Anomeric Spironucleosides of β-d-Glucopyranosyl Uracil as Potential Inhibitors of Glycogen Phosphorylase


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
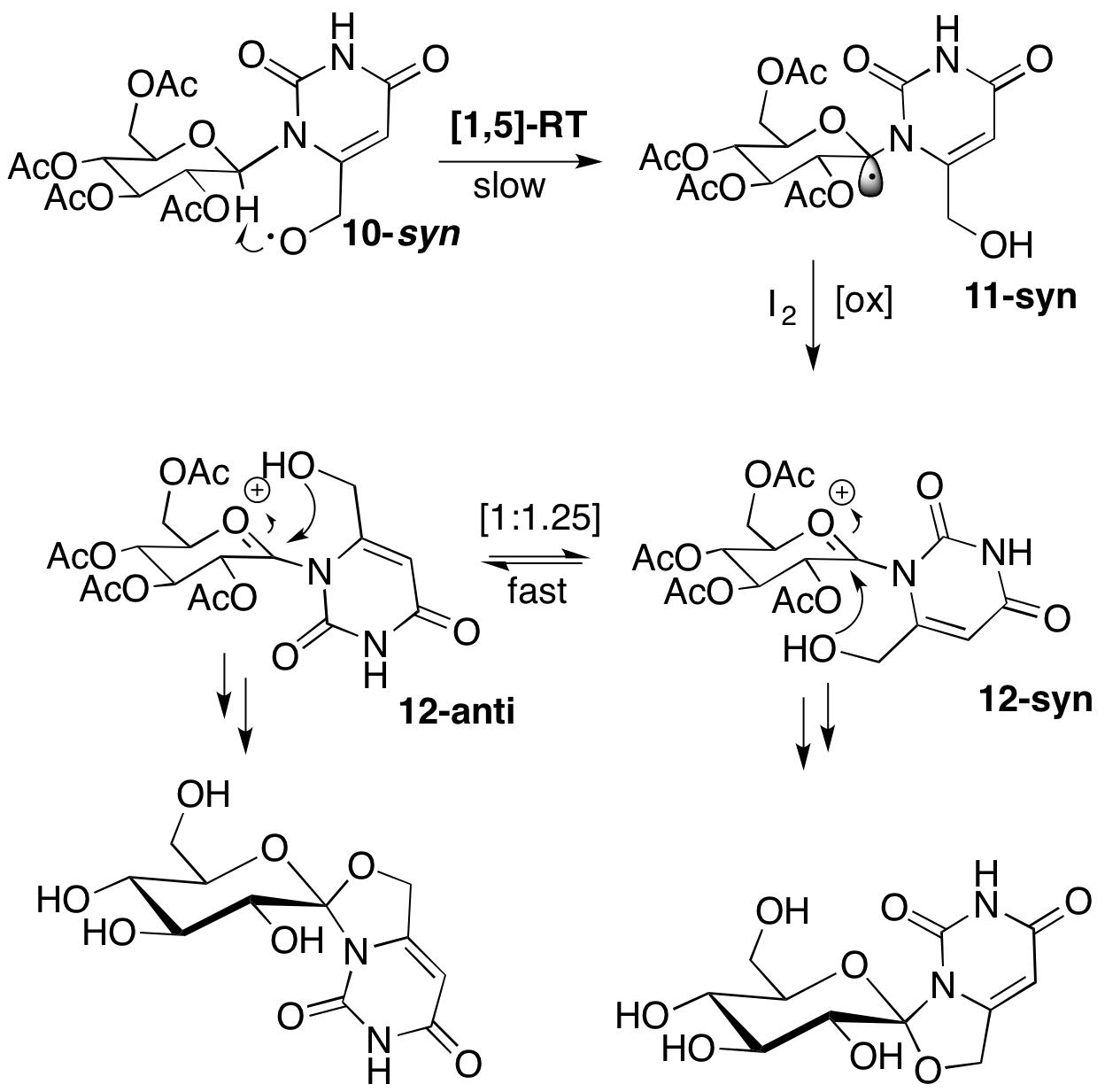
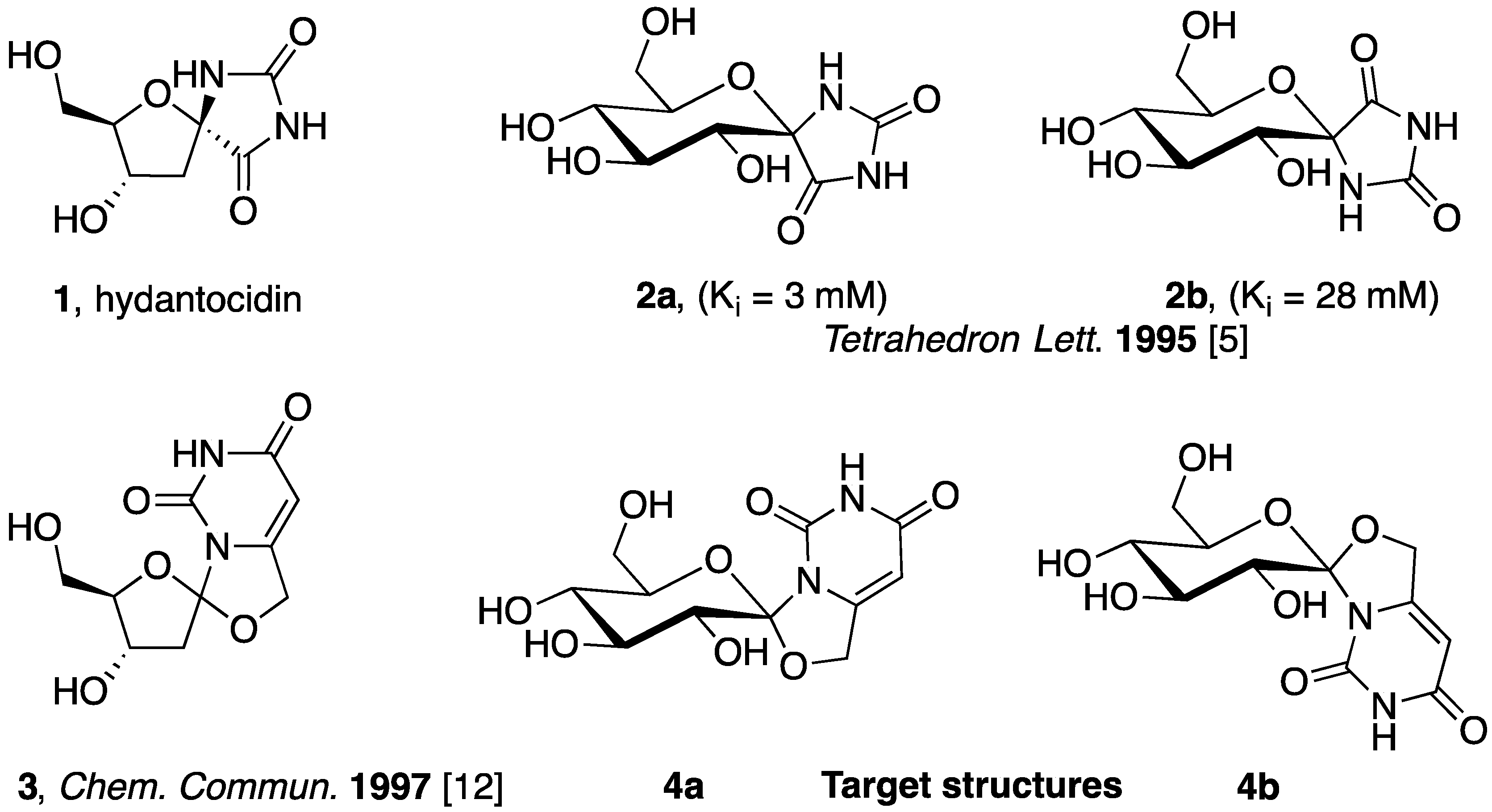
2. Results
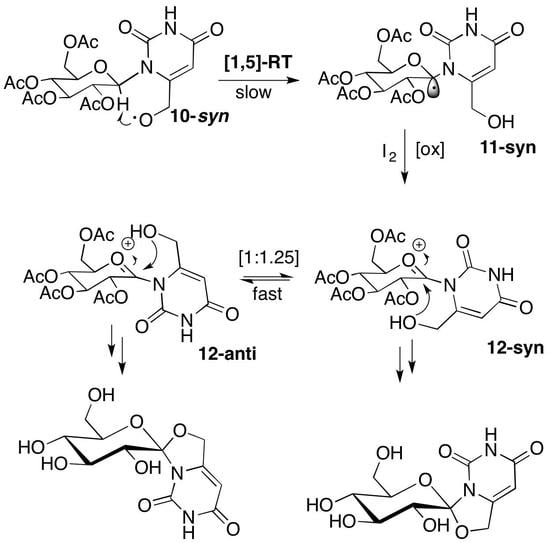
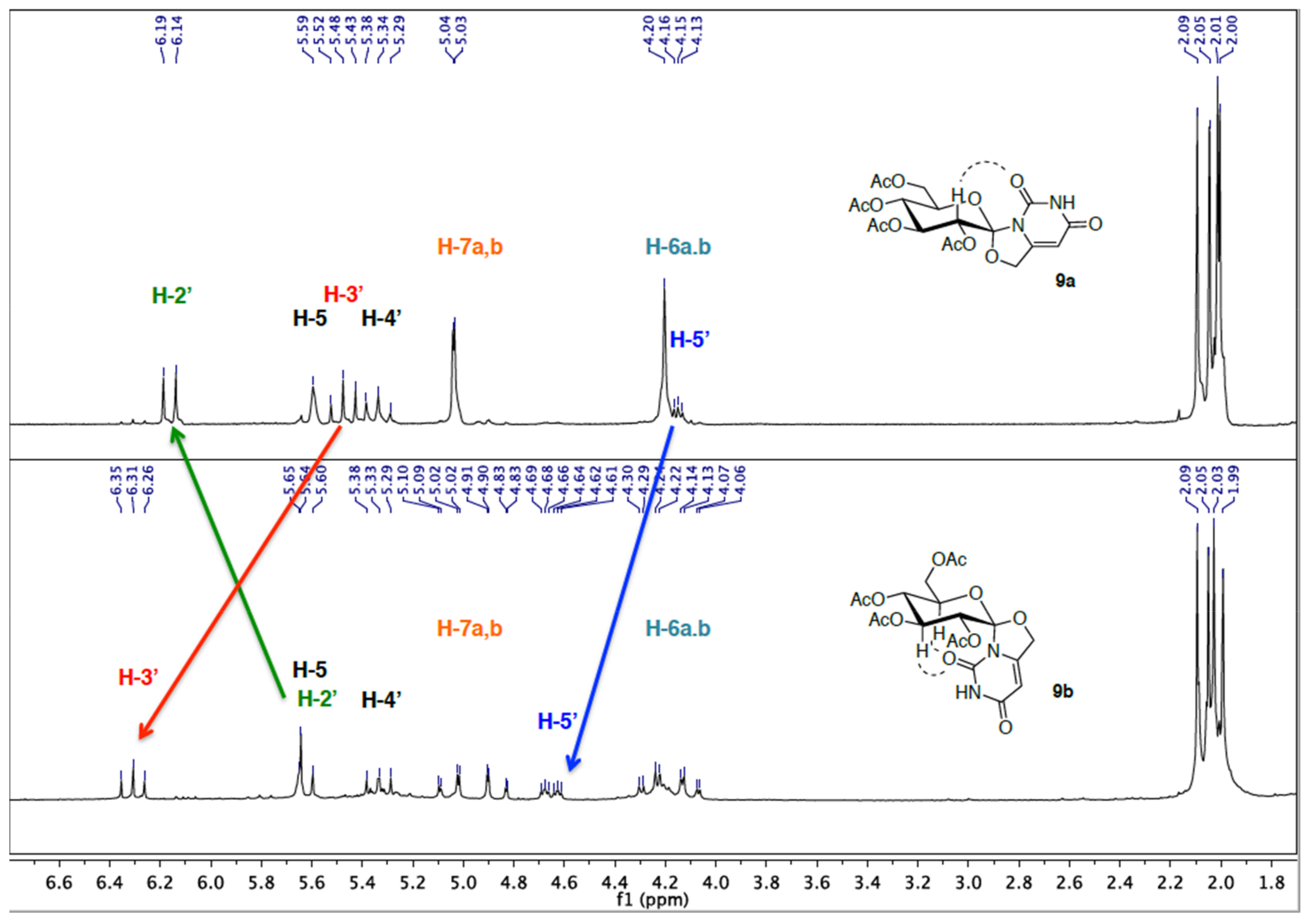
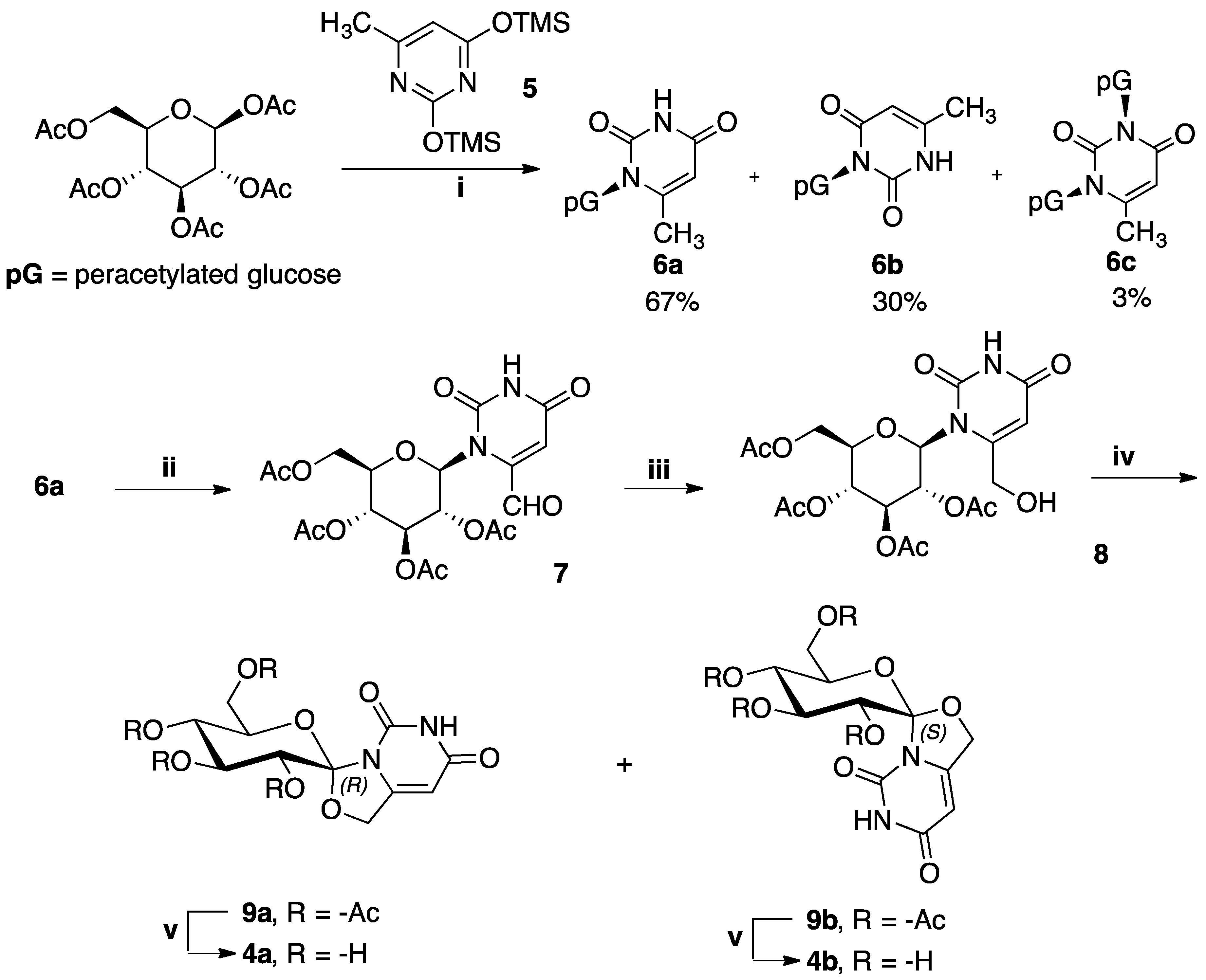
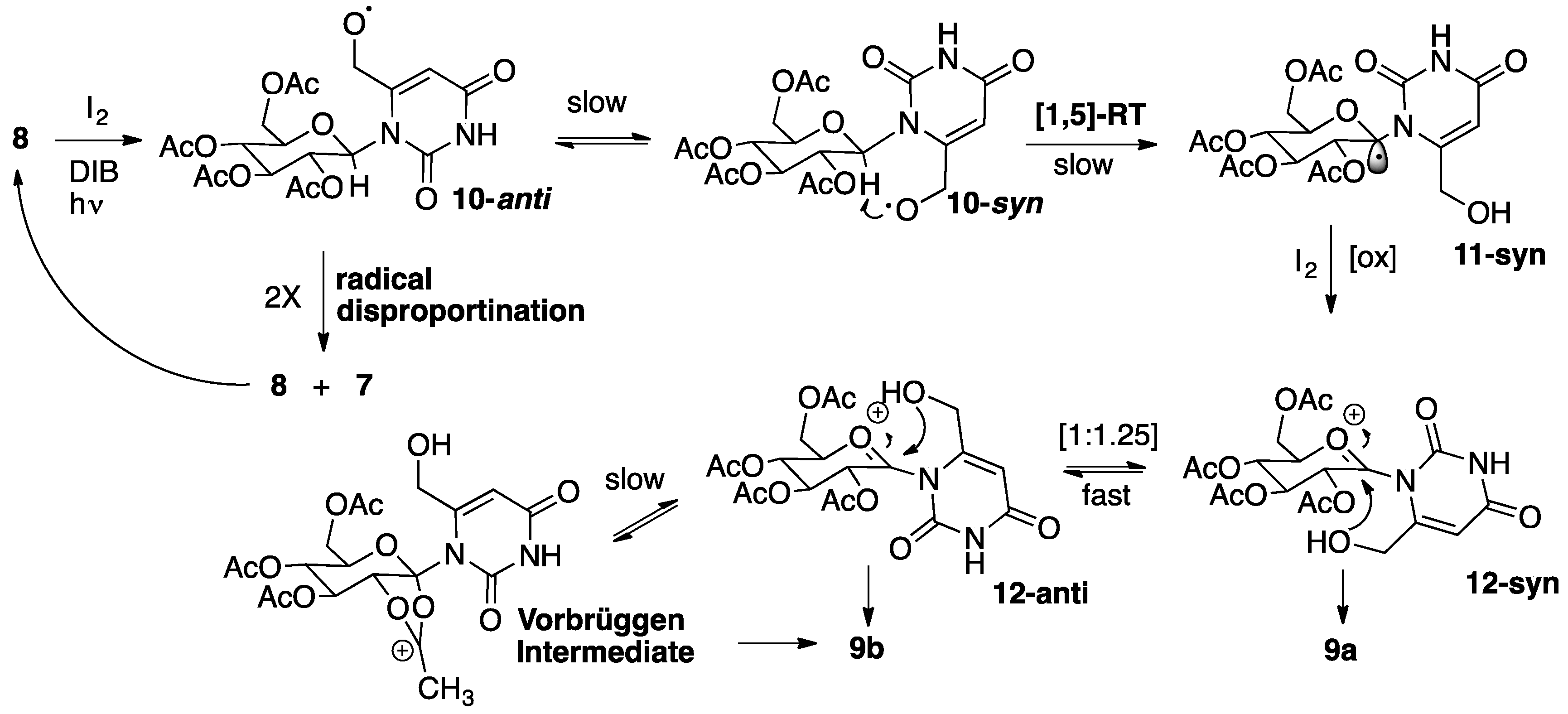
2.1. Synthesis
2.2. Kinetic Experiments
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rines, A.K.; Sharabi, K.; Tavares, C.D.J.; Puigserver, P. Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat. Rev. Drug Discov. 2016, 15, 786–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimisis, T. Synthesis of N-Glucopyranosidic Derivatives as Potential Inhibitors that Bind at the Catalytic Site of Glycogen Phosphorylase. Mini-Rev. Med. Chem. 2010, 10, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Donnier-Maréchal, M.; Vidal, S. Glycogen phosphorylase inhibitors: A patent review (2013–2015). Expert Opin. Ther. Pat. 2016, 26, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Chrysina, E.D.; Chajistamatiou, A.; Chegkazi, M. From structure-based to knowledge-based drug design through X-ray protein crystallography: Sketching glycogen phosphorylase binding sites. Curr. Med. Chem. 2011, 18, 2620–2629. [Google Scholar] [CrossRef] [PubMed]
- Bichard, C.J.F.; Mitchell, E.P.; Wormald, M.R.; Watson, K.A.; Johnson, L.N.; Zographos, S.E.; Koutra, D.D.; Oikonomakos, N.G.; Fleet, G.W.J. Potent inhibition of glycogen phosphorylase by a spirohydantoin of glucopyranose: First pyranose analogues of hydantocidin. Tetrahedron Lett. 1995, 36, 2145–2148. [Google Scholar] [CrossRef]
- Goyard, D.; Kónya, B.; Chajistamatiou, A.S.; Chrysina, E.D.; Leroy, J.; Balzarin, S.; Tournier, M.; Tousch, D.; Petit, P.; Duret, C.; et al. Glucose-derived spiro-isoxazolines are anti-hyperglycemic agents against type 2 diabetes through glycogen phosphorylase inhibition. Eur. J. Med. Chem. 2016, 108, 444–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamais, M.; Degli Esposti, A.; Kouloumoundra, V.; Gustavsson, T.; Monti, F.; Venturini, A.; Chrysina, E.D.; Markovitsi, D.; Gimisis, T. A New Potent Inhibitor of Glycogen Phosphorylase Reveals the Basicity of the Catalytic Site. Chem. A Eur. J. 2017, 23, 8800–8805. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.; Rodríguez-Solla, H.; Soengas, R. Recent Advances in the Chemistry and Biology of Spirocyclic Nucleosides. In Topics in Heterocyclic Chemistry; Springer: Berlin, Heidelberg, Germany, 2019; pp. 1–43. [Google Scholar]
- Chatgilialoglu, C.; Ferreri, C.; Gimisis, T.; Roberti, M.; Balzarini, J.; De Clercq, E. Synthesis and Biological Evaluation of Novel 1’-Branched and Spiro-Nucleoside Analogues. Nucleosides Nucleotides Nucleic Acids 2004, 23, 1565–1581. [Google Scholar] [CrossRef] [PubMed]
- Gómez-García, O.; Gómez, E.; Toscano, R.; Salgado-Zamora, H.; Álvarez-Toledano, C. One-Pot Synthesis of Spirotetrahydrooxino [3,4-c] pyridines and Spirotetrahydrofuro [3,2-b] pyridin-2-ones via Lactonization from Activated Pyridyldihydroozaxoles and Bis(trimethylsilyl)ketene Acetals. Synthesis 2016, 48, 1371–1380. [Google Scholar] [CrossRef]
- Martín, A.; Suárez, E. Carbohydrate Spiro-heterocycles via Radical Chemistry. In Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–54. [Google Scholar]
- Chatgilialoglu, C.; Gimisis, T.; Spada, G.P. C-1’ Radical-Based Approaches for the Synthesis of Anomeric Spironucleosides. Chem. A Eur. J. 1999, 5, 2866–2876. [Google Scholar] [CrossRef]
- Gimisis, T.; Chatgilialoglu, C.; Gimisis, T.; Castellari, C. A new class of anomeric spironucleosides. Chem. Commun. 1997, 2089–2090. [Google Scholar] [CrossRef]
- Liao, J.; Sun, J.; Yu, B. Effective synthesis of nucleosides with glycosyl trifluoroacetimidates as donors. Tetrahedron Lett. 2008, 49, 5036–5038. [Google Scholar] [CrossRef]
- Groziak, M.P.; Koohang, A. Facile addition of hydroxylic nucleophiles to the formyl group of uridine-6-carboxaldehydes. J. Org. Chem. 1992, 57, 940–944. [Google Scholar] [CrossRef]
- Tanaka, H.; Hayakawa, H.; Miyasaka, T. “Umpulong” of reactivity at the C-6 position of uridine: A simple and general method for 6-substituted uridines. Tetrahedron 1982, 38, 2635–2642. [Google Scholar] [CrossRef]
- Liu, H.-J.; Yip, J.; Shia, K.-S. Reductive cleavage of benzyl ethers with lithium naphthalenide. A convenient method for debenzylation. Tetrahedron Lett. 1997, 38, 2253–2256. [Google Scholar] [CrossRef]
- Wittenburg, E. Nucleoside und verwandte Verbindungen. VII. Alkylierung und Glykosidierung der Silyl-derivate 6-substituierter Uracile. Collect. Czechoslov. Chem. Commun. 1971, 36, 246–261. [Google Scholar] [CrossRef]
- Felczak, K.; Drabikowska, A.K.; Vilpo, J.A.; Kulikowski, T.; Shugar, D. 6-Substituted and 5,6-Disubstituted Derivatives of Uridine: Stereoselective Synthesis, Interaction with Uridine Phosphorylase, and In Vitro Antitumor Activity. J. Med. Chem. 1996, 39, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Warpehoski, M.A.; Chabaud, B.; Sharpless, K.B. Selenium dioxide oxidation of endocyclic olefins. Evidence for a dissociation-recombination pathway. J. Org. Chem. 1982, 47, 2897–2900. [Google Scholar] [CrossRef]
- Młochowski, J.; Brząszcz, M.; Giurg, M.; Palus, J.; Wójtowicz, H. Selenium-Promoted Oxidation of Organic Compounds: Reactions and Mechanisms. Eur. J. Org. Chem. 2003, 2003, 4329–4339. [Google Scholar] [CrossRef]
- Florent, J.-C.; Dong, X.; Gaudel, G.; Mitaku, S.; Monneret, C.; Gesson, J.-P.; Jacquesy, J.-C.; Mondon, M.; Renoux, B.; Andrianomenjanahary, S.; et al. Prodrugs of Anthracyclines for Use in Antibody-Directed Enzyme Prodrug Therapy. J. Med. Chem. 1998, 41, 3572–3581. [Google Scholar] [CrossRef]
- Francisco, C.G.; Freire, R.; Herrera, A.J.; Pérez-Martín, I.; Suárez, E. Intramolecular 1,5-versus 1,6-hydrogen abstraction reaction promoted by alkoxyl radicals in pyranose and furanose models. Tetrahedron 2007, 63, 8910–8920. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Beaton, J.M.; Geller, L.E.; Pechet, M.M. A New Photochemical Reaction 1. J. Am. Chem. Soc. 1961, 83, 4076–4083. [Google Scholar] [CrossRef]
- Kittaka, A.; Kato, H.; Tanaka, H.; Nonaka, Y.; Amano, M.; Nakamura, K.T.; Miyasaka, T. Face selective 6,1’-(1-oxo) ethano bridge formation of uracil nucleosides under hypoiodite reaction conditions. Tetrahedron 1999, 55, 5319–5344. [Google Scholar] [CrossRef]
- Oikonomakos, N.G.; Kontou, M.; Zographos, S.E.; Watson, K.A.; Johnson, L.N.; Bichard, C.J.F.; Fleet, G.W.J.; Acharya, K.R. N-acetyl-β-d-glucopyranosylamine: A potent T-state inhibitor of glycogen phosphorylase. A comparison with α-d-glucose. Protein Sci. 1995, 4, 2469–2477. [Google Scholar] [CrossRef] [PubMed]
- Saheki, S.; Takeda, A.; Shimazu, T. Assay of inorganic phosphate in the mild pH range, suitable for measurement of glycogen phosphorylase activity. Anal. Biochem. 1985, 148, 277–281. [Google Scholar] [CrossRef]
- Vorbrüggen, H.; Krolikiewicz, K.; Bennua, B. Nucleoside syntheses, XXII1) Nucleoside synthesis with trimethylsilyl triflate and perchlorate as catalysts. Chem. Ber. 1981, 114, 1234–1255. [Google Scholar] [CrossRef]
- Martin, J.L.; Veluraja, K.; Ross, K.; Johnson, L.N.; Fleet, G.W.J.; Ramsden, N.G.; Bruce, I.; Orchard, M.G.; Oikonomakos, N.G. Glucose analog inhibitors of glycogen phosphorylase: The design of potential drugs for diabetes. Biochemistry 1991, 30, 10101–10116. [Google Scholar] [CrossRef] [PubMed]
- Bokor, É.; Kun, S.; Docsa, T.; Gergely, P.; Somsák, L. 4(5)-Aryl-2-C-glucopyranosyl-imidazoles as New Nanomolar Glucose Analogue Inhibitors of Glycogen Phosphorylase. ACS Med. Chem. Lett. 2015, 6, 1215–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grugel, H.; Minuth, T.; Boysen, M. Novel Olefin-Phosphorus Hybrid and Diene Ligands Derived from Carbohydrates. Synthesis 2010, 19, 3248–3258. [Google Scholar]
- Helmreich, E.; Cori, C.F. The role of adenylic acid in the activation of phsphorylase. Proc. Natl. Acad. Sci. USA 1964, 51, 131–138. [Google Scholar] [CrossRef]
Sample Availability: Samples of compounds 4, 6–9 are available from the authors. |




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stathi, A.; Mamais, M.; Chrysina, E.D.; Gimisis, T. Anomeric Spironucleosides of β-d-Glucopyranosyl Uracil as Potential Inhibitors of Glycogen Phosphorylase. Molecules 2019, 24, 2327. https://doi.org/10.3390/molecules24122327
Stathi A, Mamais M, Chrysina ED, Gimisis T. Anomeric Spironucleosides of β-d-Glucopyranosyl Uracil as Potential Inhibitors of Glycogen Phosphorylase. Molecules. 2019; 24(12):2327. https://doi.org/10.3390/molecules24122327
Chicago/Turabian StyleStathi, Aggeliki, Michael Mamais, Evangelia D. Chrysina, and Thanasis Gimisis. 2019. "Anomeric Spironucleosides of β-d-Glucopyranosyl Uracil as Potential Inhibitors of Glycogen Phosphorylase" Molecules 24, no. 12: 2327. https://doi.org/10.3390/molecules24122327