Synthesis and Light-Induced Actuation of Photo-Labile 2-Pyridyl-1,2,3-Triazole Ru(bis-bipyridyl) Appended Ferrocene Rotors


Abstract
:1. Introduction
2. Results
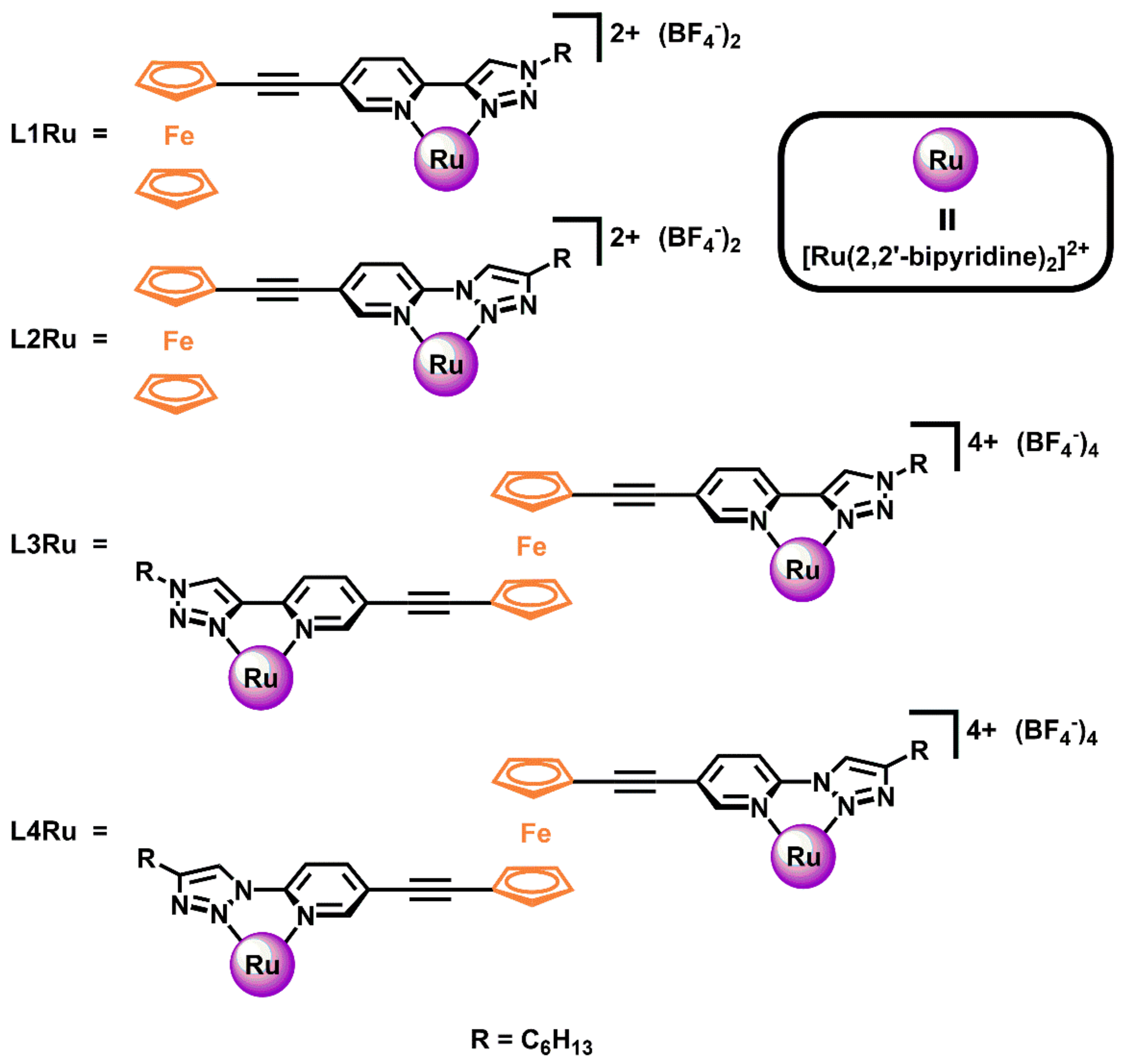
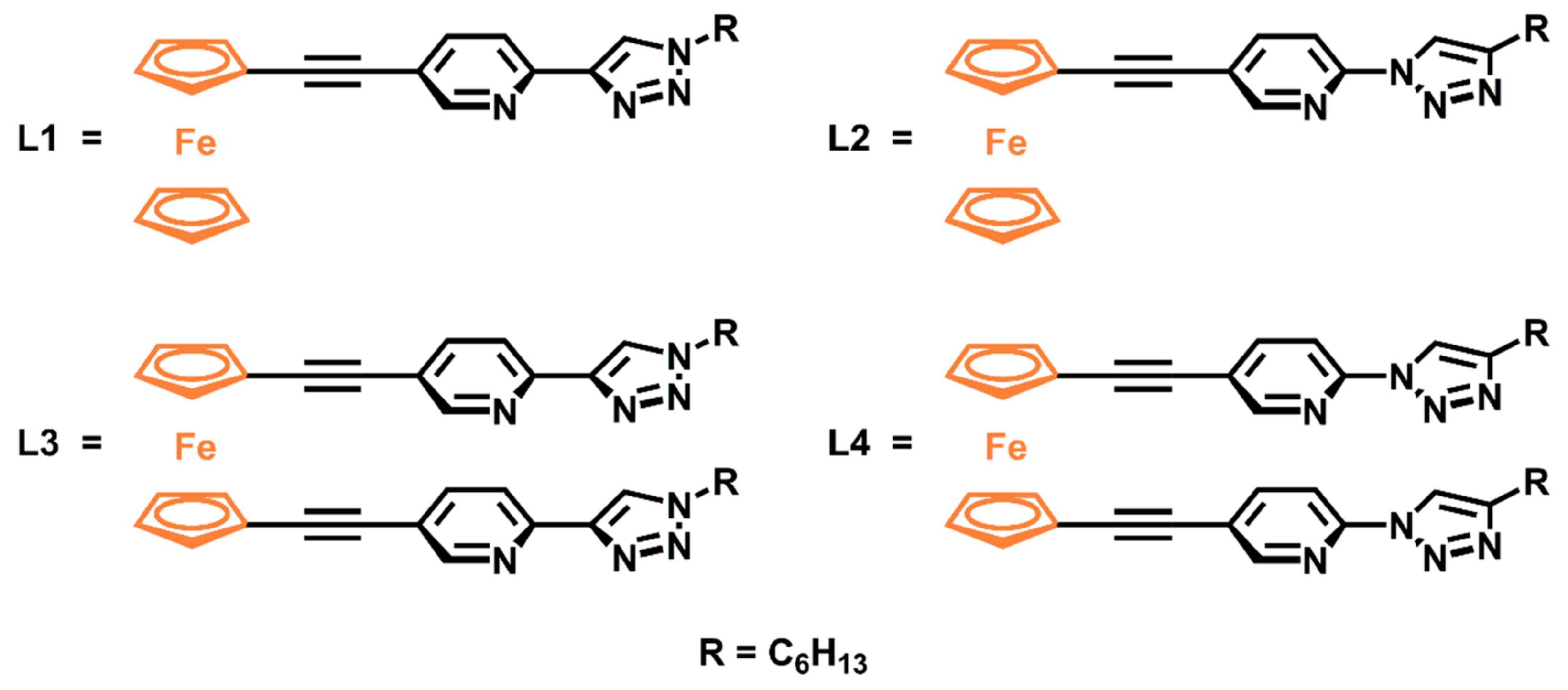
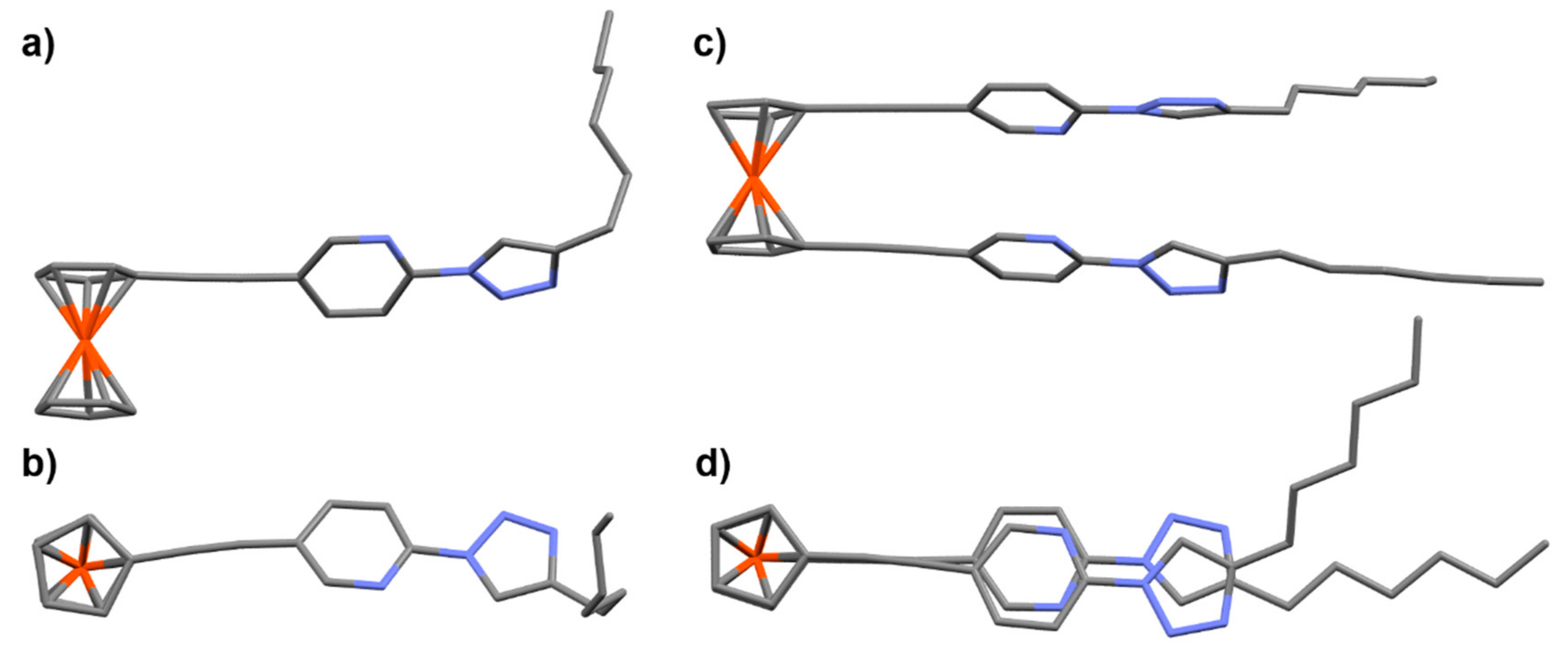
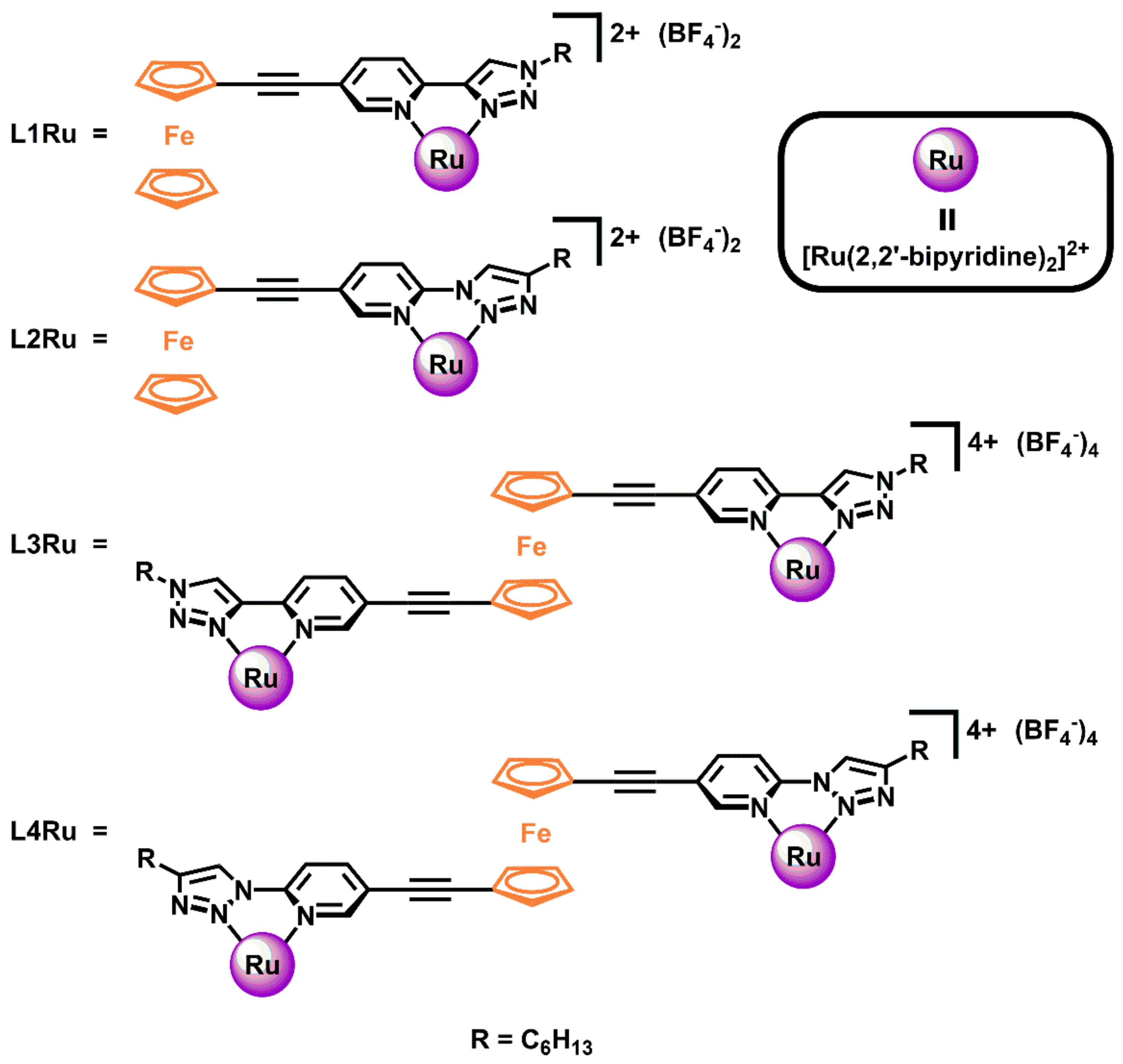
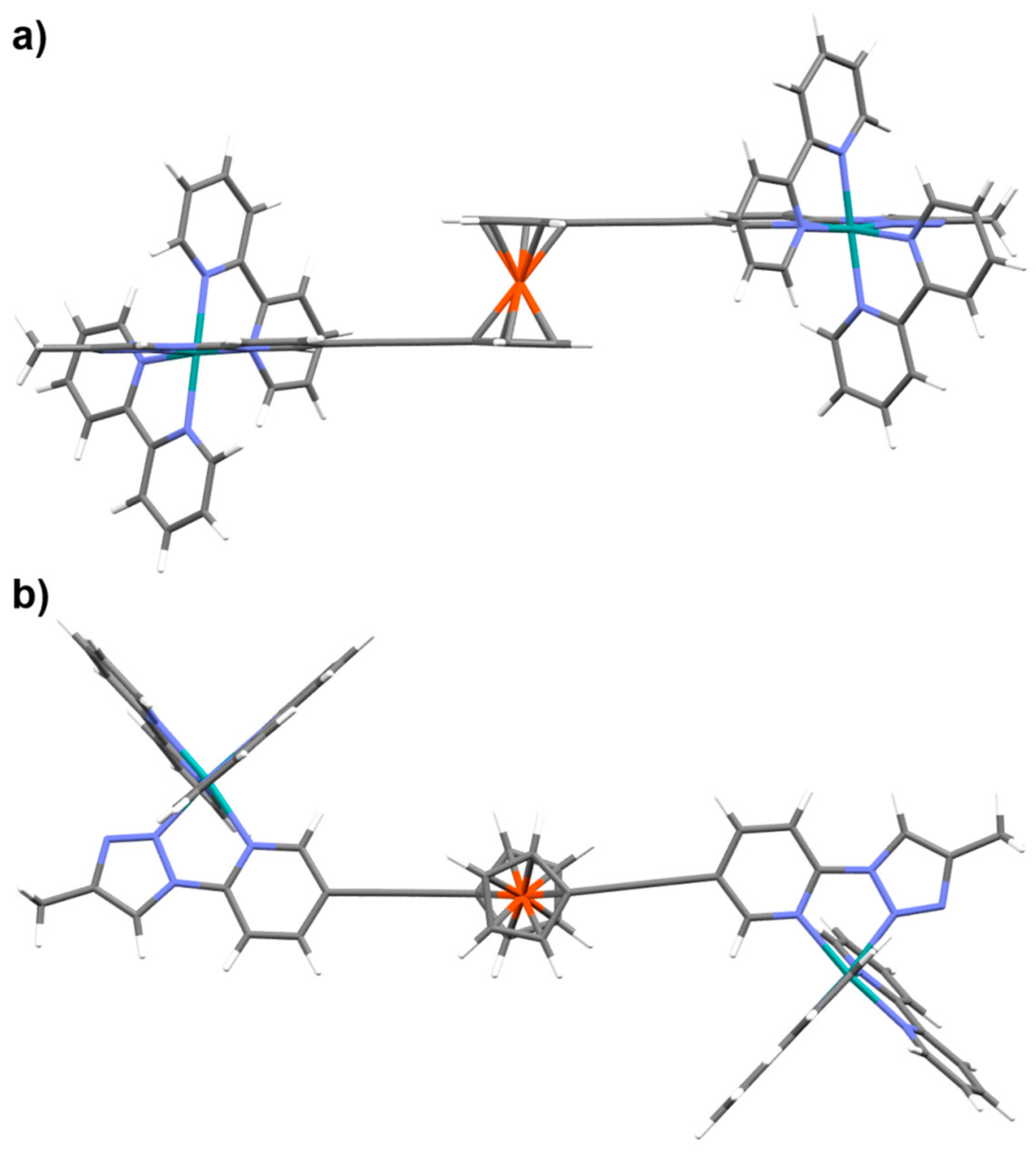
2.1. Synthesis of Ligands and Ru(II)(bipy)2 Complexes
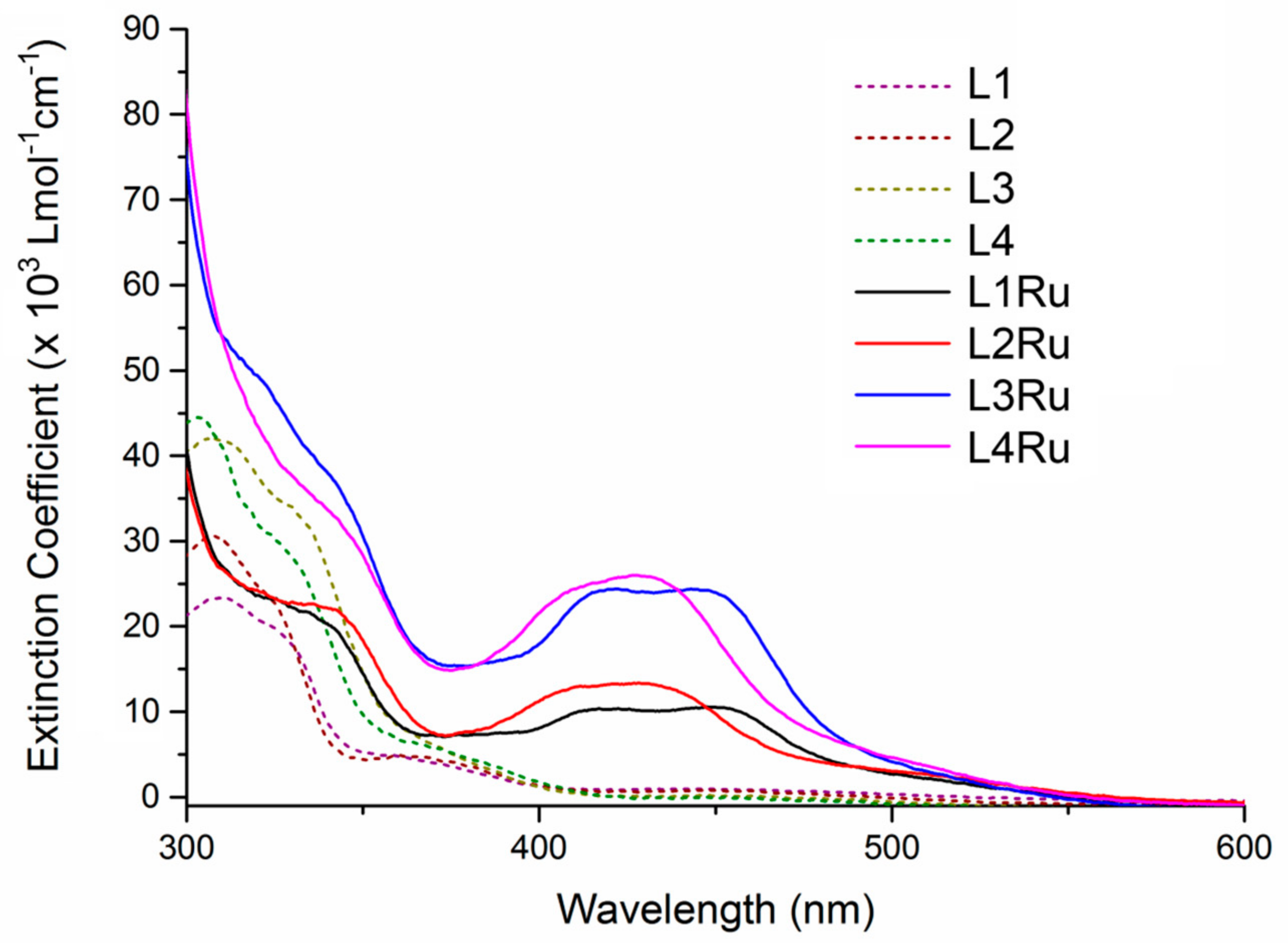
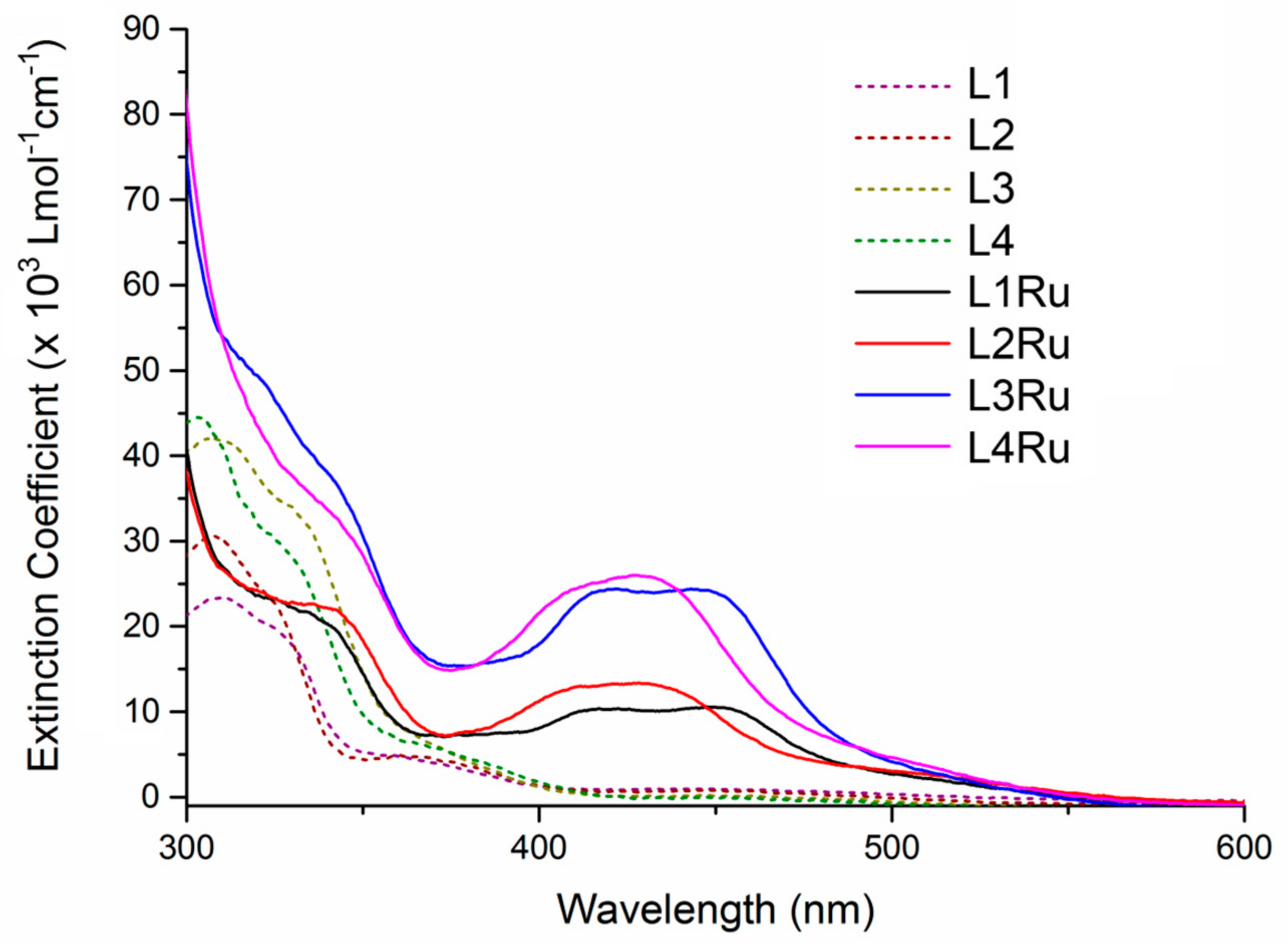
2.2. UV-Visible Spectroscopy
2.3. Electrochemistry
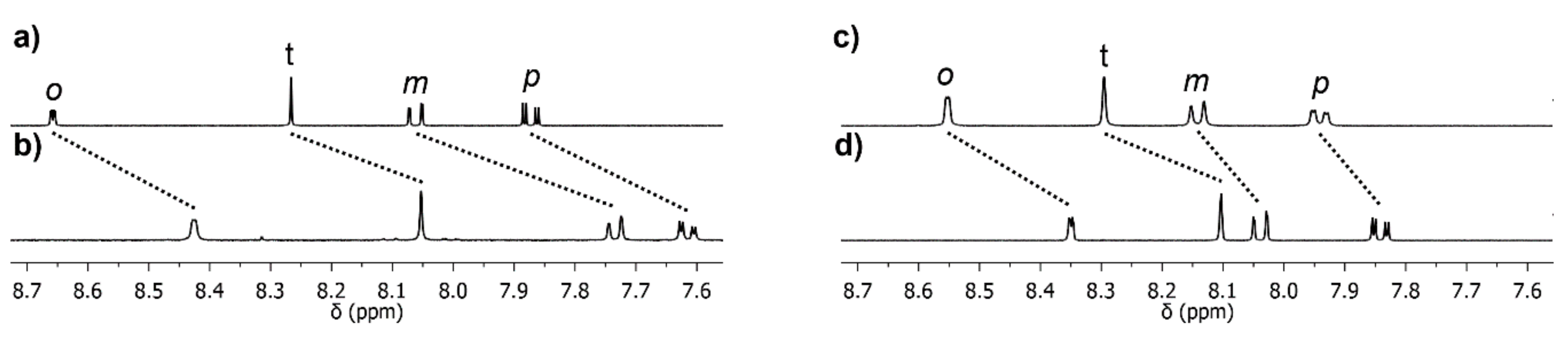
2.4. Photo-Reactivity of L1Ru−L4Ru
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kay, E.R.; Leigh, D.A. Rise of the molecular machines. Angew. Chem. Int. Ed. 2015, 54, 10080–10088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzato, C.; Cheng, C.; Stoddart, J.F.; Astumian, R.D. Mastering the non-equilibrium assembly and operation of molecular machines. Chem. Soc. Rev. 2017, 46, 5491–5507. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Stoddart, J.F. Wholly synthetic molecular machines. Chem. Phys. Chem. 2016, 17, 1780–1793. [Google Scholar] [CrossRef] [PubMed]
- Richards, V. Molecular machines. Nat. Chem. 2016, 8, 1090. [Google Scholar] [CrossRef] [PubMed]
- Feringa, B.L. The art of building small: From molecular switches to motors (Nobel lecture). Angew. Chem. Int. Ed. 2017, 56, 11060–11078. [Google Scholar] [CrossRef] [PubMed]
- Findlay, J.A.; Crowley, J.D. Functional nanomachines: Recent advances in synthetic molecular machinery. Tetrahedron Lett. 2018, 59, 334–346. [Google Scholar] [CrossRef]
- Erbas-Cakmak, S.; Fielden, S.D.P.; Karaca, U.; Leigh, D.A.; McTernan, C.T.; Tetlow, D.J.; Wilson, M.R. Rotary and linear molecular motors driven by pulses of a chemical fuel. Science 2017, 358, 340–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.R.; Solà, J.; Carlone, A.; Goldup, S.M.; Lebrasseur, N.; Leigh, D.A. An autonomous chemically fuelled small-molecule motor. Nature 2016, 534, 235–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogi, S.; Ikeda, T.; Wakabayashi, R.; Shinkai, S.; Takeuchi, M. A bevel-gear-shaped rotor bearing a double-decker porphyrin complex. Chem. Eur. J. 2010, 16, 8285–8290. [Google Scholar] [CrossRef] [PubMed]
- Durot, S.; Reviriego, F.; Sauvage, J.-P. Copper-complexed catenanes and rotaxanes in motion: 15 years of molecular machines. Dalton Trans. 2010, 39, 10557–10570. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, S.; Laursen, B.W.; Flood, A.H.; Hansen, C.N.; Jeppesen, J.O.; Stoddart, J.F. Quantifying the working stroke of tetrathiafulvalene-based electrochemically-driven linear motor-molecules. Chem. Commun. 2006, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Le Poul, N.; Colasson, B. Electrochemically and chemically induced redox processes in molecular machines. ChemElectroChem 2015, 2, 475–496. [Google Scholar] [CrossRef]
- Saha, S.; Stoddart, J.F. Photo-driven molecular devices. Chem. Soc. Rev. 2007, 36, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Balzani, V.; Bergamini, G.; Ceroni, P. From the photochemistry of coordination compounds to light-powered nanoscale devices and machines. Coord. Chem. Rev. 2008, 252, 2456–2469. [Google Scholar] [CrossRef]
- Ceroni, P.; Credi, A.; Venturi, M. Light to investigate (read) and operate (write) molecular devices and machines. Chem. Soc. Rev. 2014, 43, 4068–4083. [Google Scholar] [CrossRef] [PubMed]
- Irie, M.; Fukaminato, T.; Matsuda, K.; Kobatake, S. Photochromism of diarylethene molecules and crystals: Memories, switches and actuators. Chem. Rev. 2014, 114, 12174–12277. [Google Scholar] [CrossRef] [PubMed]
- Bléger, D.; Hecht, S. Visible-light-activated molecular switches. Angew. Chem. Int. Ed. 2015, 54, 11338–11349. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Moscoso, A.; Ballester, P. Light-responsive molecular containers. Chem. Commun. 2017, 53, 4635–4652. [Google Scholar] [CrossRef] [PubMed]
- Tron, A.; Jacquot de Rouville, H.-P.; Ducrot, A.; Tucker, J.H.R.; Baroncini, M.; Credi, A.; McClenaghan, N.D. Photodriven [2]rotaxane–[2]catenane interconversion. Chem. Commun. 2015, 51, 2810–2813. [Google Scholar] [CrossRef] [PubMed]
- Tron, A.; Thornton, P.J.; Lincheneau, C.; Desvergne, J.-P.; Spencer, N.; Tucker, J.H.R.; McClenaghan, N.D. Reversible photocapture of a [2]rotaxane harnessing a barbiturate template. J. Org. Chem. 2015, 80, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Tron, A.; Pianet, I.; Martinez-Cuezva, A.; Tucker, J.H.R.; Pisciottani, L.; Alajarin, M.; Berna, J.; McClenaghan, N.D. Remote photoregulated ring gliding in a [2]rotaxane via a molecular effector. Org. Lett. 2017, 19, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Koumura, N.; Zijlstra, R.W.J.; van Delden, R.A.; Harada, N.; Feringa, B.L. Light-driven monodirectional molecular rotor. Nature 1999, 401, 152–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ter Wiel, M.K.J.; van Delden, R.A.; Meetsma, A.; Feringa, B.L. Light-driven molecular motors: Stepwise thermal helix inversion during unidirectional rotation of sterically overcrowded biphenanthrylidenes. J. Am. Chem. Soc. 2005, 127, 14208–14222. [Google Scholar] [CrossRef] [PubMed]
- Kazaryan, A.; Kistemaker, J.C.M.; Schäfer, L.V.; Browne, W.R.; Feringa, B.L.; Filatov, M. Understanding the dynamics behind the photoisomerization of a light-driven fluorene molecular rotary motor. J. Phys. Chem. A 2010, 114, 5058–5067. [Google Scholar] [CrossRef] [PubMed]
- Kistemaker, J.C.M.; Štacko, P.; Roke, D.; Wolters, A.T.; Heideman, G.H.; Chang, M.-C.; van der Meulen, P.; Visser, J.; Otten, E.; Feringa, B.L. Third-generation light-driven symmetric molecular motors. J. Am. Chem. Soc. 2017, 139, 9650–9661. [Google Scholar] [CrossRef] [PubMed]
- Credi, A.; Silvi, S.; Venturi, M. Light-operated machines based on threaded molecular structures. In Molecular Machines and Motors: Recent Advances and Perspectives; Credi, A., Silvi, S., Venturi, M., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 1–34. [Google Scholar]
- Berná, J.; Leigh, D.A.; Lubomska, M.; Mendoza, S.M.; Pérez, E.M.; Rudolf, P.; Teobaldi, G.; Zerbetto, F. Macroscopic transport by synthetic molecular machines. Nat. Mater. 2005, 4, 704–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, K.-S.; Chang, K.-J.; An, Y.-J. A pseudorotaxane-based molecular machine controlled by light and thermal stimuli. Chem. Commun. 2003, 1450–1451. [Google Scholar] [CrossRef]
- Colasson, B.; Credi, A.; Ragazzon, G. Light-driven molecular machines based on ruthenium(II) polypyridine complexes: Strategies and recent advances. Coord. Chem. Rev. 2016, 325, 125–134. [Google Scholar] [CrossRef]
- Armaroli, N.; Balzani, V.; Collin, J.-P.; Gaviña, P.; Sauvage, J.-P.; Ventura, B. Rotaxanes incorporating two different coordinating units in their thread: Synthesis and electrochemically and photochemically induced molecular motions. J. Am. Chem. Soc. 1999, 121, 4397–4408. [Google Scholar] [CrossRef]
- Livoreil, A.; Sauvage, J.-P.; Armaroli, N.; Balzani, V.; Flamigni, L.; Ventura, B. Electrochemically and photochemically driven ring motions in a disymmetrical copper [2]-catenate. J. Am. Chem. Soc. 1997, 119, 12114–12124. [Google Scholar] [CrossRef] [PubMed]
- Laemmel, A.C.; Collin, J.P.; Sauvage, J.P. Efficient and selective photochemical labilization of a given bidentate ligand in mixed ruthenium(II) complexes of the Ru(phen)2L2+ and Ru(bipy)2L2+ family (L = sterically hindering chelate). Eur. J. Inorg. Chem. 1999, 1999, 383–386. [Google Scholar] [CrossRef]
- Collin, J.-P.; Laemmel, A.-C.; Sauvage, J.-P. Photochemical expulsion of a Ru(phen)2 unit from a macrocyclic receptor and its thermal recoordination. New J. Chem. 2001, 25, 22–24. [Google Scholar] [CrossRef]
- Mobian, P.; Kern, J.-M.; Sauvage, J.-P. Light-driven machine prototypes based on dissociative excited states: Photoinduced decoordination and thermal recoordination of a ring in a ruthenium(I)-containing [2]catenane. Angew. Chem. Int. Ed. 2004, 43, 2392–2395. [Google Scholar] [CrossRef] [PubMed]
- Collin, J.-P.; Jouvenot, D.; Koizumi, M.; Sauvage, J.-P. A ruthenium(II)-complexed rotaxane whose ring incorporates a 6,6′-diphenyl-2,2′-bipyridine: Synthesis and light-driven motions. Eur. J. Inorg. Chem. 2005, 2005, 1850–1855. [Google Scholar] [CrossRef]
- Scattergood, P.A.; Ross, D.A.W.; Rice, C.R.; Elliott, P.I.P. Labilizing the photoinert: Extraordinarily facile photochemical ligand ejection in an [Os(N^N)3]2+ complex. Angew. Chem. Int. Ed. 2016, 55, 10697–10701. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Renfrew, A.K. Photolabile ruthenium complexes to cage and release a highly cytotoxic anticancer agent. J. Inorg. Biochem. 2018, 179, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Havrylyuk, D.; Heidary, D.K.; Nease, L.; Parkin, S.; Glazer, E.C. Photochemical properties and structure—Activity relationships of Ru(II) complexes with pyridylbenzazole ligands as promising anticancer agents. Eur. J. Inorg. Chem. 2017, 2017, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Wachter, E.; Moyá, D.; Parkin, S.; Glazer, E.C. Ruthenium complex “light switches” that are selective for different g-quadruplex structures. Chem. Eur. J. 2016, 22, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Kohler, L.; Nease, L.; Vo, P.; Garofolo, J.; Heidary, D.K.; Thummel, R.P.; Glazer, E.C. Photochemical and photobiological activity of Ru(II) homoleptic and heteroleptic complexes containing methylated bipyridyl-type ligands. Inorg. Chem. 2017, 56, 12214–12223. [Google Scholar] [CrossRef] [PubMed]
- Abreu, F.D.; Paulo, T.D.F.; Gehlen, M.H.; Ando, R.A.; Lopes, L.G.F.; Gondim, A.C.S.; Vasconcelos, M.A.; Teixeira, E.H.; Sousa, E.H.S.; de Carvalho, I.M.M. Aryl-substituted ruthenium(II) complexes: A strategy for enhanced photocleavage and efficient DNA binding. Inorg. Chem. 2017, 56, 9084–9096. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Rees, T.W.; Ke, L.; Ji, L.; Chao, H. Harnessing ruthenium(II) as photodynamic agents: Encouraging advances in cancer therapy. Coord. Chem. Rev. 2018, 363, 17–28. [Google Scholar] [CrossRef]
- Alfredo, N.V.; Jalapa, N.E.; Morales, S.L.; Ryabov, A.D.; Le Lagadec, R.; Alexandrova, L. Light-driven living/controlled radical polymerization of hydrophobic monomers catalyzed by ruthenium(II) metallacycles. Macromolecules 2012, 45, 8135–8146. [Google Scholar] [CrossRef]
- Laramée-Milette, B.; Nastasi, F.; Puntoriero, F.; Campagna, S.; Hanan, G.S. Cover feature: Photo-induced assembly of a luminescent tetraruthenium square. Chem. Eur. J. 2017, 23, 16410. [Google Scholar] [CrossRef]
- Bonnet, S.; Collin, J.-P. Ruthenium-based light-driven molecular machine prototypes: Synthesis and properties. Chem. Soc. Rev. 2008, 37, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Collin, J.-P.; Jouvenot, D.; Koizumi, M.; Sauvage, J.-P. Light-driven expulsion of the sterically hindering ligand L in tris-diimine ruthenium(II) complexes of the Ru(phen)2(L)2+ family: A pronounced ring effect. Inorg. Chem. 2005, 44, 4693–4698. [Google Scholar] [CrossRef] [PubMed]
- Hammarström, L.; Alsins, J.; Börje, A.; Norrby, T.; Zhang, L.; Åkermark, B. Structure and photophysical properties of novel ruthenium(II) complexes containing 6-substituted bipyridines. J. Photochem. Photobiol. A 1997, 102, 139–150. [Google Scholar] [CrossRef]
- Welby, C.E.; Armitage, G.K.; Bartley, H.; Sinopoli, A.; Uppal, B.S.; Elliott, P.I.P. Photochemical ligand ejection from non-sterically promoted Ru(II)bis(diimine) 4,4′-bi-1,2,3-triazolyl complexes. Photochem. Photobiol. Sci. 2014, 13, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.K.C.; Huff, G.S.; Cubanski, J.R.; Kennedy, A.D.W.; McAdam, C.J.; McMorran, D.A.; Gordon, K.C.; Crowley, J.D. Comparison of inverse and regular 2-pyridyl-1,2,3-triazole “click” complexes: Structures, stability, electrochemical, and photophysical properties. Inorg. Chem. 2015, 54, 1572–1587. [Google Scholar] [CrossRef] [PubMed]
- Hohloch, S.; Schweinfurth, D.; Sommer, M.G.; Weisser, F.; Deibel, N.; Ehret, F.; Sarkar, B. The redox series [Ru(bpy)2(L)]n, n = +3, +2, +1, 0, with L = bipyridine, “click” derived pyridyl-triazole or bis-triazole: A combined structural, electrochemical, spectroelectrochemical and DFT investigation. Dalton Trans. 2014, 43, 4437–4450. [Google Scholar] [CrossRef] [PubMed]
- Dixon, I.M.; Heully, J.-L.; Alary, F.; Elliott, P.I.P. Theoretical illumination of highly original photoreactive 3MC states and the mechanism of the photochemistry of Ru(II) tris(bidentate) complexes. Phys. Chem. Chem. Phys. 2017, 19, 27765–27778. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Miyazaki, T.; Tanaka, N.; Tatsuma, T. Three-dimensional motion and transformation of a photoelectrochemical actuator. Chem. Commun. 2006, 2024–2026. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Fuks, G.; Moulin, E.; Maaloum, M.; Rawiso, M.; Kulic, I.; Foy, J.T.; Giuseppone, N. Macroscopic contraction of a gel induced by the integrated motion of light-driven molecular motors. Nat. Nanotechnol. 2015, 10, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Foy, J.T.; Li, Q.; Goujon, A.; Colard-Itté, J.-R.; Fuks, G.; Moulin, E.; Schiffmann, O.; Dattler, D.; Funeriu, D.P.; Giuseppone, N. Dual-light control of nanomachines that integrate motor and modulator subunits. Nat. Nanotechnol. 2017, 12, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-L.; Liebig, T.; Hecht, S.; Bléger, D.; Rabe, J.P. Light-induced contraction and extension of single macromolecules on a modified graphite surface. ACS Nano 2014, 8, 11987–11993. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-B.; Dai, B.; Woo, H.-K.; Wang, L.-S. Intramolecular rotation through proton transfer: [Fe(η5-C5H4CO2−)2] versus [(η5-C5H4CO2−)Fe(η5-C5H4CO2H)]. Angew. Chem. Int. Ed. 2005, 44, 6022–6024. [Google Scholar] [CrossRef] [PubMed]
- Pisciottani, L.; Douarre, M.; Bibal, B.; Davies, C.; Roberts, H.; Kauffmann, B.; Horswell, S.L.; Tucker, J.H.R.; McClenaghan, N.D. Macrocyclic hamilton-type receptors comprising a ferrocene pivot. Supramol. Chem. 2018, 30, 869–875. [Google Scholar] [CrossRef]
- Heinze, K.; Schlenker, M. Anion-induced motion in a ferrocene diamide. Eur. J. Inorg. Chem. 2005, 2005, 66–71. [Google Scholar] [CrossRef]
- Takai, A.; Yasuda, T.; Ishizuka, T.; Kojima, T.; Takeuchi, M. A directly linked ferrocene–naphthalenediimide conjugate: Precise control of stacking structures of π-systems by redox stimuli. Angew. Chem. Int. Ed. 2013, 52, 9167–9171. [Google Scholar] [CrossRef] [PubMed]
- Iordache, A.; Oltean, M.; Milet, A.; Thomas, F.; Baptiste, B.; Saint-Aman, E.; Bucher, C. Redox control of rotary motions in ferrocene-based elemental ball bearings. J. Am. Chem. Soc. 2012, 134, 2653–2671. [Google Scholar] [CrossRef] [PubMed]
- Scottwell, S.O.; Elliott, A.B.S.; Shaffer, K.J.; Nafady, A.; McAdam, C.J.; Gordon, K.C.; Crowley, J.D. Chemically and electrochemically induced expansion and contraction of a ferrocene rotor. Chem. Commun. 2015, 51, 8161–8164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scottwell, S.O.; Barnsley, J.E.; McAdam, C.J.; Gordon, K.C.; Crowley, J.D. A ferrocene based switchable molecular folding ruler. Chem. Commun. 2017, 53, 7628–7631. [Google Scholar] [CrossRef] [PubMed]
- Collin, J.-P.; Gavinã, P.; Sauvage, J.-P. Electrochemically induced molecular motions in a copper(I) complex pseudorotaxane. Chem. Commun. 1996, 2005–2006. [Google Scholar] [CrossRef]
- Collin, J.-P.; Gavina, P.; Sauvage, J.-P. Electrochemically induced molecular motions in copper-complexed threaded systems: From the unstoppered compound to the semi-rotaxane and the fully blocked rotaxane. New J. Chem. 1997, 21, 525–528. [Google Scholar]
- Schmittel, M.; Ganz, A.; Schenk, W.A.; Hagel, M. Synthesis and coordination properties of 6,6′-dimesityl-2,2′-bipyridine. Z. Naturforsch. B 1999, 54, 559–564. [Google Scholar] [CrossRef]
- Schmittel, M.; He, B.; Fan, J.; Bats, J.W.; Engeser, M.; Schlosser, M.; Deiseroth, H.-J. Cap for copper(I) ions! Metallosupramolecular solid and solution state structures on the basis of the dynamic tetrahedral [Cu(phenar2)(py)2]+ motif. Inorg. Chem. 2009, 48, 8192–8200. [Google Scholar] [CrossRef] [PubMed]
- Findlay, J.A.; McAdam, C.J.; Sutton, J.J.; Preston, D.; Gordon, K.C.; Crowley, J.D. Metallosupramolecular architectures formed with ferrocene-linked bis-bidentate ligands: Synthesis, structures, and electrochemical studies. Inorg. Chem. 2018, 57, 3602–3614. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K. Ultraviolet spectra and structures of 2,2′-bipyridine and 2,2′,2′′-terpyridine in aqueous solution. J. Phys. Chem. 1960, 64, 1420–1425. [Google Scholar] [CrossRef]
- Bozak, R.E. Photochemistry in the metallocenes. In Advances in Photochemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1971; pp. 227–244. [Google Scholar]
- Inkpen, M.S.; Du, S.; Driver, M.; Albrecht, T.; Long, N.J. Oxidative purification of halogenated ferrocenes. Dalton Trans. 2013, 42, 2813–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noviandri, I.; Brown, K.N.; Fleming, D.S.; Gulyas, P.T.; Lay, P.A.; Masters, A.F.; Phillips, L. The decamethylferrocenium/decamethylferrocene redox couple: A superior redox standard to the ferrocenium/ferrocene redox couple for studying solvent effects on the thermodynamics of electron transfer. J. Phys. Chem. B 1999, 103, 6713–6722. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors upon request. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E° (ΔE) (V (mV)) a | ||||
|---|---|---|---|---|
| Compound | Bipy 1(0/-) | Bipy 2(0/-) | Fc(+/0) | Ru(III/II) |
| L1 | - | - | 0.63 (88) | - |
| L2 | - | - | 0.62 (86) | - |
| L3 | - | - | 0.69 (90) | - |
| L4 | - | - | 0.75 (84) | - |
| L1Ru | −1.51 (150) | −1.22 (80) | 0.69 (79) | 1.51 (110) |
| L2Ru | −1.38 (150) | −1.14 (86) | 0.70 (76) | 1.60 b |
| L3Ru | −1.50 (200) | −1.21 (84) | 0.83 (72) | 1.48 (84) |
| L4Ru | −1.41 (200) | −1.15 (120) | 0.83 (88) | 1.57 (170) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Findlay, J.A.; Barnsley, J.E.; Gordon, K.C.; Crowley, J.D. Synthesis and Light-Induced Actuation of Photo-Labile 2-Pyridyl-1,2,3-Triazole Ru(bis-bipyridyl) Appended Ferrocene Rotors. Molecules 2018, 23, 2037. https://doi.org/10.3390/molecules23082037
Findlay JA, Barnsley JE, Gordon KC, Crowley JD. Synthesis and Light-Induced Actuation of Photo-Labile 2-Pyridyl-1,2,3-Triazole Ru(bis-bipyridyl) Appended Ferrocene Rotors. Molecules. 2018; 23(8):2037. https://doi.org/10.3390/molecules23082037
Chicago/Turabian StyleFindlay, James A., Jonathan E. Barnsley, Keith C. Gordon, and James D. Crowley. 2018. "Synthesis and Light-Induced Actuation of Photo-Labile 2-Pyridyl-1,2,3-Triazole Ru(bis-bipyridyl) Appended Ferrocene Rotors" Molecules 23, no. 8: 2037. https://doi.org/10.3390/molecules23082037