamczyk-Woźniak, A.; Cyrański, M.K.; Jakubczyk, M.; Klimentowska, P.; Koll, A.; Kołodziejczak, J.; Pojmaj, G.; Żubrowska, A.; Żukowska, G.Z.; Sporzyński, A. Influence of the substituents on the structure and properties of benzoxaboroles. J. Phys. Chem. A 2010, 114, 2324–2330. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.A.; da Silva, F.; de Souza, M.V.N.; Lourenco, M.C.S.; Vincente, F.R. Synthesis and antimycobacterial evaluation of trans-cinnamic acid hydrazide derivatives. Bioorg. Med. Chem. Lett. 2008, 18, 538–541. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Yoya, G.K.; Constant, P.; Bedos-Belval, F.; Duran, H.; Saffon, N.; Daffé, M.; Baltas, M. Design, synthesis, and biological evaluation of new cinnamic derivatives as antituberculosis agents. J. Med. Chem. 2011, 54, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Kakwani, M.D.; Suryavanshi, P.; Ray, M.; Rajan, M.G.R.; Majee, S.; Samad, A.; Devarajan, P.; Degani, M.S. Design, synthesis and antimycobacterial activity of cinnamide derivatives: A molecular hybridization approach. Bioorg. Med. Chem. Lett. 2011, 21, 1997–1999. [Google Scholar] [CrossRef] [PubMed]
- Pérez, B.; Teixeira, C.; Gut, J.; Rosenthal, P.J.; Gomes, J.R.B.; Gomes, P. Cinnamic acid/chloroquinoline conjugates as potent agents against chloroquine-resistant Plasmodium falciparum. Chem. Med. Chem. 2012, 7, 1537–1540. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Yin, S.L.; Wang, J.; Jing, Y.K.; Dong, J.H. Design and synthesis of novel 2-phenylaminopyrimidine (PAP) derivatives and their antiproliferative effects in human chronic myeloid leukemia cells. Molecules 2009, 14, 4166–4179. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.Z.; Zhang, Y.K.; Li, X.; Liu, Y.; Zhang, S.; Zhou, Y.; Plattner, J.J.; Baker, S.J.; Liu, L.; Duan, M.; et al. Synthesis and biological evaluation of P4-benzoxaborole-substituted macrocyclic inhibitors of HCV NS3 protease. Bioorg. Med. Chem. Lett. 2010, 20, 7317–7322. [Google Scholar] [CrossRef] [PubMed]
- Hirymi, H.; Hirumi, K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989, 75, 985–989. [Google Scholar] [CrossRef]
- Oderinlo, O.O.; Tukulula, M.; Isaacs, M.; Hoppe, H.C.; Taylor, D.; Smith, V.J.; Khanye, S.D. New thiazolidine-2,4-dione derivatives compbined with organometallic ferrocene: Synthesis, structure and antiparasitic activity. Appl. Organomet. Chem. 2018, 32, e4385. [Google Scholar] [CrossRef]
- Abrahams, G.L.; Kumar, A.; Savvi, S.; Hung, A.W.; Wen, S.; Abell, C.; Barry, C.E., 3rd; Sherman, D.R.; Boshoff, H.I.; Mizrahi, V. Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem. Biol. 2012, 19, 844–854. [Google Scholar] [CrossRef] [PubMed]
- De Voss, J.J.; Rutter, K.; Schroeder, B.G.; Su, H.; Zhu, Y.; Barry III, C.E. The salicylate-derived mycobactin siderophores of mycobacterium tuberculosis are essential for growth in macrophages. Proc. Nat. Acad. Sci. USA 2000, 97, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Franzblau, S.G.; DeGroote, M.A.; Cho, S.H.; Andries, K.; Nuermberger, E.; Orme, I.M.; Mdluli, K.; Angulo-Barturen, I.; Dick, T.; Dartois, W.; et al. Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis 2012, 92, 453–488. [Google Scholar] [CrossRef] [PubMed]
- Ollinger, J.; Bailey, A.; Moraski, C.; Casey, A.; Florio, S.; Alling, T.; Miller, J.; Parish, T. A dual read-out assay to evaluate the potency of compounds active against Mycobacterium tuberculosis. PLoS ONE 2013, 8, e60531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of all the compounds 5a–g are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) | MIC99 (μM) | %Viability |
|---|---|---|---|
| T. vaginalis | H37Rv | HeLa Cells | |
| 5a | 15.3 ± 0.33 | 61.8 | 108.5 ± 5.35 |
| 5b | 41.7 ± 0.23 | 107 | 87.0 ± 1.59 |
| 5c | 10.2 ± 0.56 | 26.5 | 94.3 ± 28.8 |
| 5d | 12.6 ± 0.28 | 10.7 | 89.9 ± 5.12 |
| 5e | 11.7 ± 0.28 | 18.7 | 102.5 ± 2.34 |
| 5f | ˃50 | ˃125 | 109.9 ± 0.84 |
| 5g | 22.8 ± 1.02 | 40.7 | 131.5 ± 26.6 |
| EM | - | - | 3.195 |
| ME | 0.53 | - | - |
| RE | - | 0.003 | - |
| Compound | IC50 (μM) |
|---|---|
| 1 | 0.292 ± 0.019 a |
| 2 | 0.14 b |
| 5a | 0.13 ± 0.02 |
| 5b | 0.13 ± 0.01 |
| 5c | 0.92 ± 0.04 |
| 5d | 0.47 ± 0.04 |
| 5e | 0.66 ± 0.03 |
| 5f | 8.71 ± 0.65 |
| 5g | 0.086 ± 0.002 |
| PE | 0.0012 ± 0.0001 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gumbo, M.; Beteck, R.M.; Mandizvo, T.; Seldon, R.; Warner, D.F.; Hoppe, H.C.; Isaacs, M.; Laming, D.; Tam, C.C.; Cheng, L.W.; et al. Cinnamoyl-Oxaborole Amides: Synthesis and Their in Vitro Biological Activity. Molecules 2018, 23, 2038. https://doi.org/10.3390/molecules23082038
Gumbo M, Beteck RM, Mandizvo T, Seldon R, Warner DF, Hoppe HC, Isaacs M, Laming D, Tam CC, Cheng LW, et al. Cinnamoyl-Oxaborole Amides: Synthesis and Their in Vitro Biological Activity. Molecules. 2018; 23(8):2038. https://doi.org/10.3390/molecules23082038
Chicago/Turabian StyleGumbo, Maureen, Richard M. Beteck, Tawanda Mandizvo, Ronnett Seldon, Digby F. Warner, Heinrich C. Hoppe, Michelle Isaacs, Dustin Laming, Christina C. Tam, Luisa W. Cheng, and et al. 2018. "Cinnamoyl-Oxaborole Amides: Synthesis and Their in Vitro Biological Activity" Molecules 23, no. 8: 2038. https://doi.org/10.3390/molecules23082038