Chiroptical Symmetry Analysis: Exciton Chirality-Based Formulae to Understand the Chiroptical Responses of Cn and Dn Symmetric Systems

 ,
, 
Abstract
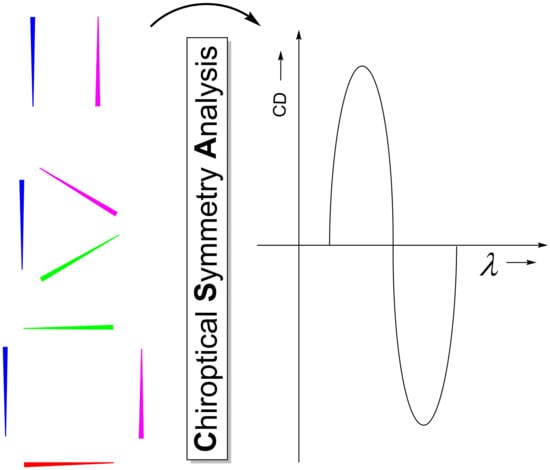
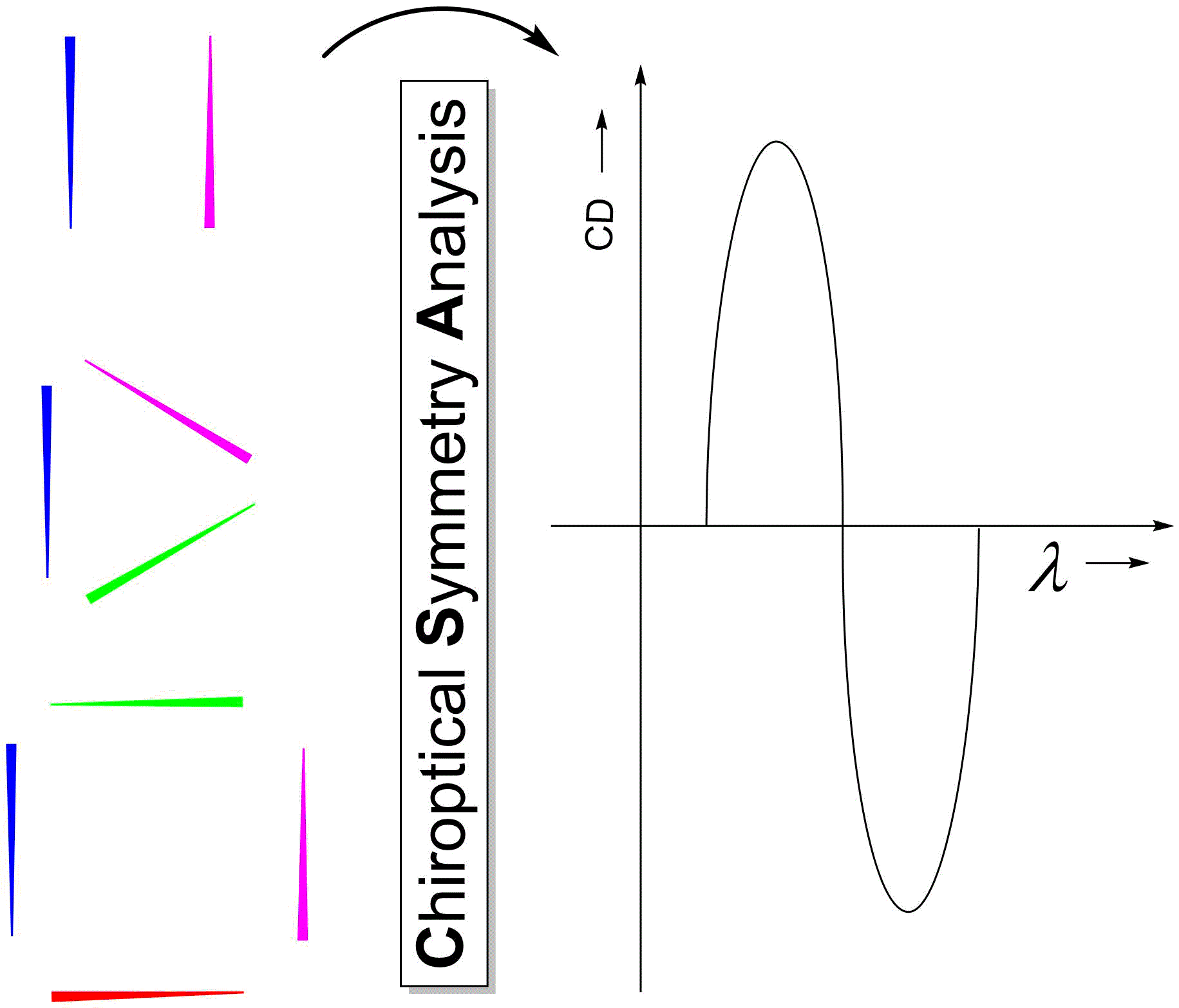{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
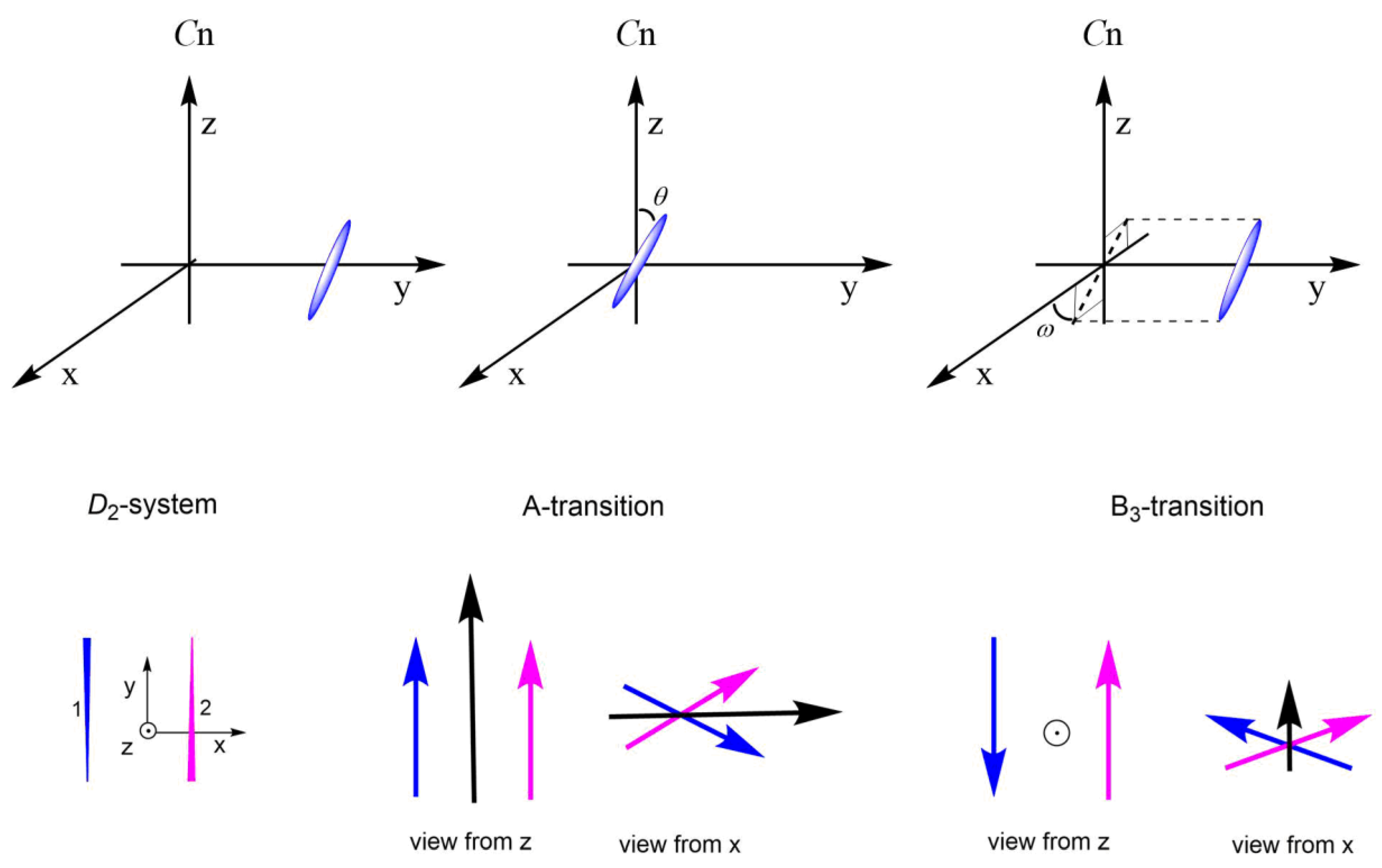
2.1. Theoretical Background (Binary Systems)
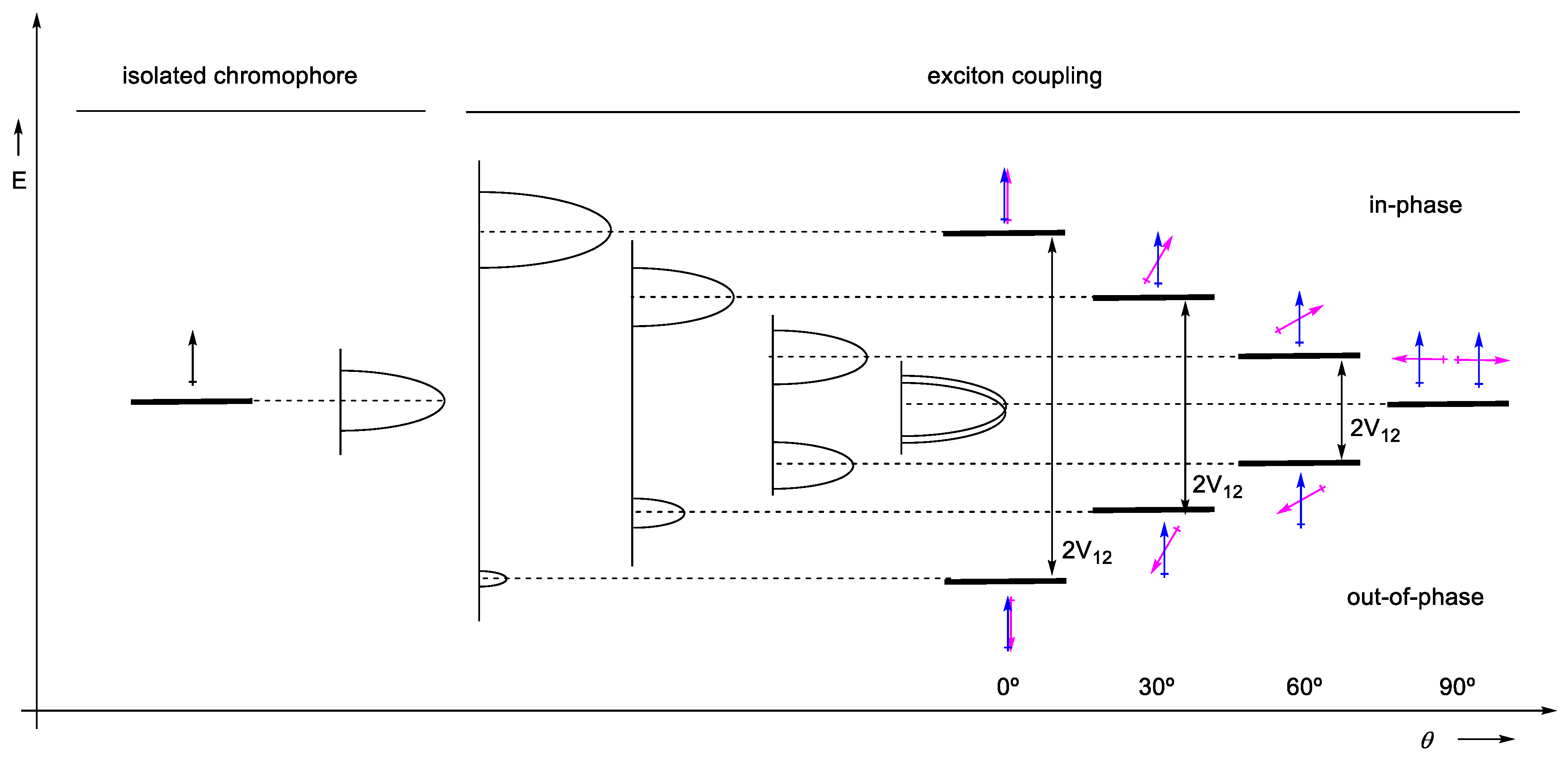
2.1.1. Approximated Wave Functions and Energy Levels
2.1.2. Rotatory Strength
2.1.3. Electric Dipole Transition Moment
2.1.4. Magnetic Dipole Transition Moment
2.1.5. Expressions for Rotatory Strength
2.2. Extending the Exciton-Independent Model to Systems with Three and Four Chromophores
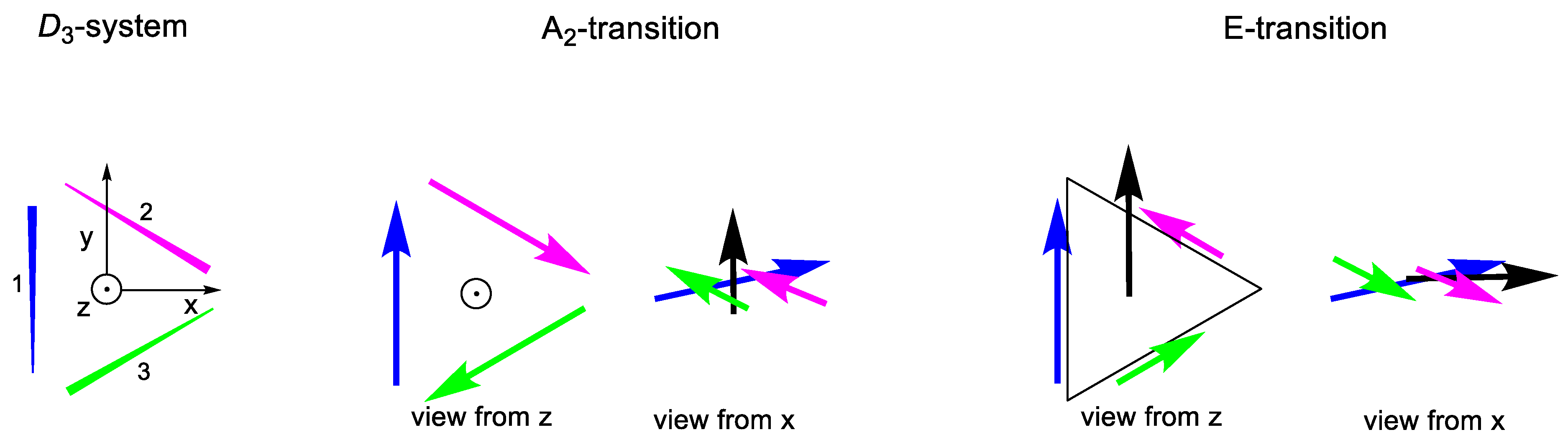
2.2.1. C3 Geometries
2.2.2. D3 Geometries
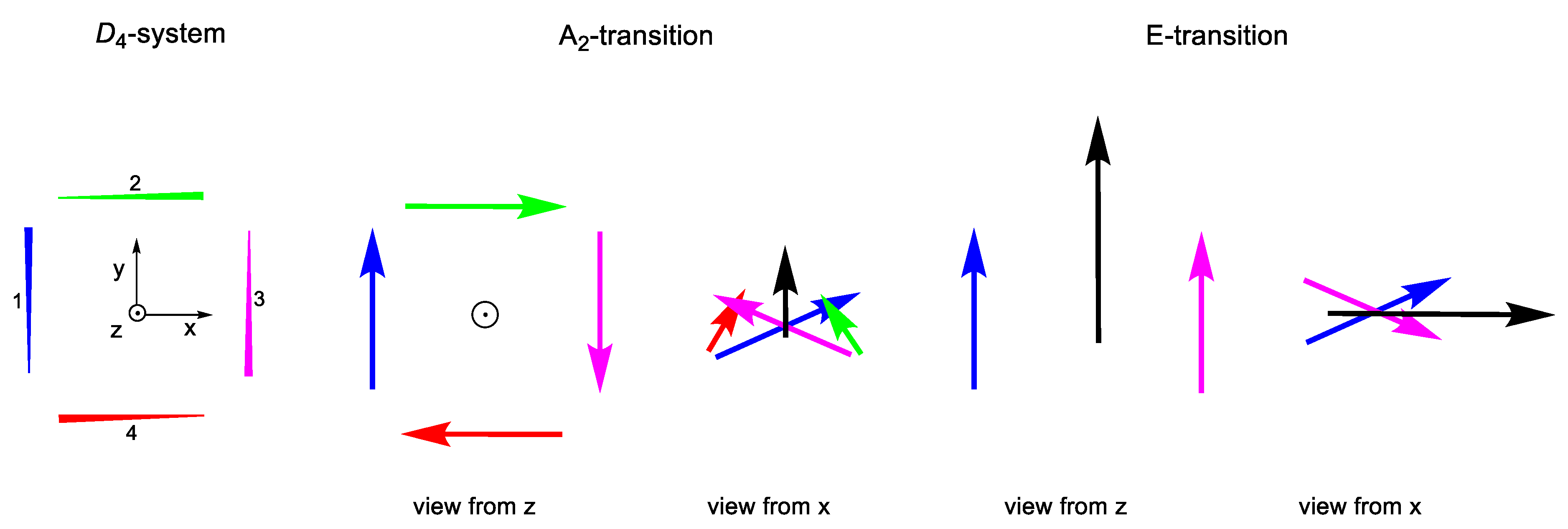
2.2.3. C4 and D4 Geometries
2.3. Comparison with DFT Calculations
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bentley:, R. From Optical Activity in Quartz to Chiral Drugs: Molecular Handedness in Biology and Medicine. Perspect. Biol. Med. 1995, 38, 188–229. [Google Scholar] [CrossRef]
- Gal, J. History Molecular Chirality in Chemistry and Biology: Historical Milestones. Helv. Chimica Acta. 2013, 96, 1617–1657. [Google Scholar] [CrossRef]
- Kelly, T.R.; De Silva, H.; Silva, R.A. Unidirectional rotary motion in a molecular system. Nature 1999, 401, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, J.-P.; Amendola, V.; Ballardini, R.; Balzani, V.; Credi, A.; Fabbrizzi, L.; Gandolfi, M.T.; Gimzewski, J.K.; Gómez-Kaifer, M.; Joachim, C.; et al. (Eds.) Molecular Machines and Motors. In Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 2001; Volume 99, ISBN 978-3-540-41382-0. [Google Scholar]
- Zsila, F. Circular Dichroism Spectroscopy Is a Sensitive Tool for Investigation of Bilirubin-Enzyme Interactions. Biomacromolecules 2011, 12, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Barron, L.D. Molecular Light Scattering and Optical Activity; Cambridge University Press: New York, NY, USA, 2004. [Google Scholar]
- Pescitelli, G.; Di Bari, L.; Berova, N. Application of electronic circular dichroism in the study of supramolecular systems. Chem. Soc. Rev. 2014, 43, 5211–5233. [Google Scholar] [CrossRef]
- Polavarapu, P.L. Chiroptical Spectroscopy: Fundamentals and Applications; CRC Press: Boca Raton, FL, USA, 2016; ISBN 9781420092462. [Google Scholar]
- Nakanishi, K.; Berova, N.; Polavarapu, P.L.; Woody, R.W. Comprehensive Chiroptical Spectroscopy, Volume 1, Instrumentation, Methodologies, and Theoretical Simulations; Wiley-VCH Verlag: Weinheim, Germany, 2013. [Google Scholar]
- Petrovic, A.G.A.G.; Navarro-Vázquez, A.; Alonso-Gómez, J.L.J.L.; Navarro-Vazquez, A.; Alonso-Gomez, J.L. From Relative to Absolute Configuration of Complex Natural Products: Interplay Between NMR, ECD, VCD, and ORD Assisted by ab initio Calculations. Curr. Org. Chem. 2010, 14, 1612–1628. [Google Scholar] [CrossRef]
- Alonso-Gómez, J.L.; Petrovic, A.G.; Harada, N.; Rivera-Fuentes, P.; Berova, N.; Diederich, F. Chiral induction from allenes into twisted 1,1,4,4-tetracyanobuta-1,3-dienes (TCBDs): Conformational assignment by circular dichroism spectroscopy. Chem. Eur. J. 2009, 15, 8396–8400. [Google Scholar] [CrossRef]
- Rivera-Fuentes, P.; Alonso-Gómez, J.L.J.L.; Petrovic, A.G.A.G.; Santoro, F.; Harada, N.; Berova, N.; Diederich, F. Amplification of chirality in monodisperse, enantiopure alleno-acetylenic oligomers. Angew. Chem. Int. Ed. 2010, 49, 2247–2250. [Google Scholar] [CrossRef]
- Petrovic, A.G.; Berova, N.; Alonso-Gómez, J.L. Structure Elucidation in Organic Chemistry. In Structure Elucidation in Organic Chemistry: The Search for the Right Tools; Cid, M.-M., Bravo, J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; pp. 65–104. ISBN 9783527664610. [Google Scholar]
- Harada, N.; Nakanishi, K. Exciton chirality method and its application to configurational and conformational studies of natural products. Acc. Chem. Res. 1972, 5, 257–263. [Google Scholar] [CrossRef]
- Davydov, A.S. The theory of molecular excitons. Sov. Phys. Usp. 1964, 7, 145–178. [Google Scholar] [CrossRef]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Gómez, J.L.J.L.; Rivera-Fuentes, P.; Harada, N.; Berova, N.; Diederich, F. An enantiomerically pure alleno-acetylenic macrocycle: Synthesis and rationalization of its outstanding chiroptical response. Angew. Chem. Int. Ed. 2009, 48, 5545–5548. [Google Scholar] [CrossRef] [PubMed]
- Míguez-Lago, S.; Llamas-Saiz, A.L.; Magdalena Cid, M.; Alonso-Gómez, J.L. A Covalent Organic Helical Cage with Remarkable Chiroptical Amplification. Chem. A Eur. J. 2015, 21, 18085–18088. [Google Scholar] [CrossRef] [PubMed]
- Míguez-Lago, S.; Cid, M.M.M.; Alonso-Gómez, J.L.; Míguez-Lago, S.; Cid, M.M.M.; Alonso-Gómez, J.L. Covalent Organic Helical Cages as Sandwich Compound Containers. Eur.J. Org. Chem. 2016, 2016, 5716–5721. [Google Scholar] [CrossRef]
- Castro-Fernández, S.; Yang, R.; García, A.P.; Garzón, I.L.; Xu, H.; Petrovic, A.G.; Alonso-Gómez, J.L. Diverse Chiral Scaffolds from Diethynylspiranes: All-Carbon Double Helices and Flexible Shape-Persistent Macrocycles. Chem. A Eur. J. 2017, 23, 11747–11751. [Google Scholar] [CrossRef]
- Ozcelik, A.; Pereira-Cameselle, R.; Von Weber, A.; Paszkiewicz, M.; Carlotti, M.; Paintner, T.; Zhang, L.; Lin, T.; Zhang, Y.-Q.; Barth, J.V.V.; et al. Device-Compatible Chiroptical Surfaces through Self-Assembly of Enantiopure Allenes. Langmuir 2018, 34, 4548–4553. [Google Scholar] [CrossRef]
- Rivera-Fuentes, P.; Alonso-Gómez, J.L.J.L.; Petrovic, A.G.A.G.; Seiler, P.; Santoro, F.; Harada, N.; Berova, N.; Rzepa, H.S.H.S.; Diederich, F. Enantiomerically pure alleno-acetylenic macrocycles: Synthesis, solid-state structures, chiroptical properties, and electron localization function analysis. Chem. Eur. J. 2010, 16, 9796–9807. [Google Scholar] [CrossRef]
- Castro-Fernández, S.; Cid, M.M.; López, C.S.; Alonso-Gómez, J.L. Opening Access to New Chiral Macrocycles: From Allenes to Spiranes. J. Phys. Chem. A 2015, 119, 1747–1753. [Google Scholar] [CrossRef]
- Condon, E.U. Theories of Optical Rotatory Power. Rev. Mod. Phys. 1937, 9, 432–457. [Google Scholar] [CrossRef]
- Kirkwood, J.G. On the theory of optical rotatory power. J. Chem. Phys. 1937, 5, 479–491. [Google Scholar] [CrossRef]
- Schellman, J.A. Circular Dichroism and Optical Rotation. Chem. Rev. 1974, 75, 323–331. [Google Scholar] [CrossRef]
- Taniguchi, T.; Monde, K. Exciton chirality method in vibrational circular dichroism. J. Am. Chem. Soc. 2012, 134, 3695–3698. [Google Scholar] [CrossRef] [PubMed]
- Covington, C.L.; Nicu, V.P.; Polavarapu, P.L. Determination of the Absolute Configurations Using Exciton Chirality Method for Vibrational Circular Dichroism: Right Answers for the Wrong Reasons? J. Phys. Chem. A 2015, 119, 10589–10601. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, L. Quantenmechanische Theorie der natürlichen optischen Aktivität von Flüssigkeiten und Gasen. Z. Phys. 1929, 52, 161–174. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro-Fernández, S.; Peña-Gallego, Á.; Mosquera, R.A.; Alonso-Gómez, J.L. Chiroptical Symmetry Analysis: Exciton Chirality-Based Formulae to Understand the Chiroptical Responses of Cn and Dn Symmetric Systems. Molecules 2019, 24, 141. https://doi.org/10.3390/molecules24010141
Castro-Fernández S, Peña-Gallego Á, Mosquera RA, Alonso-Gómez JL. Chiroptical Symmetry Analysis: Exciton Chirality-Based Formulae to Understand the Chiroptical Responses of Cn and Dn Symmetric Systems. Molecules. 2019; 24(1):141. https://doi.org/10.3390/molecules24010141
Chicago/Turabian StyleCastro-Fernández, Silvia, Ángeles Peña-Gallego, Ricardo A. Mosquera, and José Lorenzo Alonso-Gómez. 2019. "Chiroptical Symmetry Analysis: Exciton Chirality-Based Formulae to Understand the Chiroptical Responses of Cn and Dn Symmetric Systems" Molecules 24, no. 1: 141. https://doi.org/10.3390/molecules24010141
APA StyleCastro-Fernández, S., Peña-Gallego, Á., Mosquera, R. A., & Alonso-Gómez, J. L. (2019). Chiroptical Symmetry Analysis: Exciton Chirality-Based Formulae to Understand the Chiroptical Responses of Cn and Dn Symmetric Systems. Molecules, 24(1), 141. https://doi.org/10.3390/molecules24010141