Asymmetric Synthesis of (−)-6-Desmethyl-Fluvirucinine A1 via Conformationally-Controlled Diastereoselective Lactam-Ring Expansions

 , and
, and
Abstract
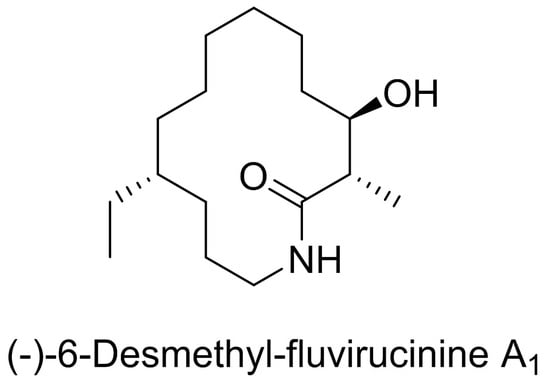
:
{kind=link}
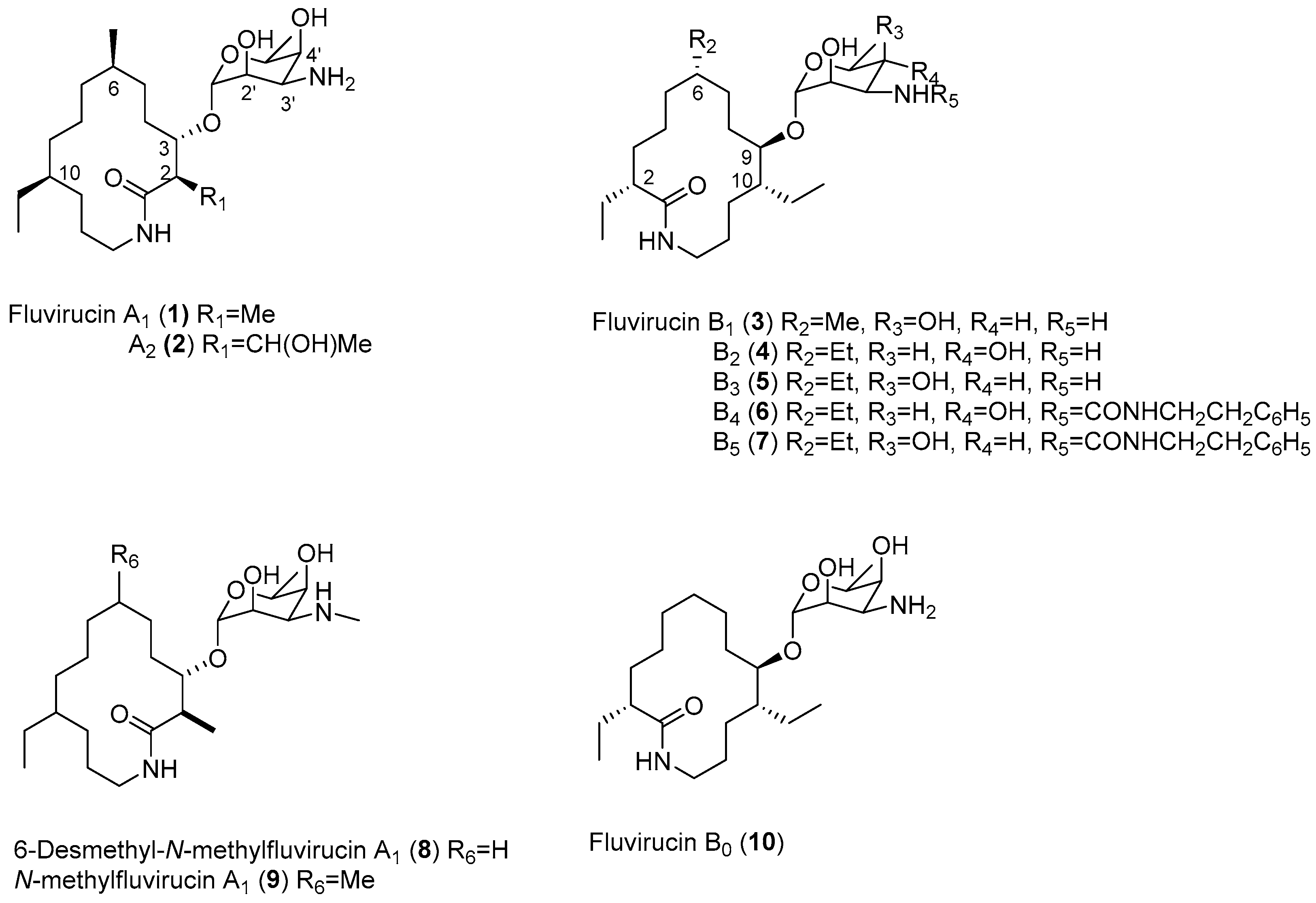{kind=link}
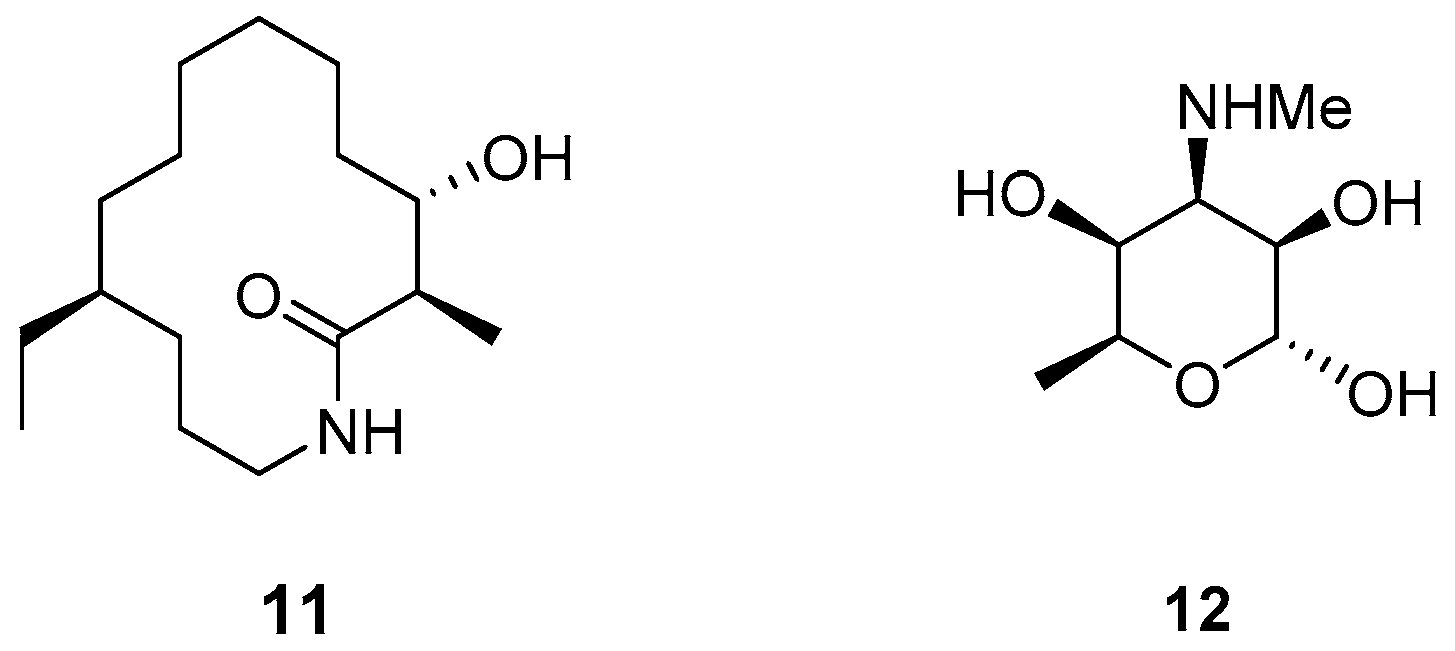{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
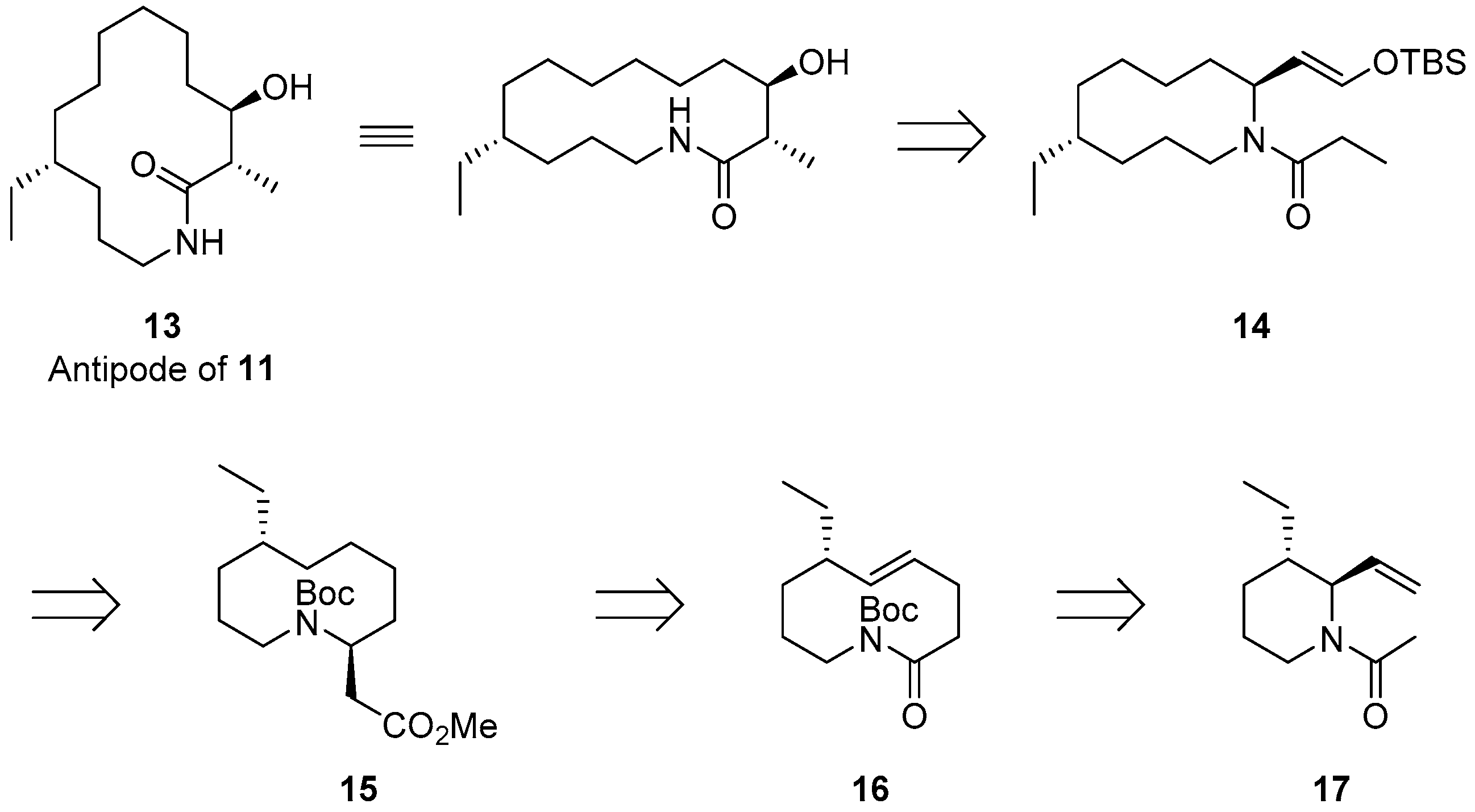
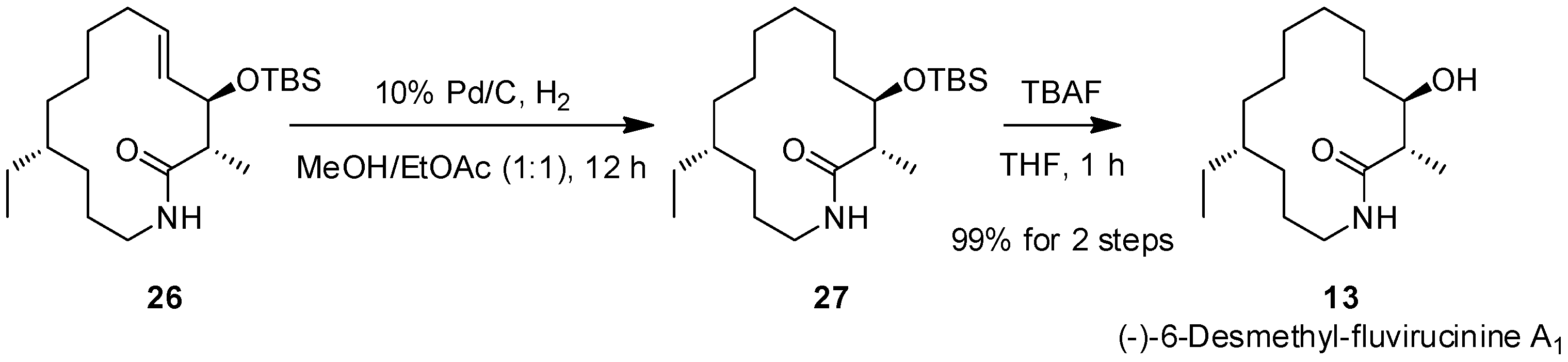
2.1. Synthetic Strategy for (−)-6-Desmethyl-Fluvirucinine A1 (13)
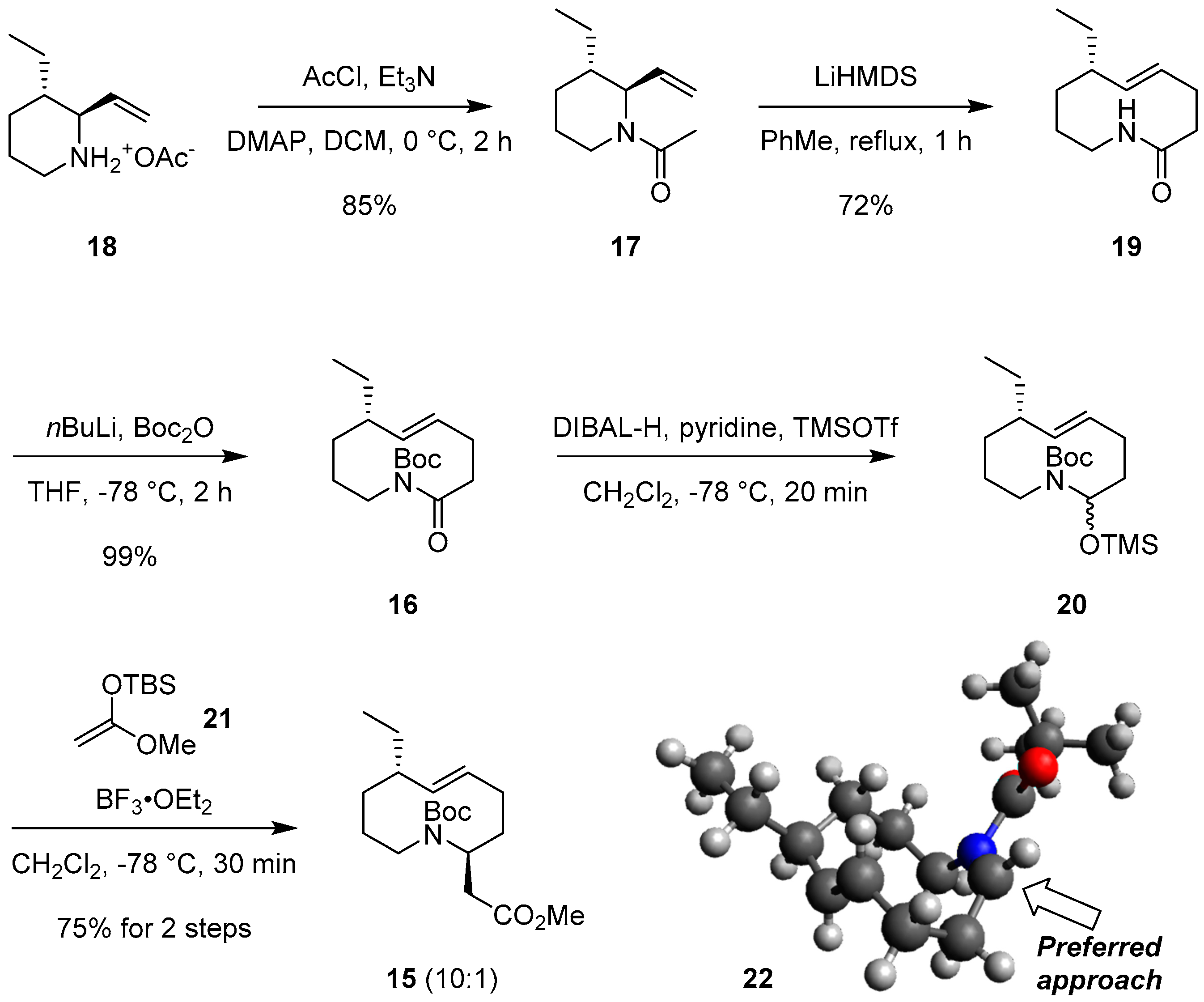
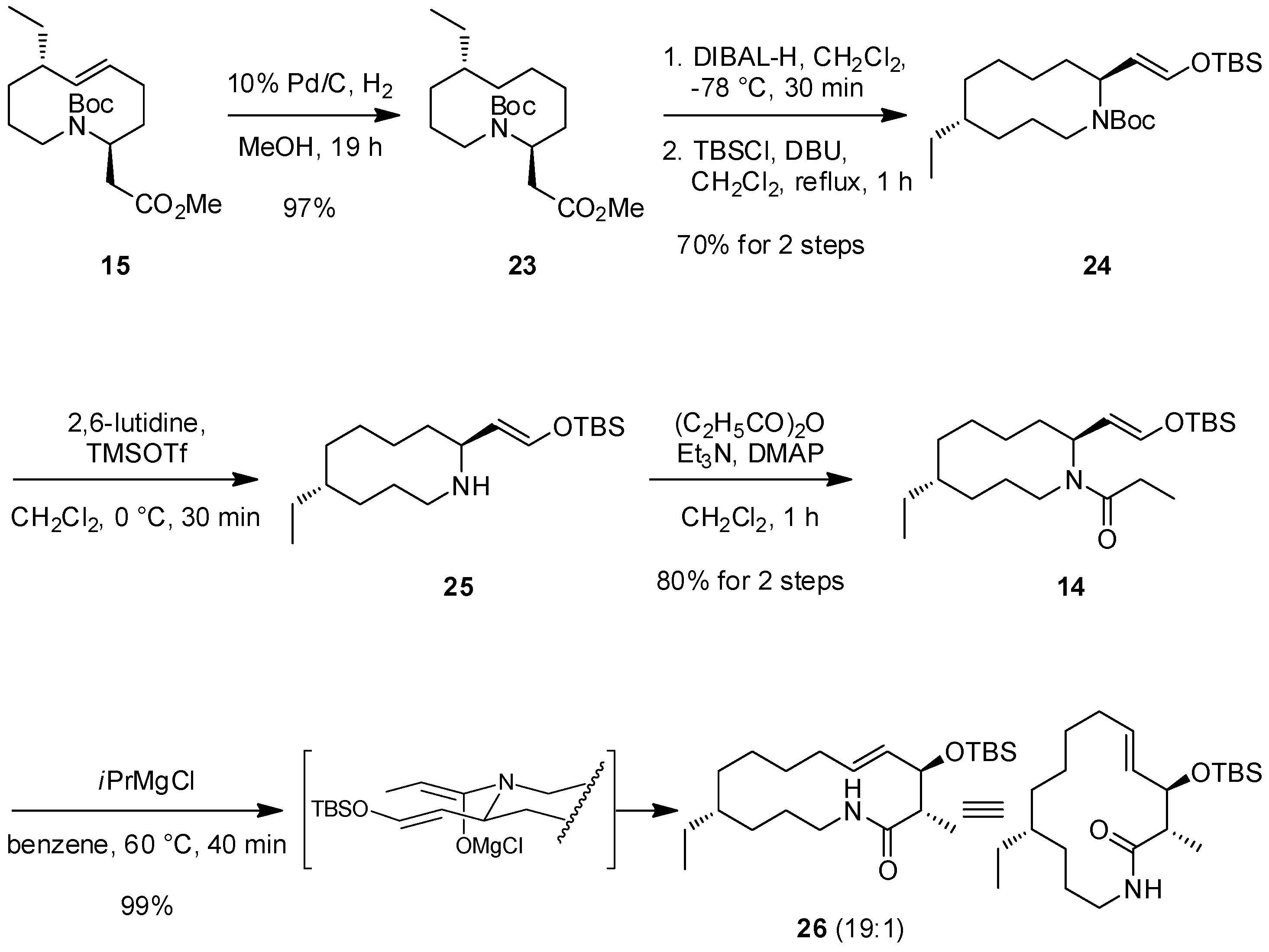
2.2. The First ACR and Diastereoselective Amidoalkylation for Synthesis of Ester 15
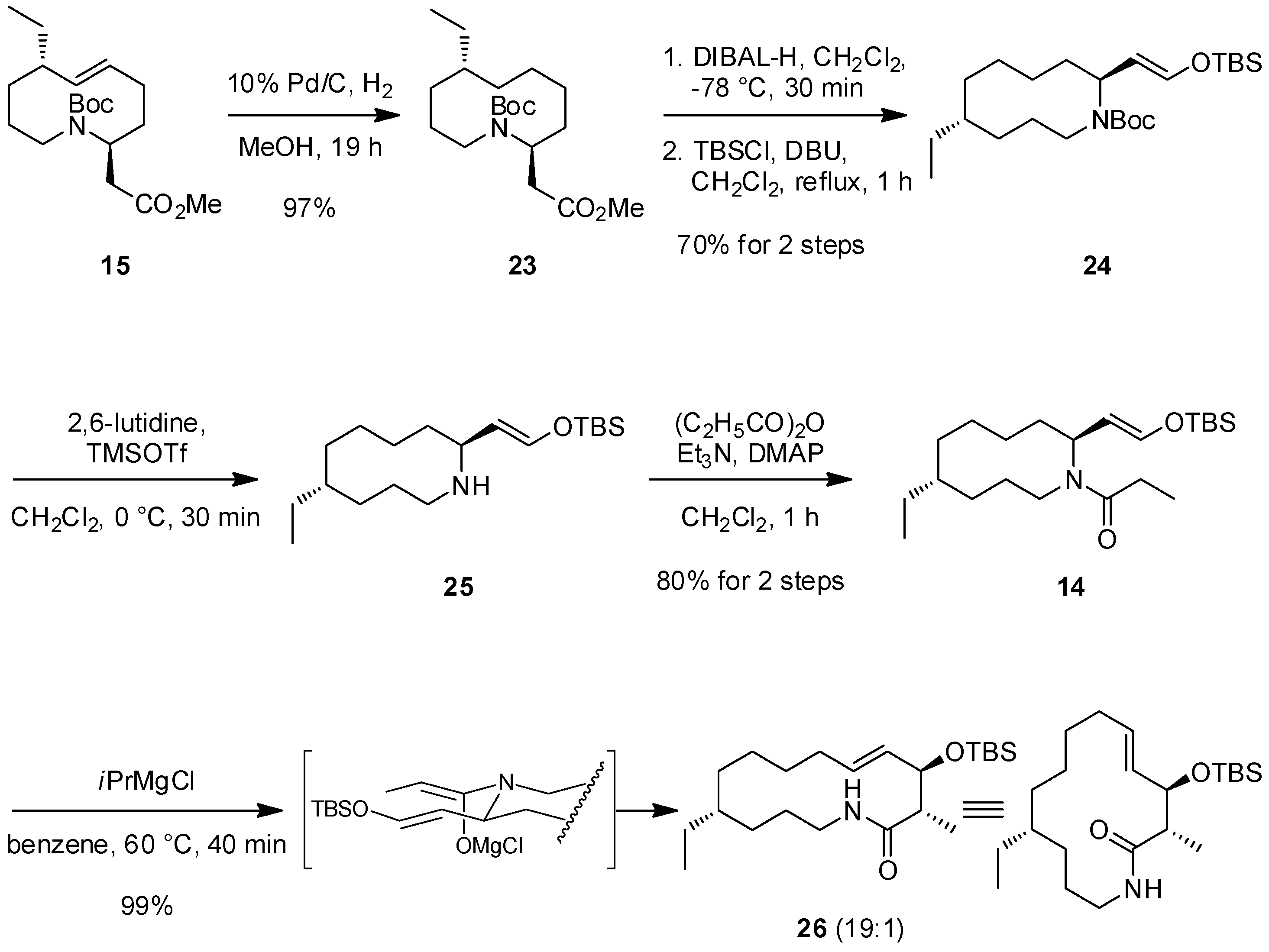
2.3. The Second ACR and Completion of the Synthesis
3. Materials and Methods
3.1. General Information
3.2. Experimental Part
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Naruse, N.; Tenmyo, O.; Kawano, K.; Tomita, K.; Ohgusa, N.; Miyaki, T.; Konishi, M.; Oki, T. Fluvirucins A1, A2, B1, B2, B3, B4 and B5, new antibiotics active against influenza A virus. I. Production, isolation, chemical properties and biological activities. J. Antibiot. 1991, 44, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Naruse, N.; Tsuno, T.; Sawada, Y.; Konishi, M.; Oki, T. Fluvirucins A1, A2, B1, B2, B3, B4 and B5, new antibiotics active against influenza A virus. II. Structure determination. J. Antibiot. 1991, 44, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Naruse, N.; Konishi, M.; Oki, T.; Inouye, Y.; Kakisawa, H. Fluvirucins A1, A2, B1, B2, B3, B4 and B5, new antibiotics active against influenza A virus. III. The stereochemistry and absolute configuration of fluvirucin A1. J. Antibiot. 1991, 44, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Oda, N.; Hoshino, Y.; Ohkusa, N.; Chikazawa, H. Fluvirucins A1, A2, B1, B2, B3, B4 and B5, new antibiotics active against influenza A virus. IV. Taxonomy on the producing organisms. J. Antibiot. 1991, 44, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Hegde, V.R.; Patel, M.G.; Gullo, V.P.; Ganguly, A.K.; Sarre, O.; Puar, M.S.; McPhail, A.T. Macrolactams: A new class of antifungal agents. J. Am. Chem. Soc. 1990, 112, 6403–6405. [Google Scholar] [CrossRef]
- Hegde, V.; Patel, M.; Horan, A.; Gullo, V.; Marquez, J.; Gunnarsson, I.; Gentile, F.; Loebenberg, D.; King, A.; Puar, M.; et al. Macrolactams: A novel class of antifungal antibiotics produced by Actinomadura spp. SCC 1776 and SCC 1777. J. Antibiot. 1992, 45, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Houri, A.F.; Xu, Z.M.; Cogan, D.A.; Hoveyda, A.H. Cascade catalysis in synthesis. an enantioselective route to Sch-38516 (and fluvirucin B1) aglycon macrolactam. J. Am. Chem. Soc. 1995, 117, 2943–2944. [Google Scholar] [CrossRef]
- Xu, Z.M.; Johannes, C.W.; Salman, S.S.; Hoveyda, A.H. Enantioselective total synthesis of antifungal agent Sch 38516. J. Am. Chem. Soc. 1996, 118, 10926–10927. [Google Scholar] [CrossRef]
- Xu, Z.M.; Johannes, C.W.; Houri, A.F.; La, D.S.; Cogan, D.A.; Hofilena, G.E.; Hoveyda, A.H. Applications of Zr-catalyzed carbomagnesation and Mo-catalyzed macrocyclic ring closing metathesis in asymmetric synthesis. Enantioselective total synthesis of Sch 38516 (Fluvirucin B1). J. Am. Chem. Soc. 1997, 119, 10302–10316. [Google Scholar] [CrossRef]
- Trost, B.M.; Ceschi, M.A.; Konig, B. Palladium-catalyzed additions of alkenyl epoxides to pronucleophiles: A synthesis of the macrolactam aglycone of fluviricin B1. Angew. Chem. Int. Ed. 1997, 36, 1486–1489. [Google Scholar] [CrossRef]
- Martin, M.; Mas, G.; Urpi, F.; Vilarrasa, J. High-yielding enantioselective synthesis of the macrolactam aglycon of Sch 38516 from two units of (2R)-2-ethyl-4-penten-1-ol. Angew. Chem. Int. Ed. 1999, 38, 3086–3089. [Google Scholar] [CrossRef]
- Liang, B.; Negishi, E. Highly efficient asymmetric synthesis of fluvirucinine A1, via Zr-catalyzed asymmetric carboalumination of alkenes (ZACA)-lipase-catalyzed acetylation tandem process. Org. Lett. 2008, 10, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Llacer, E.; Urpi, F.; Vilarrasa, J. Efficient approach to fluvirucins B2, B5, Sch 38518, and Sch 39185. First synthesis of their aglycon, via CM and RCM reactions. Org. Lett. 2009, 11, 3198–3201. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.-G.; Kim, S.-A.; Jung, J.-K.; Shin, D.-Y.; Min, K.-H.; Koo, B.-A.; Kim, H.-S. Asymmetric total synthesis of fluvirucinine A1. Angew. Chem. Int. Ed. 1999, 38, 3545–3547. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Jung, J.-W.; Kim, S.-H.; Jung, J.-K.; Paek, S.-M.; Kim, N.-J.; Chang, D.-J.; Lee, J.; Suh, Y.-G. First total synthesis and structural confirmation of fluvirucinine A2 via an iterative ring expansion strategy. Org. Lett. 2010, 12, 2040–2043. [Google Scholar] [CrossRef] [PubMed]
- Ayers, S.; Zink, D.L.; Mohn, K.; Powell, J.S.; Brown, C.M.; Murphy, T.; Grund, A.; Genilloud, O.; Salazar, O.; Thompson, D.; et al. Anthelmintic macrolactams from Nonomuraea turkmeniaca MA7364. J. Nat. Prod. 2007, 70, 1371–1373. [Google Scholar] [CrossRef] [PubMed]
- Ayers, S.; Zink, D.L.; Powell, J.S.; Brown, C.M.; Grund, A.; Genilloud, O.; Salazar, O.; Thompson, D.; Singh, S.B. Anthelmintic macrolactams from Nonomuraea turkmeniaca MA7381. J. Antibiot. 2008, 61, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, K.C.; Bhattacharyya, T.; Chattopadhyay, B.; Sinha, B. Recent advances in the aza-Claisen rearrangement. Synthesis 2009, 13, 2117–2142. [Google Scholar] [CrossRef]
- Jung, J.-W.; Kim, S.-H.; Suh, Y.-G. Advances in aza-Claisen-rearrangement-induced ring-expansion strategies. Asian J. Org. Chem. 2017, 6, 1117–1129. [Google Scholar] [CrossRef]
- Castro, A.M.M. Claisen rearrangement over the past nine decades. Chem. Rev. 2004, 104, 2939–3002. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.-G.; Lee, J.-Y.; Kim, S.-A.; Jung, J.-K. A new ring expansion reaction of 1-acyl-2-vinylpiperidine and 1-acyl-2-vinylpiperazine via aza-Claisen rearrangement of amide enolate. Synth. Commun. 1996, 26, 1675–1680. [Google Scholar] [CrossRef]
- Jung, J.-K.; Choi, N.-S.; Suh, Y.-G. Functional divergency oriented synthesis of azoninones as the key intermediates for bioactive indolizidine alkaloids analogs. Arch. Pharm. Res. 2004, 27, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.-G.; Lee, Y.-S.; Kim, S.-H.; Jung, J.-K.; Yun, H.; Jang, J.; Kim, N.-J.; Jung, J.-W. A stereo-controlled access to functionalized macrolactams via an aza-Claisen rearrangement. Org. Biomol. Chem. 2012, 10, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.; Kim, J.; Sim, J.; Lee, S.; Han, Y.T.; Chang, D.-J.; Kim, D.-D.; Suh, Y.-G. Asymmetric syntheses of 1-deoxy-6,8a-di-epi-castanospermine and 1-deoxy-6-epi-castanospermine. J. Org. Chem. 2012, 77, 5389–5393. [Google Scholar] [CrossRef] [PubMed]
- Paek, S.-M.; Kim, N.-J.; Shin, D.; Jung, J.-K.; Jung, J.-W.; Chang, D.-J.; Moon, H.; Suh, Y.-G. A concise total synthesis of (+)-tetrabenazine and (+)-α-dihydrotetrabenazine. Chem. Eur. J. 2010, 16, 4623–4628. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Jung, J.-W.; Ahn, J.; Sim, J.; Chang, D.-J.; Kim, D.-D.; Suh, Y.-G. Asymmetric formal synthesis of schulzeines A and C. Org. Biomol. Chem. 2012, 10, 5202–5204. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.; Yun, H.; Jung, J.-W.; Lee, S.; Kim, N.-J.; Suh, Y.-G. Achiral auxiliary-assisted chiral transfer via microwave-accelerated aza-Claisen rearrangement: A short synthesis of (+)-1-hydroxyquinolizidinone. Tetrahedron Lett. 2012, 53, 4813–4815. [Google Scholar] [CrossRef]
- Moon, H.; An, H.; Sim, J.; Kim, K.; Paek, S.-M.; Suh, Y.-G. Efficient strategy for the stereoselective synthesis of 2,3-disubstituted benzo[α]quinolizidine alkaloids: Concise synthesis of (−)-protoemetinol. Tetrahedron Lett. 2015, 56, 608–611. [Google Scholar] [CrossRef]
- Suh, Y.-G.; Lim, C.; Sim, J.; Lee, J.K.; Surh, Y.-J.; Paek, S.-M. Construction of the azacyclic core of tabernaemontanine-related alkaloids via tandem Reformatsky aza-Claisen rearrangement. J. Org. Chem. 2017, 82, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.-G.; Shin, D.-Y.; Jung, J.-K.; Kim, S.-H. The versatile conversion of acyclic amides to α-alkylated amines. Chem. Commun. 2002, 1064–1065. [Google Scholar] [CrossRef]
- Suh, Y.-G.; Kim, S.-H.; Jung, J.-K.; Shin, D.-Y. The versatile conversion of lactams to the α-alkylated azacycles via cyclic N,O-acetal TMS ether. Tetrahedron Lett. 2002, 43, 3165–3167. [Google Scholar] [CrossRef]
- Jung, J.-W.; Shin, D.-Y.; Seo, S.-Y.; Kim, S.-H.; Paek, S.-M.; Jung, J.-K.; Suh, Y.-G. A new entry to functionalized cycloalkylamines: Diastereoselective intramolecular amidoalkylation of N,O-acetal TMS ether possessing allylsilane. Tetrahedron Lett. 2005, 46, 573–575. [Google Scholar] [CrossRef]
- Suh, Y.-G.; Jang, J.; Yun, H.; Han, S.M.; Shin, D.; Jung, J.-K.; Jung, J.-W. Expedient synthesis of chiral homoallylamines via N,O-acetal TMS ethers and its application. Org. Lett. 2011, 13, 5920–5923. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-Y.; Jung, J.-K.; Seo, S.-Y.; Lee, Y.-S.; Paek, S.-M.; Chung, Y.K.; Shin, D.M.; Suh, Y.-G. Stereoselective formation of N-acyliminium ion via chiral N,O-acetal TMS ether and its application to the synthesis of β-amino acids. Org. Lett. 2003, 5, 3635–3638. [Google Scholar] [CrossRef] [PubMed]
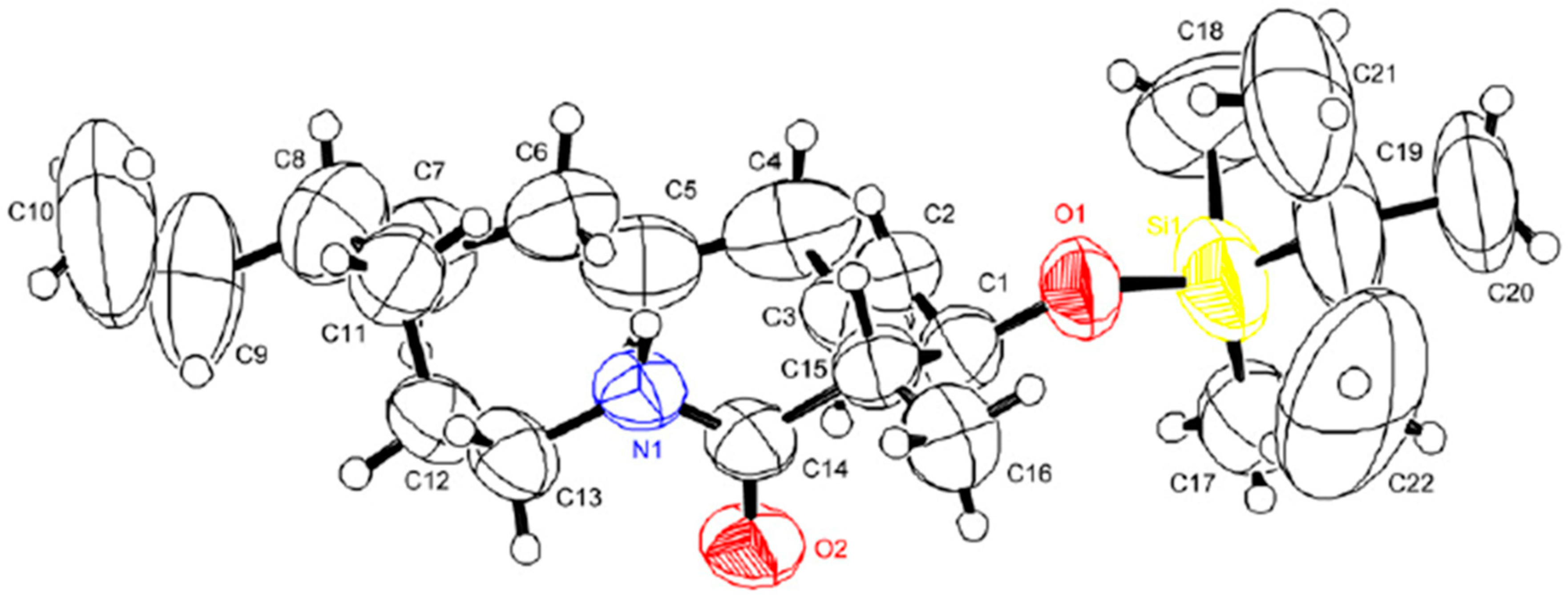
- Geometry optimization of (Z)-N-acyl iminium intermediate 22 was performed using ChemBio3D (Ver 14.0), Avogadro, and MOPAC. Details of the geometry opimization are provided in the Supplementary Materials.
- Taniguchi, Y.; Inanaga, J.; Yamaguchi, M. Use of 1,8-diazabicyclo[5.4.0]undec-7-ene in preparation of trimethylsilyl enol ethers and trimethylsilylacetylenes. Bull. Chem. Soc. Jpn. 1981, 54, 3229–3230. [Google Scholar] [CrossRef]
- Fleming, I.; Barbero, A.; Walter, D. Stereochemical control in organic synthesis using silicon-containing compounds. Chem. Rev. 1997, 97, 2063–2192. [Google Scholar] [CrossRef] [PubMed]
- Cambridge Crystallographic Data Centre; CCDC 1864701 (Online); 12 Union Road, Cambridge CB2 1EZ, UK. Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 10 September 2018).
Sample Availability: Samples of compounds are available from the authors. |






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, H.; Yoon, H.; Lim, C.; Jang, J.; Yi, J.-J.; Lee, J.K.; Lee, J.; Na, Y.; Son, W.S.; Kim, S.-H.; et al. Asymmetric Synthesis of (−)-6-Desmethyl-Fluvirucinine A1 via Conformationally-Controlled Diastereoselective Lactam-Ring Expansions. Molecules 2018, 23, 2351. https://doi.org/10.3390/molecules23092351
Moon H, Yoon H, Lim C, Jang J, Yi J-J, Lee JK, Lee J, Na Y, Son WS, Kim S-H, et al. Asymmetric Synthesis of (−)-6-Desmethyl-Fluvirucinine A1 via Conformationally-Controlled Diastereoselective Lactam-Ring Expansions. Molecules. 2018; 23(9):2351. https://doi.org/10.3390/molecules23092351
Chicago/Turabian StyleMoon, Hyunyoung, Hojong Yoon, Changjin Lim, Jaebong Jang, Jong-Jae Yi, Jae Kyun Lee, Jeeyeon Lee, Younghwa Na, Woo Sung Son, Seok-Ho Kim, and et al. 2018. "Asymmetric Synthesis of (−)-6-Desmethyl-Fluvirucinine A1 via Conformationally-Controlled Diastereoselective Lactam-Ring Expansions" Molecules 23, no. 9: 2351. https://doi.org/10.3390/molecules23092351
APA StyleMoon, H., Yoon, H., Lim, C., Jang, J., Yi, J.-J., Lee, J. K., Lee, J., Na, Y., Son, W. S., Kim, S.-H., & Suh, Y.-G. (2018). Asymmetric Synthesis of (−)-6-Desmethyl-Fluvirucinine A1 via Conformationally-Controlled Diastereoselective Lactam-Ring Expansions. Molecules, 23(9), 2351. https://doi.org/10.3390/molecules23092351