
Accelerating Drug Discovery by Early Protein Drug Target Prediction Based on a Multi-Fingerprint Similarity Search †

 , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
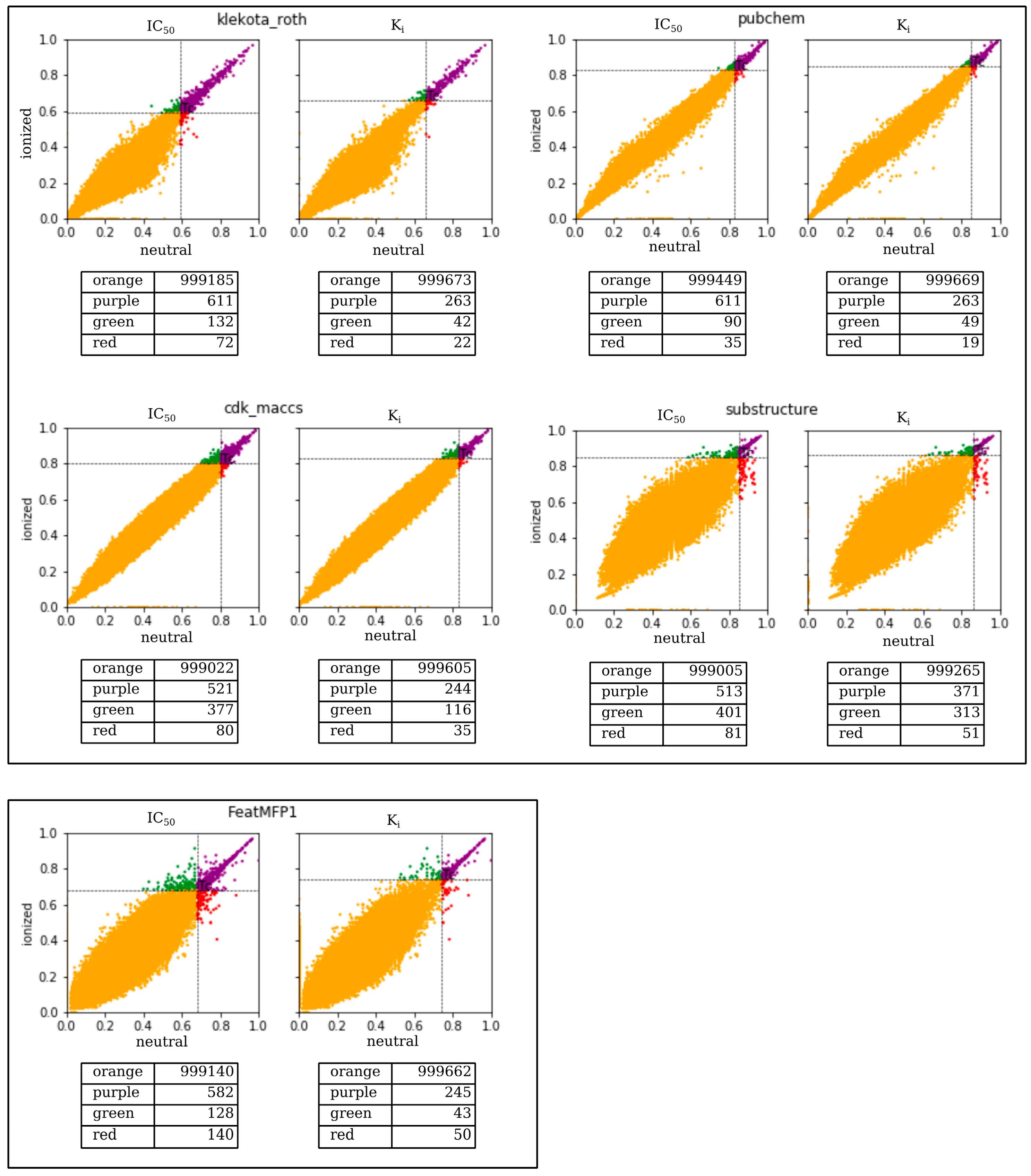
2.1. A Multi-Fingerprints Similarity Analysis Comparing Ionized and Neutral Molecular Pairs
2.2. Ki and IC50 based Protein Drug Target Predictions
2.3. Case Studies
3. Materials and Methods
3.1. Construction of the Ki and IC50 Database
3.2. Canonicalization and Correction of Chemical Structures
3.3. Generation of Dominant Ionized Species at a Physiological pH and Neutral Forms
3.4. Fingerprints Generation
3.5. Construction of the External Sets
3.6. Selection of Prospective Queries From Recently Published Scientific Articles
3.7. Protein Drug Target Multi-FPs Similarity Search Algorithm
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, C. Drug target validation: Hitting the target. Nature 2003, 422, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Alberga, D.; Trisciuzzi, D.; Montaruli, M.; Leonetti, F.; Mangiatordi, G.F.; Nicolotti, O. A new approach for drug target and bioactivity prediction: The Multifingerprint Similarity Search Algorithm (MuSSeL). J. Chem. Inf. Model. 2019, 59, 586–596. [Google Scholar] [CrossRef] [PubMed]
- ChEMBL v.24.1. Available online: http://ftp.ebi.ac.uk/pub/databases/chembl/ChEMBLdb/latest/ (accessed on 11 February 2019).
- ChEMBL v.22.1. Available online: http://ftp.ebi.ac.uk/pub/databases/chembl/ChEMBLdb/releases/chembl_22_1/ (accessed on 11 February 2019).
- Xia, J.; Tilahun, E.L.; Reid, T.E.; Zhang, L.; Wang, X.S. Benchmarking methods and data sets for ligand enrichment assessment in virtual screening. Methods San. Diego. Calif. 2015, 71, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.W.; Sadowski, J. Structure Modification in Chemical Databases. In Chemoinformatics in Drug Discovery; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2005; pp. 271–285. [Google Scholar]
- Nicolotti, O.; Miscioscia, T.F.; Leonetti, F.; Muncipinto, G.; Carotti, A. Screening of matrix metalloproteinases available from the protein data bank: Insights into biological functions, domain organization, and zinc binding groups. J. Chem. Inf. Model. 2007, 47, 2439–2448. [Google Scholar] [CrossRef] [PubMed]
- Floris, M.; Manganaro, A.; Nicolotti, O.; Medda, R.; Mangiatordi, G.F.; Benfenati, E. A generalizable definition of chemical similarity for read-across. J. Cheminform. 2014, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. RDKit: Open-Source Cheminformatics. 2006. Available online: https://www.rdkit.org/ (accessed on 11 February 2019).
- O’Boyle, N.M.; Morley, C.; Hutchison, G.R. Pybel: A Python wrapper for the OpenBabel cheminformatics toolkit. Chem. Cent. J. 2008, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An open-source java library for chemo- and bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef]
- Steinbeck, C.; Hoppe, C.; Kuhn, S.; Floris, M.; Guha, R.; Willighagen, E.L. Recent developments of the Chemistry Development Kit (CDK)—An open-source java library for chemo- and bioinformatics. Curr. Pharm. Des. 2006, 12, 2111–2120. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Gobbi, A.; Poppinger, D. Genetic optimization of combinatorial libraries. Biotechnol. Bioeng. 1998, 61, 47–54. [Google Scholar] [CrossRef]
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom pairs as molecular features in structure-activity studies: Definition and applications. J. Chem. Inf. Comput. Sci. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Nilakantan, R.; Bauman, N.; Dixon, J.S.; Venkataraghavan, R. Topological torsion: A new molecular descriptor for SAR applications. Comparison with other descriptors. J. Chem. Inf. Comput. Sci. 1987, 27, 82–85. [Google Scholar] [CrossRef]
- PubChem Substructure Fingerprint v1.3. Available online: ftp://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.txt (accessed on 11 February 2019).
- Klekota, J.; Roth, F.P. Chemical substructures that enrich for biological activity. Bioinforma. Oxf. Engl. 2008, 24, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Riniker, S.; Landrum, G.A. Open-source platform to benchmark fingerprints for ligand-based virtual screening. J. Cheminform. 2013, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular similarity in medicinal chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef]
- Jiang, F.; Guo, A.; Xu, J.; You, Q.D.; Xu, X.L. Discovery of a Potent Grp94 Selective inhibitor with anti-inflammatory efficacy in a mouse model of ulcerative colitis. J. Med. Chem. 2018, 61, 9513–9533. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Fan, J.; Dai, Y.; Jiang, A.; Geng, M.; Ai, J.; Duan, W. Discovery of potent irreversible pan-fibroblast growth factor receptor (FGFR) inhibitors. J. Med. Chem. 2018, 61, 9085–9104. [Google Scholar] [CrossRef]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef] [PubMed]
- Awale, M.; Reymond, J.L. Polypharmacology Browser PPB2: Target prediction combining nearest neighbors with machine learning. J. Chem. Inf. Model. 2019, 59, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, Z.; Hutle, M.; Schiper, A. Unifying byzantine consensus algorithms with weak interactive consistency. In Proceedings of the Principles of Distributed Systems; Springer: Berlin/Heidelberg, Germany, 2009; pp. 300–314. [Google Scholar]
- Ghosh, A.K.; Williams, J.N.; Ho, R.Y.; Simpson, H.M.; Hattori, S.; Hayashi, H.; Agniswamy, J.; Wang, Y.-F.; Weber, I.T.; Mitsuya, H. Design and synthesis of potent HIV-1 protease inhibitors containing bicyclic oxazolidinone scaffold as the P2 ligands: Structure–activity studies and biological and X-ray structural studies. J. Med. Chem. 2018, 61, 9722–9737. [Google Scholar] [CrossRef] [PubMed]
- Bergkemper, M.; Kronenberg, E.; Thum, S.; Börgel, F.; Daniliuc, C.; Schepmann, D.; Nieto, F.R.; Brust, P.; Reinoso, R.F.; Alvarez, I.; et al. Synthesis, receptor affinity, and antiallodynic activity of spirocyclic σ receptor ligands with exocyclic amino moiety. J. Med. Chem. 2018, 61, 9666–9690. [Google Scholar] [CrossRef] [PubMed]
- Brnardic, E.J.; Ye, G.; Brooks, C.; Donatelli, C.; Barton, L.; McAtee, J.; Sanchez, R.M.; Shu, A.; Erhard, K.; Terrell, L.; et al. Discovery of pyrrolidine sulfonamides as selective and orally bioavailable antagonists of transient receptor potential vanilloid-4 (TRPV4). J. Med. Chem. 2018, 61, 9738–9755. [Google Scholar] [CrossRef]
- Pulido, D.; Casadó-Anguera, V.; Pérez-Benito, L.; Moreno, E.; Cordomí, A.; López, L.; Cortés, A.; Ferré, S.; Pardo, L.; Casadó, V.; et al. Design of a true bivalent ligand with picomolar binding affinity for a G protein-coupled receptor homodimer. J. Med. Chem. 2018, 61, 9335–9346. [Google Scholar] [CrossRef]
- Peng, P.; Chen, H.; Zhu, Y.; Wang, Z.; Li, J.; Luo, R.-H.; Wang, J.; Chen, L.; Yang, L.M.; Jiang, H.; et al. Structure-based design of 1-Heteroaryl-1,3-propanediamine derivatives as a novel series of CC-chemokine receptor 5 antagonists. J. Med. Chem. 2018, 61, 9621–9636. [Google Scholar] [CrossRef]
- Pan, P.; Chen, J.; Li, X.; Li, M.; Yu, H.; Zhao, J.J.; Ni, J.; Wang, X.; Sun, H.; Tian, S.; et al. Structure-based drug design and identification of H2O-soluble and low toxic hexacyclic camptothecin derivatives with improved efficacy in cancer and lethal inflammation models in vivo. J. Med. Chem. 2018, 61, 8613–8624. [Google Scholar] [CrossRef]
- Kawahata, W.; Asami, T.; Kiyoi, T.; Irie, T.; Taniguchi, H.; Asamitsu, Y.; Inoue, T.; Miyake, T.; Sawa, M. Design and synthesis of novel amino-triazine analogues as selective Bruton’s tyrosine kinase inhibitors for treatment of rheumatoid arthritis. J. Med. Chem. 2018, 61, 8917–8933. [Google Scholar] [CrossRef]
- Dutour, R.; Roy, J.; Cortés-Benítez, F.; Maltais, R.; Poirier, D. Targeting Cytochrome P450 (CYP) 1B1 Enzyme with four series of a-ring substituted estrane derivatives: Design, synthesis, inhibitory activity, and selectivity. J. Med. Chem. 2018, 61, 9229–9245. [Google Scholar] [CrossRef]
- Davoren, J.E.; Nason, D.; Coe, J.; Dlugolenski, K.; Helal, C.; Harris, A.R.; LaChapelle, E.; Liang, S.; Liu, Y.; O’Connor, R.; et al. Discovery and lead optimization of atropisomer D1 agonists with reduced desensitization. J. Med. Chem. 2018, 61, 11384–11397. [Google Scholar] [CrossRef] [PubMed]
- Hobson, A.D.; Judge, R.A.; Aguirre, A.L.; Brown, B.S.; Cui, Y.; Ding, P.; Dominguez, E.; DiGiammarino, E.; Egan, D.A.; Freiberg, G.M.; et al. Identification of selective dual ROCK1 and ROCK2 inhibitors using structure-based drug design. J. Med. Chem. 2018, 61, 11074–11100. [Google Scholar] [CrossRef]
- Cao, X.; Zhang, Y.; Chen, Y.; Qiu, Y.; Yu, M.; Xu, X.; Liu, X.; Liu, B.F.; Zhang, L.; Zhang, G. Synthesis and biological evaluation of fused tricyclic heterocycle piperazine (piperidine) derivatives as potential multireceptor atypical antipsychotics. J. Med. Chem. 2018, 61, 10017–10039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Murugan, N.A.; Tian, Y.; Bertagnin, C.; Fang, Z.; Kang, D.; Kong, X.; Jia, H.; Sun, Z.; Jia, R.; et al. Structure-based optimization of N-substituted oseltamivir derivatives as potent anti-Influenza A virus agents with significantly improved potency against oseltamivir-resistant N1-H274Y variant. J. Med. Chem. 2018, 61, 9976–9999. [Google Scholar] [CrossRef] [PubMed]
- Rageot, D.; Bohnacker, T.; Melone, A.; Langlois, J.-B.; Borsari, C.; Hillmann, P.; Sele, A.M.; Beaufils, F.; Zvelebil, M.; Hebeisen, P.; et al. Discovery and preclinical characterization of 5-[4,6-Bis({3-oxa-8-azabicyclo[3.2.1]octan-8-yl})-1,3,5-triazin-2-yl]-4-(difluoromethyl)pyridin-2-amine (PQR620), a highly potent and selective mTORC1/2 inhibitor for cancer and neurological disorders. J. Med. Chem. 2018, 61, 10084–10105. [Google Scholar] [CrossRef] [PubMed]
- Damalanka, V.C.; Han, Z.; Karmakar, P.; O’Donoghue, A.J.; La Greca, F.; Kim, T.; Pant, S.M.; Helander, J.; Klefström, J.; Craik, C.S.; et al. Discovery of selective matriptase and hepsin Serine protease inhibitors: Useful chemical tools for cancer cell biology. J. Med. Chem. 2019, 62, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, J.; Aguilar, A.; McEachern, D.; Przybranowski, S.; Liu, L.; Yang, C.-Y.; Wang, M.; Han, X.; Wang, S. Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J. Med. Chem. 2019, 62, 448–466. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Wu, J.; Yuan, X.; Zhao, L.; Zhang, G.; Li, C.; Qiao, R. Design and evaluation of potent EGFR inhibitors through the incorporation of macrocyclic polyamine moieties into the 4-anilinoquinazoline scaffold. J. Med. Chem. 2018, 61, 11372–11383. [Google Scholar] [CrossRef]
- ChEMBL. Available online: https://www.ebi.ac.uk/chembl/faq (accessed on 11 February 2019).
- Glaab, E. Building a virtual ligand screening pipeline using free software: A survey. Brief. Bioinform. 2016, 17, 352–366. [Google Scholar] [CrossRef]
- González-Medina, M.; Naveja, J.J.; Sánchez-Cruz, N.; Medina-Franco, J.L. Open chemoinformatic resources to explore the structure, properties and chemical space of molecules. RSC Adv. 2017, 7, 54153–54163. [Google Scholar] [CrossRef] [Green Version]
- Chemaxon. Available online: https://chemaxon.com/ (accessed on 11 February 2019).
- Antonacci, M.; Bellotti, R.; Cafagna, F.; de Palma, M.; Diacono, D.; Donvito, G.; Italiano, A.; Gervasoni, R.; Maggi, G.; Miniello, G.; et al. The ReCaS Project: The Bari Infrastructure. In High Performance Scientific Computing Using Distributed Infrastructures; World Scientific: Singapore, 2017; pp. 17–33. ISBN 978-981-4759-70-0. [Google Scholar]
- Ziegler, S.; Pries, V.; Hedberg, C.; Waldmann, H. Target identification for small bioactive molecules: Finding the needle in the haystack. Angew. Chem. Int. Ed. Engl. 2013, 52, 2744–2792. [Google Scholar] [CrossRef] [PubMed]
- Alberga, D.; Trisciuzzi, D.; Mansouri, K.; Mangiatordi, G.F.; Nicolotti, O. Prediction of acute oral systemic toxicity using a multifingerprint similarity approach. Toxicol. Sci. Off. J. Soc. Toxicol. 2019, 167, 484–495. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
| Fingerprints Name | Description | Package | Reference |
|---|---|---|---|
| MFP1 | Morgan connectivity invariants (ECFP-like) with radius = 1 | RDKit | [13] |
| FeatMFP1 | Morgan feature invariants (FCFP-like) with radius = 1 | RDKit | [13,14] |
| AP_bits | Atom pairs fingerprint | RDKit | [15] |
| Pattern | SMARTS Pattern fingerprint | RDKit | [9] |
| RDKit7 | Daylight-like topological fingerprint | RDKit | [9] |
| TT_bits | Topological torsion fingerprint | RDKit | [16] |
| FP2 | Indexes linear fragments up to 7 atoms | Pybel | [10] |
| pubchem | Pubchem fingerprints | CDK | [17] |
| cdk_maccs | MACCS fingerprint that generates 166-bit MACCS keys | CDK | [11,12] |
| klekota_roth | Klekota-Roth fingerprints based on 4860 substructures | CDK | [18] |
| graph | Graph fingerprint which does not take bond orders into account | CDK | [11,12] |
| substructure | Bit set type fingerprint based on 307 substructures | CDK | [11,12] |
| hybridization | Fingerprint based on hybridization state of atoms | CDK | [11,12] |
| Ki MuSSel Data1 | IC50 MuSSel Data1 | |||
|---|---|---|---|---|
| p1 | p5 | p1 | p5 | |
| Neutral database | 89.72% | 92.82% | 86.80% | 90.20% |
| Ionized database | 91.08% | 93.16% | 88.72% | 92.24% |
| Ki MuSSel Data1 | IC50 MuSSel Data1 | |||
|---|---|---|---|---|
| p1 | p5 | p1 | p5 | |
| Ext1 (n = 300) | 90.67% | 96.00% (56.20%) * | 88.00% | 93.33% (35.00%) * |
| Ext2 (n = 300) | 90.33% | 96.00% (48.60%) * | 92.00% | 95.00% (31.70%) * |
| Ext3 (n = 300) | 93.67% | 97.33% (51.40%) * | 89.33% | 92.00% (29.30%) * |
| Ext4 (n = 1000) | 90.77% | 94.32% | 90.10% | 93.20% |
 |  |  |
| 1 HIV-1 Protease CHEMBL2366517, n = 997 [29] | 2 Heat shock protein 90 kDa beta member 1 CHEMBL4303, n = 538 [24] | 3 Sigma opioid receptor CHEMBL4153, n = 1426 [30] |
 |  |  |
| 4 Transient receptor potential cation channel subfamily V 4 CHEMBL3119, n = 50 [31] | 5 Dopamine D2 receptor CHEMBL339, n = 3934 [32] | 6 CC-Chemokine Receptor 5 CHEMBL274, n = 2051 [33] |
 |  |  |
| 7 DNA topoisomerase I CHEMBL1781, n = 347 [34] | 8 Tyrosine-protein kinase BTK CHEMBL5251, n = 808 [35] | 9 Cytochrome P450 (CYP) 1B1 CHEMBL1978, n = 1858 [36] |
 |  |  |
| 10 Fibroblast growth factor receptor 1 CHEMBL4142, n = 288 [25] | 11 Dopamine D1 receptor CHEMBL2056, n = 986 [37] | 12 Rho-associated protein kinase 2 CHEMBL2973, n = 1687 [38] |
 |  |  |
| 13 Dopamine D2 receptor CHEMBL339, n = 3934 [39] | 14 Neuraminidase - Influenza A virus CHEMBL1667684, n = 35 [40] | 15 Serine/threonine-protein kinase mTOR CHEMBL2842, n = 3087 [41] |
 |  |  |
| 16 Hepsin serine protease CHEMBL204, n = 4774 [42] | 17 p53-binding protein Mdm-2 CHEMBL5023, n = 1830 [43] | 18 Epidermal growth factor receptor CHEMBL203, n = 5187 [44] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montaruli, M.; Alberga, D.; Ciriaco, F.; Trisciuzzi, D.; Tondo, A.R.; Mangiatordi, G.F.; Nicolotti, O. Accelerating Drug Discovery by Early Protein Drug Target Prediction Based on a Multi-Fingerprint Similarity Search †. Molecules 2019, 24, 2233. https://doi.org/10.3390/molecules24122233
Montaruli M, Alberga D, Ciriaco F, Trisciuzzi D, Tondo AR, Mangiatordi GF, Nicolotti O. Accelerating Drug Discovery by Early Protein Drug Target Prediction Based on a Multi-Fingerprint Similarity Search †. Molecules. 2019; 24(12):2233. https://doi.org/10.3390/molecules24122233
Chicago/Turabian StyleMontaruli, Michele, Domenico Alberga, Fulvio Ciriaco, Daniela Trisciuzzi, Anna Rita Tondo, Giuseppe Felice Mangiatordi, and Orazio Nicolotti. 2019. "Accelerating Drug Discovery by Early Protein Drug Target Prediction Based on a Multi-Fingerprint Similarity Search †" Molecules 24, no. 12: 2233. https://doi.org/10.3390/molecules24122233