
New Alkoxy Flavone Derivatives Targeting Caspases: Synthesis and Antitumor Activity Evaluation


 , , , , and
, , , , and
Abstract
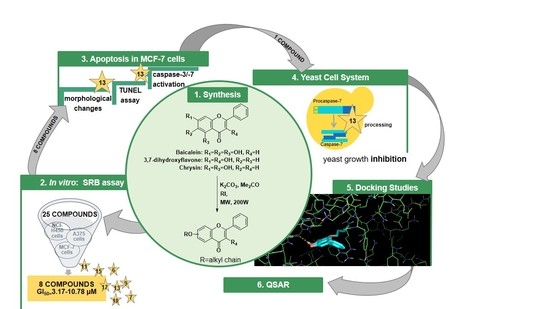
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Structure Elucidation
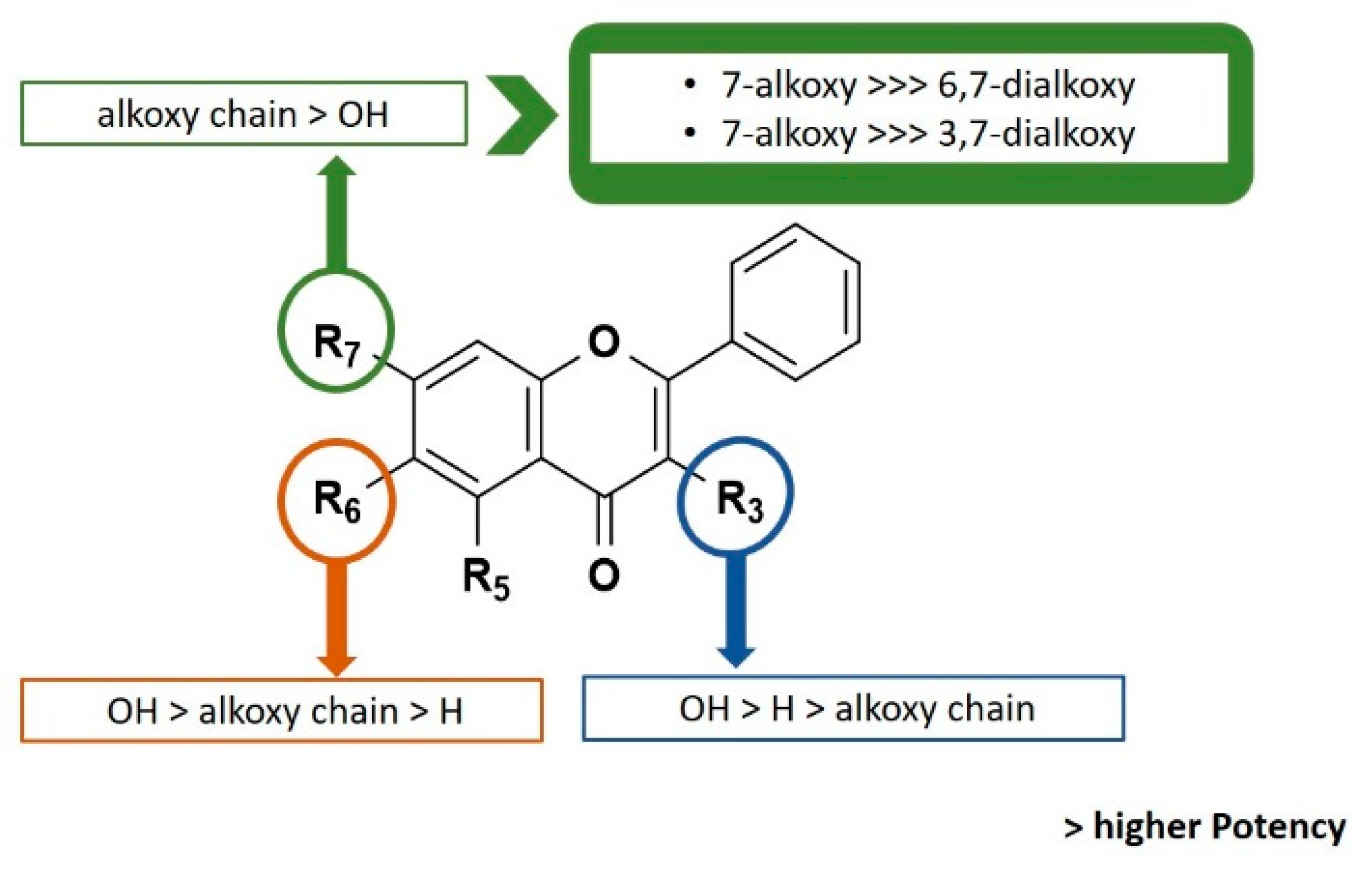
2.3. Biological Activity
Growth Inhibitory Activity in Human Tumor Cell Lines
2.4. Docking Studies
2.5. QSAR Model
3. Material and Methods
3.1. Synthesis
3.1.1. General Procedure for the Synthesis of Baicalein Derivatives (4–14)
3.1.2. General Procedure for the Synthesis of 3,7-Dihydroxyflavone Derivatives (15–21)
3.1.3. General Procedure for the Synthesis of Chrysin Derivatives (22–28)
3.2. Biological Activity
3.2.1. Chemicals
3.2.2. Tumor Cell Growth Assay
3.2.3. TUNEL Assay
3.2.4. Caspase-Glo 3/7 Assay
3.2.5. Microscopy Analysis and Image Processing
3.2.6. Yeast Caspase Assay
3.2.7. Western Blotting
3.2.8. Statistical Analysis
3.3. Virtual Screening and Docking Studies
3.4. QSAR Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect Biol. 2013, 5, 8656. [Google Scholar] [CrossRef] [PubMed]
- Peterson, Q.P.; Hsu, D.C.; Goode, D.R.; Novotny, C.J.; Totten, R.K.; Hergenrother, P.J. Procaspase-3 activation as an anti-cancer strategy: Structure−activity relationship of procaspase-activating compound 1 (PAC-1) and Its cellular co-localization with caspase-3. J. Med. Chem. 2009, 52, 5721–5731. [Google Scholar] [CrossRef] [PubMed]
- Cidade, H.; Pinto, M.; Saraiva, L. Flavones: Promising Basis for Drug Development of Pro-Apoptotic Caspase Activators. In Apoptosis; Avid Science: Telangana, India, 2017; ISBN 978-93-86337-72-6. [Google Scholar]
- Chang, W.-H.; Chen, C.-H.; Gau, R.-J.; Lin, C.-C.; Tsai, C.-L.; Tsai, K.; Lu, F.-J. Effect of baicalein on apoptosis of the human Hep G2 cell line was induced by mitochondrial dysfunction. Planta Med. 2002, 68, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Monasterio, A.; Urdaci, M.C.; Pinchuk, I.V.; Lopez-Moratalla, N.; Martinez-Irujo, J.J. Flavonoids induce apoptosis in human leukemia U937 cells through caspase-and caspase-calpain-dependent pathways. Nutr. Cancer 2004, 50, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Li-Weber, M. Targeting apoptosis pathways in cancer by Chinese medicine. Cancer Lett. 2013, 332, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Kasala, E.R.; Bodduluru, L.N.; Madana, R.M.; Gogoi, R.; Barua, C.C. Chemopreventive and therapeutic potential of chrysin in cancer: Mechanistic perspectives. Toxicol. Lett. 2015, 233, 214–225. [Google Scholar] [CrossRef]
- Leão, M.; Soares, J.; Gomes, S.; Raimundo, L.; Ramos, H.; Bessa, C.; Queiroz, G.; Domingos, S.; Pinto, M.; Inga, A. Enhanced cytotoxicity of prenylated chalcone against tumour cells via disruption of the p53–MDM2 interaction. Life Sci. 2015, 142, 60–65. [Google Scholar] [CrossRef]
- Neves, M.P.; Lima, R.T.; Choosang, K.; Pakkong, P.; de São José Nascimento, M.; Vasconcelos, M.H.; Pinto, M.; Silva, A.M.; Cidade, H. Synthesis of a natural chalcone and its prenyl analogs—Evaluation of tumor cell growth-inhibitory activities, and effects on cell cycle and apoptosis. Chem. Biodivers. 2012, 9, 1133–1143. [Google Scholar] [CrossRef]
- Neves, M.P.; Cidade, H.; Pinto, M.; Silva, A.M.; Gales, L.; Damas, A.M.; Lima, R.T.; Vasconcelos, M.H.; Nascimento, M.d.S.J. Prenylated derivatives of baicalein and 3, 7-dihydroxyflavone: Synthesis and study of their effects on tumor cell lines growth, cell cycle and apoptosis. Eur. J. Med. Chem. 2011, 46, 2562–2574. [Google Scholar] [CrossRef]
- Brandão, P.; Loureiro, J.B.; Carvalho, S.; Hamadou, M.H.; Cravo, S.; Moreira, J.; Pereira, D.; Palmeira, A.; Pinto, M.; Saraiva, L. Targeting the MDM2-p53 protein-protein interaction with prenylchalcones: Synthesis of a small library and evaluation of potential antitumor activity. Eur. J. Med. Chem. 2018, 156, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Lopes-Rodrigues, V.; Coutinho, I.; Neves, M.P.; Lima, R.T.; Pinto, M.; Cidade, H.; Vasconcelos, M.H.; Saraiva, L. Potential small-molecule activators of caspase-7 identified using yeast-based caspase-3 and-7 screening assays. Eur. J. Pharm. Sci. 2014, 54, 8–16. [Google Scholar] [CrossRef]
- Lee, Y.; Yeo, H.; Liu, S.-H.; Jiang, Z.; Savizky, R.M.; Austin, D.J.; Cheng, Y.-c. Increased anti-P-glycoprotein activity of baicalein by alkylation on the A ring. J. Med. Chem. 2004, 47, 5555–5566. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-H.; Chen, C.-H.; Lo, C.-Y.; Feng, J.-Z.; Lin, H.-J.; Chang, P.-Y.; Yang, L.-L.; Chen, L.-G.; Liu, Y.-W.; Kuo, C.-D. Synthesis and biological evaluation of novel 7-O-lipophilic substituted baicalein derivatives as potential anticancer agents. MedChemComm 2015, 6, 1864–1873. [Google Scholar] [CrossRef]
- Chang, H.; Lei, L.; Zhou, Y.; Ye, F.; Zhao, G. Dietary flavonoids and the risk of colorectal cancer: An updated meta-analysis of epidemiological studies. Nutrients 2018, 10, 950. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Kasum, C.M. Dietary flavonoids: Bioavailability, metabolic effects, and safety. Annu. Rev. Nutr. 2002, 22, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Walle, T. Methylation of dietary flavones greatly improves their hepatic metabolic stability and intestinal absorption. Mol. Pharm. 2007, 4, 826–832. [Google Scholar] [CrossRef]
- Kim, H.; Lim, D.; Shin, I.; Lee, D. Gram-scale synthesis of anti-pancreatic flavonoids (±)-8-[1-(4′-hydroxy-3′-methoxyphenyl) prop-2-en-1-yl]-chrysin and-galangin. Tetrahedron 2014, 70, 4738–4744. [Google Scholar] [CrossRef]
- Cheng, N.; Yi, W.-B.; Wang, Q.-Q.; Peng, S.-M.; Zou, X.-Q. Synthesis and α-glucosidase inhibitory activity of chrysin, diosmetin, apigenin, and luteolin derivatives. Chin. Chem. Lett. 2014, 25, 1094–1098. [Google Scholar] [CrossRef]
- Wang, J.F.; Ding, N.; Zhang, W.; Wang, P.; Li, Y.X. Synthesis of ring A-modified baicalein derivatives. Helv. Chim. Acta 2011, 94, 2221–2230. [Google Scholar] [CrossRef]
- Ghani, N.A.; Ahmat, N.; Ismail, N.H.; Zakaria, I. Flavonoid constituents from the stem bark of polyalthia cauliflora var. Cauliflora. Aust. J. Basic Appl. Sci. 2011, 5, 154–158. [Google Scholar]
- Sutthanut, K.; Sripanidkulchai, B.; Yenjai, C.; Jay, M. Simultaneous identification and quantitation of 11 flavonoid constituents in Kaempferia parviflora by gas chromatography. J. Chromatogr. A 2007, 1143, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Barron, D.; Mariotte, A.-M. Syntheses of 8-C-(1, 1-dimethylallyl) flavones and 3-methyl flavonols. Nat. Prod. Lett. 1994, 4, 21–28. [Google Scholar] [CrossRef]
- Gunduz, S.; Goren, A.C.; Ozturk, T. Facile syntheses of 3-hydroxyflavones. Org. Lett. 2012, 14, 1576–1579. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhou, Q.; Xiong, W.; Pu, W.; Zhang, W.; Zhang, G.; Wang, C. Synthesis of 5-subsituted flavonols via the Algar-Flynn-Oyamada (AFO) reaction: The mechanistic implication. Tetrahedron 2017, 73, 4822–4829. [Google Scholar] [CrossRef]
- Chen, H.; Hu, J.; Chen, Y.; Wu, J.; Liu, X.; Li, C. Baicalein Derivative and Preparation Method Thereof. CN 201610685066, 19 August 2016. [Google Scholar]
- Samarghandian, S.; Azimi-Nezhad, M.; Borji, A.; Hasanzadeh, M.; Jabbari, F.; Farkhondeh, T.; Samini, M. Inhibitory and cytotoxic activities of chrysin on human breast adenocarcinoma cells by induction of apoptosis. Pharmacogn. Mag. 2016, 12, 436–440. [Google Scholar] [CrossRef]
- Schipper, J.L.; MacKenzie, S.H.; Sharma, A.; Clark, A.C. A bifunctional allosteric site in the dimer interface of procaspase-3. Biophys. Chem. 2011, 159, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Vickers, C.J.; González-Páez, G.E.; Umotoy, J.C.; Cayanan-Garrett, C.; Brown, S.J.; Wolan, D.W. Small-molecule procaspase activators identified using fluorescence polarization. ChemBioChem 2013, 14, 1419–1422. [Google Scholar] [CrossRef]
- Dudek, A.Z.; Arodz, T.; Galvez, J. Computational methods in developing quantitative structure-activity relationships (QSAR): A review. Comb. Chem. High Throughput Screen 2006, 9, 213–228. [Google Scholar] [CrossRef]
- Dunn, W.J.; Hopfinger, A.J. 3D QSAR of flexible molecules using tensor representation. Perspect. Drug Discovery Des. 1998, 12, 167–182. [Google Scholar] [CrossRef]
- Kubinyi, H. QSAR: Hansch Analysis and Related Approaches. In Methods and Principles in Medicinal Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Alexander, D.L.J.; Tropsha, A.; Winkler, D.A. Beware of R2: Simple, unambiguous assessment of the prediction accuracy of QSAR and QSPR models. J. Chem. Inf. Model. 2015, 55, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P. On the development and validation of QSAR models. Methods Mol. Biol. 2013, 930, 499–526. [Google Scholar] [CrossRef] [PubMed]
- Veerasamy, R.; Rajak, H.; Jain, A.; Sivadasan, S.; Varghese, C.P.; Agrawal, R.K. Validation of QSAR models —Strategies and importance. Int. J. Drug Design Dis. 2011, 2, 511–519. [Google Scholar]
- Liu, P.; Long, W. Current mathematical methods used in QSAR/QSPR studies. Int. J. Mol. Sci. 2009, 10, 1978–1998. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.D.; Lee, K.H.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple method of calculating octanol/water partition coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef]
- Rohrbaugh, R.H.; Jurs, P.C. Descriptions of molecular shape applied in studies of structure/activity and structure/property relationships. Anal. Chim. Acta 1987, 199, 99–109. [Google Scholar] [CrossRef]
- Mandel, M. The mean electric moment of polar monosubstituted vinylic polymers. Mol. Phys. 1964, 7, 433–442. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, 3rd ed.; Pergamon Press: Oxford, UK, 1988. [Google Scholar]
- Scheer, J.M.; Romanowski, M.J.; Wells, J.A. A common allosteric site and mechanism in caspases. Proc. Natl. Acad. Sci. USA 2006, 103, 7595–7600. [Google Scholar] [CrossRef] [Green Version]
- CODESSA, version 2.7.10; Semichem, Inc.: Shawnee, KS, USA, 2004.
Sample Availability: Samples of the compounds 4–28 are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | GI50 (μM) | ||
|---|---|---|---|
| A375-C5 | MCF7 | NCI H460 | |
| 4 | >150 | >150 | >150 |
| 5 | 72.05 ± 5.95 | 78.05 ± 4.85 | 78.60 ± 3.40 |
| 6 | 10.78 ± 1.66 | 7.86 ± 0.87 | 7.64 ± 0.18 |
| 7 | 7.13 ± 0.76 | 4.9 ± 0.76 | 5.10 ± 0.71 |
| 8 | >150 | >150 | >150 |
| 9 | 4.92 ± 1.89 | 4.37 ± 0.6 | 4.35 ± 0.4 |
| 10 | 80.12 ± 3.73 | 89.16 ± 4.47 | 100.3 ± 0.85 |
| 11 | 3.61 ± 0.54 | 3.54 ± 0.54 | 3.39 ± 1.37 |
| 12 | 110.64 ± 19.74 | 87.07 ± 2.81 | 76.52 ± 12.83 |
| 13 | 5.6 ± 1.94 | 5.01 ± 0.84 | 5.06 ± 1.03 |
| 14 | 122.50 ± 4.50 | >150 | >150 |
| 15 | 5.68 ± 0.74 | 5.79 ± 0.04 | 5.89 ± 0.48 |
| 16 | 28.68 ± 24.01 | 31.71 ± 38.52 | 38.10 ± 45.32 |
| 17 | 9.10 ± 3.23 | 10.61 ± 1.41 | 8.26 ± 0.61 |
| 18 | 3.35 ± 0.25 | 4.10 ± 2.20 | 3.17 ± 0.64 |
| 19 | 15.08 ± 0.56 | 14.9 ± 1.32 | 14.57 ± 0.36 |
| 20 | 103.49 ± 2.84 | 94.94 ± 1.14 | 99.49 ± 7.52 |
| 21 | 80.97 ± 55.34 | 80.36 ± 56.75 | 44.92 ± 8.74 |
| 22 | >150 | >150 | >150 |
| 23 | 52.81 ± 3.17 | 32.23 ± 0.93 | 38.7 ± 2.02 |
| 24 | 124.71 ± 9.2 | 106.37 ± 21.54 | 98.08 ± 20.4 |
| 25 | 70.15 ± 18.3 | 51.59 ± 14.59 | 58.17 ± 3.01 |
| 26 | >150 | >150 | >150 |
| 27 | >150 | >150 | >150 |
| 28 | 62.2 ± 16.15 | 39.33 ± 6.04 | 33.9 ± 3.67 |
| Ligand | Docking Scores (Kcal·mol−1) |
|---|---|
| 13 | −7.7 |
| 29 * | −6.1 |
| 30 * | −6.1 |
| 31 * | −6.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira, J.; Ribeiro, D.; Silva, P.M.A.; Nazareth, N.; Monteiro, M.; Palmeira, A.; Saraiva, L.; Pinto, M.; Bousbaa, H.; Cidade, H. New Alkoxy Flavone Derivatives Targeting Caspases: Synthesis and Antitumor Activity Evaluation. Molecules 2019, 24, 129. https://doi.org/10.3390/molecules24010129
Moreira J, Ribeiro D, Silva PMA, Nazareth N, Monteiro M, Palmeira A, Saraiva L, Pinto M, Bousbaa H, Cidade H. New Alkoxy Flavone Derivatives Targeting Caspases: Synthesis and Antitumor Activity Evaluation. Molecules. 2019; 24(1):129. https://doi.org/10.3390/molecules24010129
Chicago/Turabian StyleMoreira, Joana, Diana Ribeiro, Patrícia M. A. Silva, Nair Nazareth, Madalena Monteiro, Andreia Palmeira, Lucília Saraiva, Madalena Pinto, Hassan Bousbaa, and Honorina Cidade. 2019. "New Alkoxy Flavone Derivatives Targeting Caspases: Synthesis and Antitumor Activity Evaluation" Molecules 24, no. 1: 129. https://doi.org/10.3390/molecules24010129