Probing the Anticancer Action of Novel Ferrocene Analogues of MNK Inhibitors

 , and
, and
Abstract
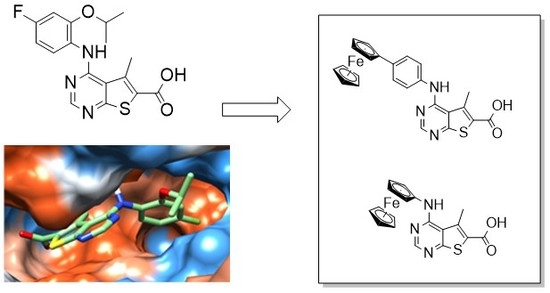
:
1. Introduction
2. Results
2.1. Molecular Modelling of Compound 1 Reveals a Large Hydrophobic Cavity in MNK2
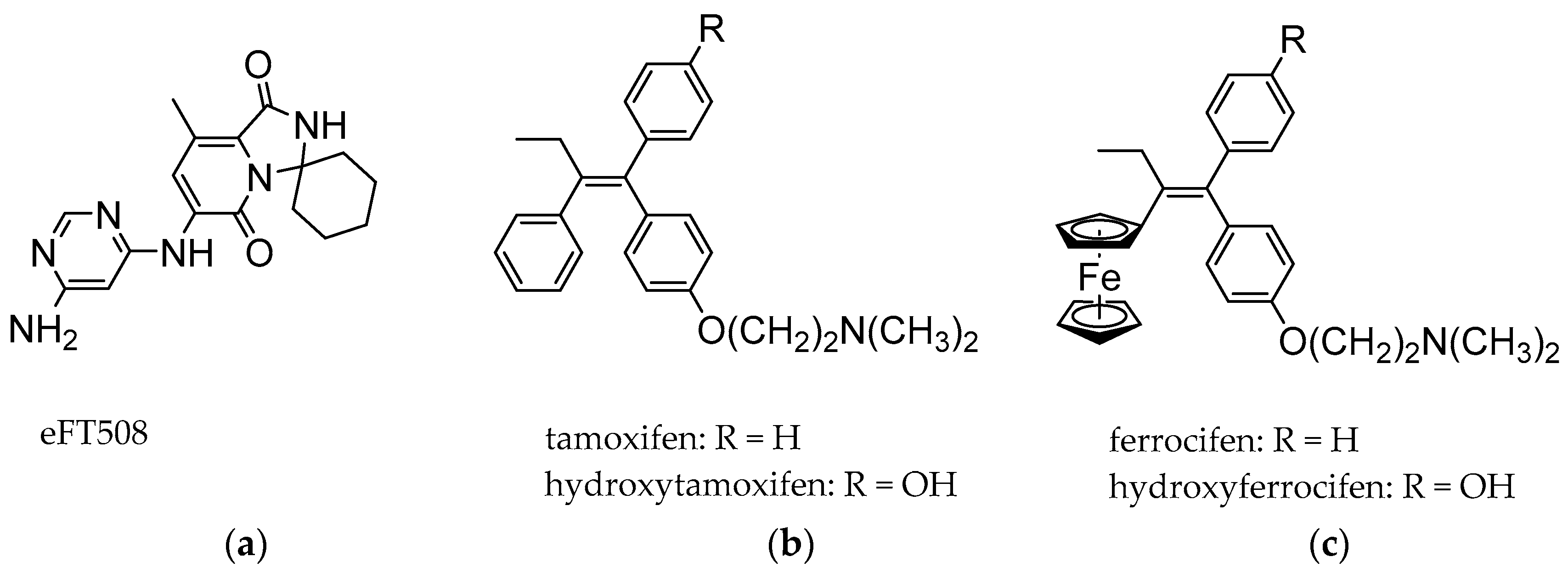
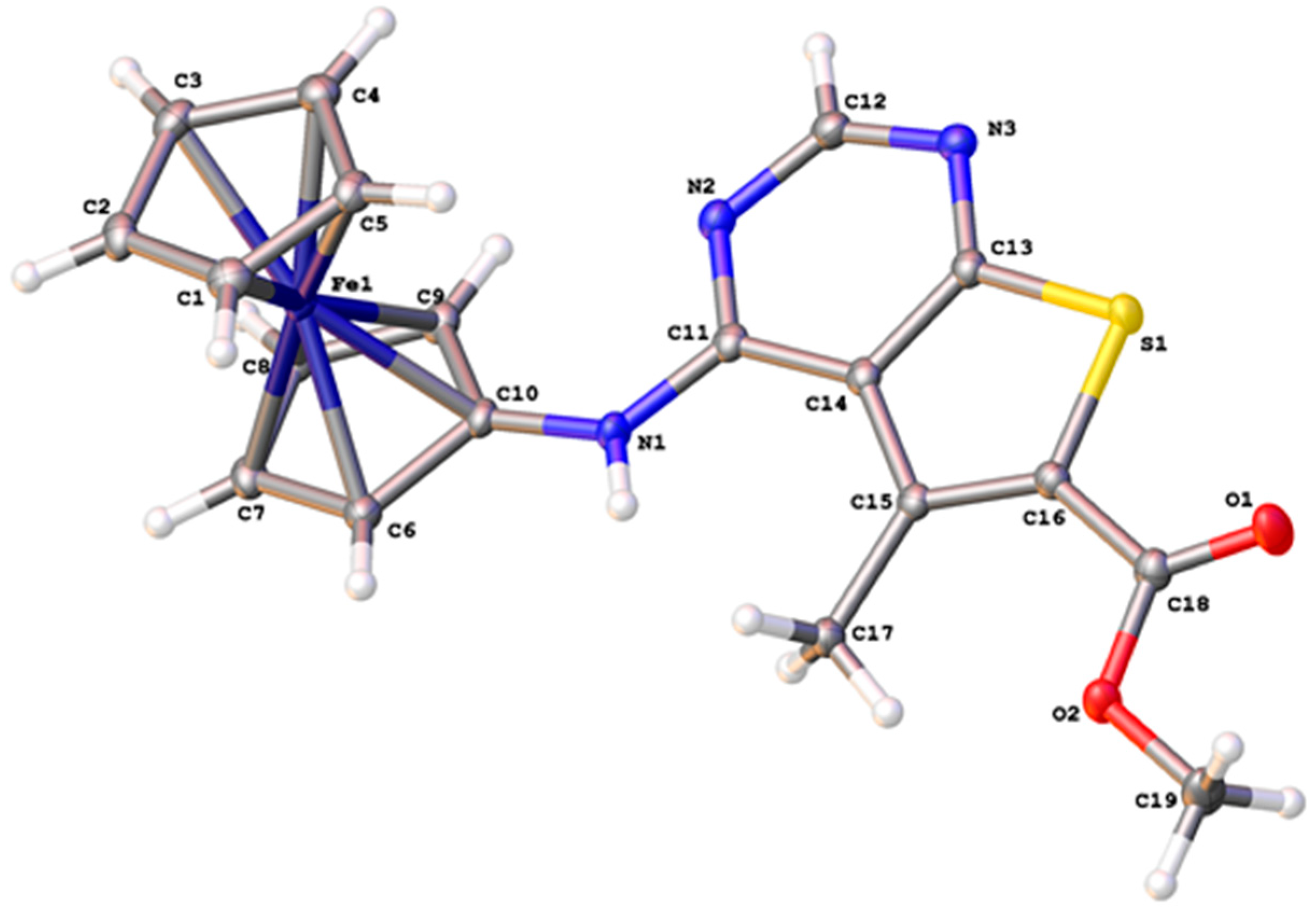
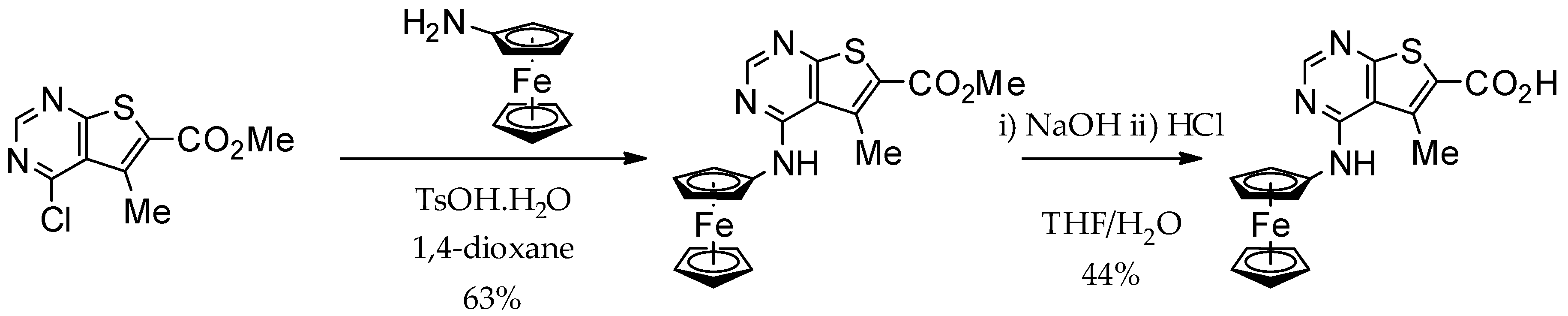
2.2. Synthesis of Ferrocene Analogues
2.3. In Vitro Analysis of Compound 1, 3, and 5 in Cancer Cell Lines
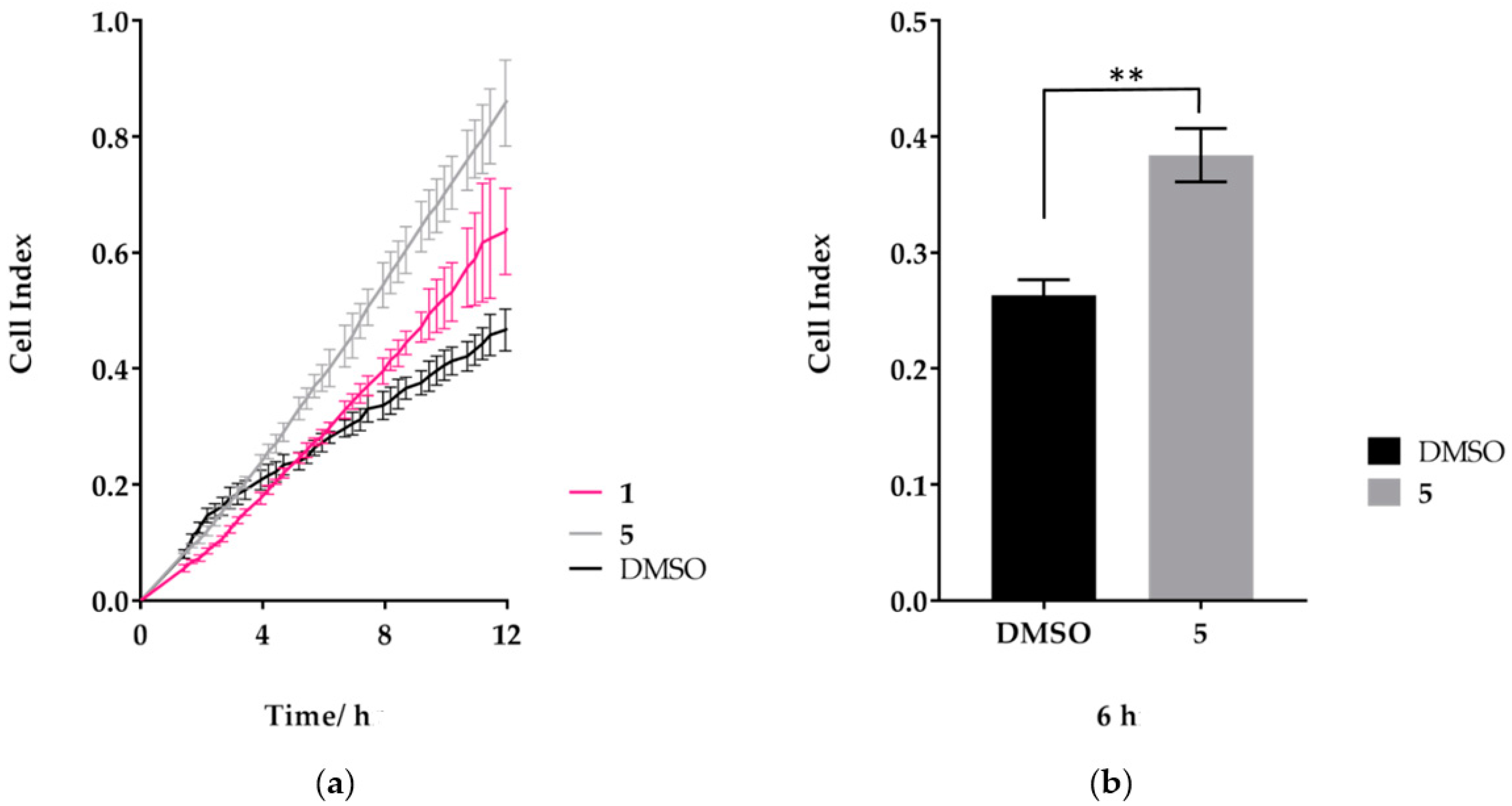
2.4. Treatment of MDA-MB-231 Cells with Compound 5 Increases the Rate of Cell Migration
2.5. Analysis of Compound 1 and 5 on MDA-MB-231 Spheroid Growth
2.6. In Vitro Kinase Assays Report That Compound 5 Has No Effect on MNK1/2 Kinase Activity
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability
4.3. Cell Migration Assay
4.4. In Vitro Kinase Assays
4.5. Three-Dimensional Cell Culture
4.6. Chemical Synthesis: General Procedures
4.7. Chemical Synthesis: Experimental
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davies, N.J.; Batehup, L.; Thomas, R. The role of diet and physical activity in breast, colorectal, and prostate cancer survivorship: A review of the literature. Br. J. Cancer 2015, 105 (Suppl. 1), S52–S73. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; Hsu, F.D.; Jensen, K.; Cheang, M.; Karaca, G.; Hu, Z.; Hernandez-Boussard, T.; Livasy, C.; Cowan, D.; Dressler, L.; et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin. Cancer Res. 2004, 10, 5367–5374. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.; Dabbs, D.J.; Schnitt, S.J.; Baehner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Basal-like and triple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2011, 24, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Mcgranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Ramon, Y.C.S.; Castellvi, J.; Hummer, S.; Peg, V.; Pelletier, J.; Sonenberg, N. Beyond molecular tumor heterogeneity: Protein synthesis takes control. Oncogene 2018, 37, 2490–2501. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Chen, A.; Bai, Z. Integrative investigation on breast cancer in ER, PR and HER2-defined subgroups using mRNA and miRNA expression profiling. Sci. Rep. 2014, 4, 6566. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D. Translational control in cancer etiology. Cold Spring Harb. Perspect. Biol. 2012, 5, a012336. [Google Scholar] [CrossRef]
- Ueda, T.; Watanabe-Fukunaga, R.; Fukuyama, H.; Nagata, S.; Fukunaga, R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol. Cell. Biol. 2004, 24, 6539–6549. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. Mnk earmarks eIF4E for cancer therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 13975–13976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beggs, J.E.; Tian, S.; Jones, G.G.; Xie, J.; Iadevaia, V.; Jenei, V.; Thomas, G.; Proud, C.G. The MAP kinase-interacting kinases regulate cell migration, vimentin expression and eIF4E/CYFIP1 binding. Biochem. J. 2015, 467, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Diab, S.; Kumarasiri, M.; Yu, M.; Teo, T.; Proud, C.; Milne, R.; Wang, S. MAP kinase-interacting kinases—Emerging targets against cancer. Chem. Biol. 2014, 21, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.H.; Sprengeler, P.A.; Chiang, G.G.; Appleman, J.R.; Chen, J.; Clarine, J.; Eam, B.; Ernst, J.T.; Han, Q.; Goel, V.K.; et al. Structure-based Design of Pyridone-Aminal eFT508 Targeting Dysregulated Translation by Selective Mitogen-activated Protein Kinase Interacting Kinases 1 and 2 (MNK1/2) Inhibition. J. Med. Chem. 2018, 61, 3516–3540. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.; Gasser, G. The medicinal chemistry of ferrocene and its derivatives. Nat. Rev. Chem. 2017, 1, 0066. [Google Scholar] [CrossRef]
- Jaouen, G.; Vessieres, A.; Top, S. Ferrocifen type anti cancer drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C.; Allen, K.E.; Dix, C.J. Pharmacology of tamoxifen in laboratory animals. Cancer Treat. Rep. 1980, 64, 745–759. [Google Scholar] [PubMed]
- Gottardis, M.M.; Jordan, V.C. Antitumor actions of keoxifene and tamoxifen in the N-nitrosomethylurea-induced rat mammary carcinoma model. Cancer Res. 1987, 47, 4020–4024. [Google Scholar] [PubMed]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 2004, 11, 643–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.; Vessières, A.; Hillard, E.; Top, S.; Pigeon, P.; Jaouen, G. Ferrocifens and ferrocifenols as new potential weapons against breast cancer. CHIMIA Int. J. Chem. 2007, 61, 716–724. [Google Scholar] [CrossRef]
- Görmen, M.; Pigeon, P.; Top, S.; Hillard, E.A.; Huché, M.; Hartinger, C.G.; de Montigny, F.; Plamont, M.A.; Vessières, A.; Jaouen, G. Synthesis, cytotoxicity, and COMPARE analysis of ferrocene and [3]ferrocenophane tetrasubstituted olefin derivatives against human cancer cells. ChemMedChem 2010, 5, 2039–2050. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L. Quinone methide bioactivation pathway: Contribution to toxicity and/or cytoprotection? Curr. Org. Chem. 2014, 18, 61–69. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, A.C.; Hillard, E.A.; Pigeon, P.; Rocha, D.D.; Rodrigues, F.A.; Montenegro, R.C.; Costa-Lotufo, L.V.; Goulart, M.O.; Jaouen, G. Biological evaluation of twenty-eight ferrocenyl tetrasubstituted olefins: Cancer cell growth inhibition, ROS production and hemolytic activity. Eur. J. Med. Chem. 2011, 46, 3778–3787. [Google Scholar] [CrossRef] [PubMed]
- Wlassoff, W.A.; Albright, C.D.; Sivashinski, M.S.; Ivanova, A.; Appelbaum, J.G.; Salganik, R.I. Hydrogen peroxide overproduced in breast cancer cells can serve as an anticancer prodrug generating apoptosis-stimulating hydroxyl radicals under the effect of tamoxifen-ferrocene conjugate. J. Pharm. Pharmacol. 2007, 59, 1549–1553. [Google Scholar] [CrossRef]
- Vessieres, A.; Corbet, C.; Heldt, J.M.; Lories, N.; Jouy, N.; Laïos, I.; Leclercq, G.; Jaouen, G.; Toillon, R.A. A ferrocenyl derivative of hydroxytamoxifen elicits an estrogen receptor-independent mechanism of action in breast cancer cell lines. J. Inorg. Biochem. 2010, 104, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.; Mendham, A.P.; Kotha, A.K.; Richardson, S.C.W.; Hillard, E.A.; Jaouen, G.; Male, L.; Hursthouse, M.B. Structural and biological investigation of ferrocene-substituted 3-methylidene-1,3-dihydro-2H-indol-2-ones. Dalton Trans. 2009, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Librizzi, M.; Longo, A.; Chiarelli, R.; Amin, J.; Spencer, J.; Luparello, C. Cytotoxic effects of Jay Amin hydroxamic acid (JAHA), a ferrocene-based class I histone deacetylase inhibitor, on triple-negative MDA-MB231 breast cancer cells. Chem. Res. Toxicol. 2012, 25, 2608–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, J.; Chuckowree, I.S.; Wang, M.; Tizzard, G.J.; Coles, S.J.; Spencer, J. Synthesis of Oxindole-Based Bioorganometallic Kinase Inhibitors Incorporating One or More Ferrocene Groups. Organometallics 2013, 32, 5818–5825. [Google Scholar] [CrossRef]
- Ocasio, C.J.; Sansook, S.; Jones, R.; Roberts, J.M.; Scott, T.G.; Tsoureas, N.; Coxhead, P.; Guille, M.; Tizzard, G.J.; Coles, S.J. Pojamide: An HDAC3-Selective Ferrocene Analogue with Remarkably Enhanced Redox-Triggered Ferrocenium Activity in Cells. Organometallics 2017, 36, 3276–3283. [Google Scholar] [CrossRef]
- Han, W.; Ding, Y.; Xu, Y.; Pfsiter, K.; Zhu, S.; Warne, R.; Doyle, M.; Aikawa, M.; Amiri, P.; Appleton, B.; et al. Discovery of a Selective and Potent Inhibitor of Mitogen-Activated Protein Kinase-Interacting Kinases 1 and 2 (MNK1/2) Utilizing Structure-Based Drug Design. J. Med. Chem. 2016, 59, 3034–3045. [Google Scholar] [CrossRef] [PubMed]
- Jauch, R.; Jakel, S.; Netter, C.; Schreiter, K.; Aicher, B.; Jäckle, H.; Wahl, M.C. Crystal structures of the Mnk2 kinase domain reveal an inhibitory conformation and a zinc binding site. Structure 2005, 13, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Poulsen, A.; Yang, H.Y.; Ho, M.; Ang, S.H.; Eldwin, T.S.; Jeyaraj, D.A.; Chennamaneni, L.R.; Liu, B.; Hill, J.; et al. Probing the binding mechanism of Mnk inhibitors by docking and molecular dynamics simulations. Biochemistry 2015, 54, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Teo, T.; Sykes, M.J.; Wang, S. Insights into the Importance of DFD-Motif and Insertion I1 in Stabilizing the DFD-Out Conformation of Mnk2 Kinase. ACS Med. Chem. Lett. 2013, 4, 736–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Teo, T.; Yu, M.; Yang, Y.; Gilliam, T.; Lam, F.; Sykes, M.J.; Wang, S. Pharmacologic co-inhibition of Mnks and mTORC1 synergistically suppresses proliferation and perturbs cell cycle progression in blast crisis-chronic myeloid leukemia cells. Cancer Lett. 2015, 357, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Podo, F.; Buydens, L.M.; Degani, H.; Hilhorst, R.; Klipp, E.; Gribbestad, I.S.; Van Huffel, S.; van Laarhoven, H.W.; Luts, J.; Monleon, D.; et al. Triple-negative breast cancer: Present challenges and new perspectives. Mol. Oncol. 2010, 4, 209–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lineham, E.; Tizzard, G.J.; Coles, S.J.; Spencer, J.; Morley, S. Synergistic effects of inhibiting the MNK-eIF4E and PI3K/AKT/mTOR pathways on cell migration in MDA-MB-231 cells. Oncotarget 2018, 9, 14148. [Google Scholar] [CrossRef] [PubMed]
- Tabbi, G.; Cassino, C.; Cavigiolio, G.; Colangelo, D.; Ghiglia, A.; Viano, I.; Osella, D. Water stability and cytotoxic activity relationship of a series of ferrocenium derivatives. ESR insights on the radical production during the degradation process. J. Med. Chem. 2002, 45, 5786–5796. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.; Amin, J.; Wang, M.; Packham, G.; Syed Alwi, S.S.; Tizzard, G.J.; Coles, J.S.; Paranal, R.M.; Bradner, J.; Heightman, T.D. Synthesis and Biological Evaluation of JAHAs: Ferrocene-Based Histone Deacetylase Inhibitors. ACS Med. Chem. Lett. 2015, 2, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Coles, S.J.; Gale, P.A. Changing and challenging times for service crystallography. Chem. Sci. 2012, 3, 683–689. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds 3, 5 are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line 1 | 1 | 3 | 5 |
|---|---|---|---|
| BT-549 | n.s | 6.67 | 1.73 |
| MDA-MB-231 | n.s | 2.83 | 0.55 |
| SK-BR-3 | n.s | 114.30 | 10.63 |
| MOLM-13 | 5.39 | n.s | 1.82 |
| MV-4-11 | 9.72 | 8.37 | 1.98 |
| Kinase 1 | 1 | 5 | ||
|---|---|---|---|---|
| Data 1 | Data 2 | Data 1 | Data 2 | |
| AKT1 | 110.03 | 105.00 | 106.92 | 105.28 |
| MNK1 | 3.64 | 2.53 | 115.13 | 111.38 |
| MNK2 | 1.87 | 0.79 | 97.56 | 96.04 |
| P38a/MAPK14 | 125.49 | 119.86 | 126.41 | 125.76 |
| PIM1 | 43.11 | 42.99 | 118.37 | 117.26 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sansook, S.; Lineham, E.; Hassell-Hart, S.; Tizzard, G.J.; Coles, S.J.; Spencer, J.; Morley, S.J. Probing the Anticancer Action of Novel Ferrocene Analogues of MNK Inhibitors. Molecules 2018, 23, 2126. https://doi.org/10.3390/molecules23092126
Sansook S, Lineham E, Hassell-Hart S, Tizzard GJ, Coles SJ, Spencer J, Morley SJ. Probing the Anticancer Action of Novel Ferrocene Analogues of MNK Inhibitors. Molecules. 2018; 23(9):2126. https://doi.org/10.3390/molecules23092126
Chicago/Turabian StyleSansook, Supojjanee, Ella Lineham, Storm Hassell-Hart, Graham J. Tizzard, Simon J. Coles, John Spencer, and Simon J. Morley. 2018. "Probing the Anticancer Action of Novel Ferrocene Analogues of MNK Inhibitors" Molecules 23, no. 9: 2126. https://doi.org/10.3390/molecules23092126