
Formation of Unstable and very Reactive Chemical Species Catalyzed by Metalloenzymes: A Mechanistic Overview




Abstract
:1. Introduction
2. Enzymes that Catalyze the Formation of Hydroxide Anions
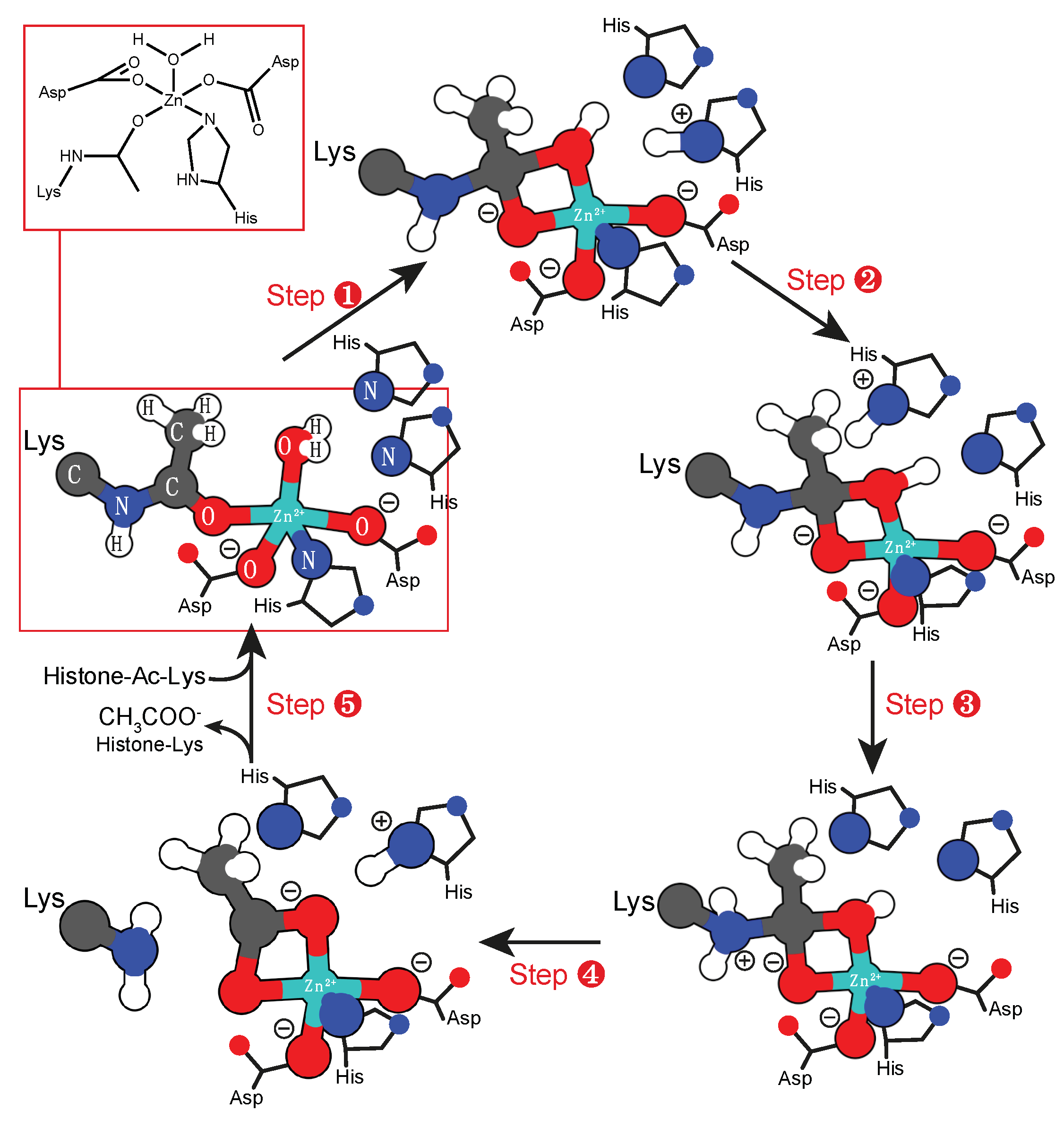
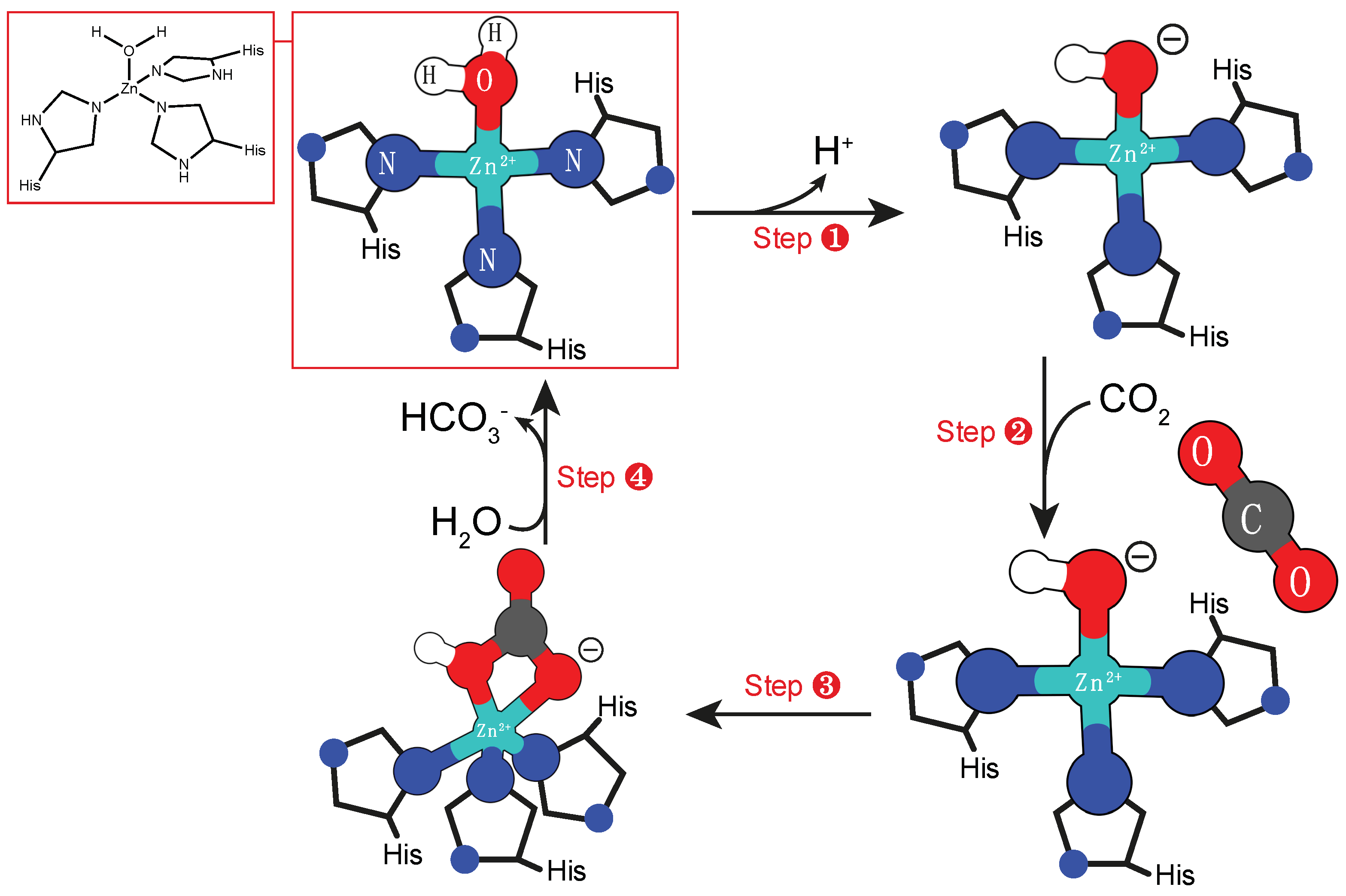
2.1. Mono-nuclear Zinc Cofactor
2.2. Binuclear Manganese Cofactor
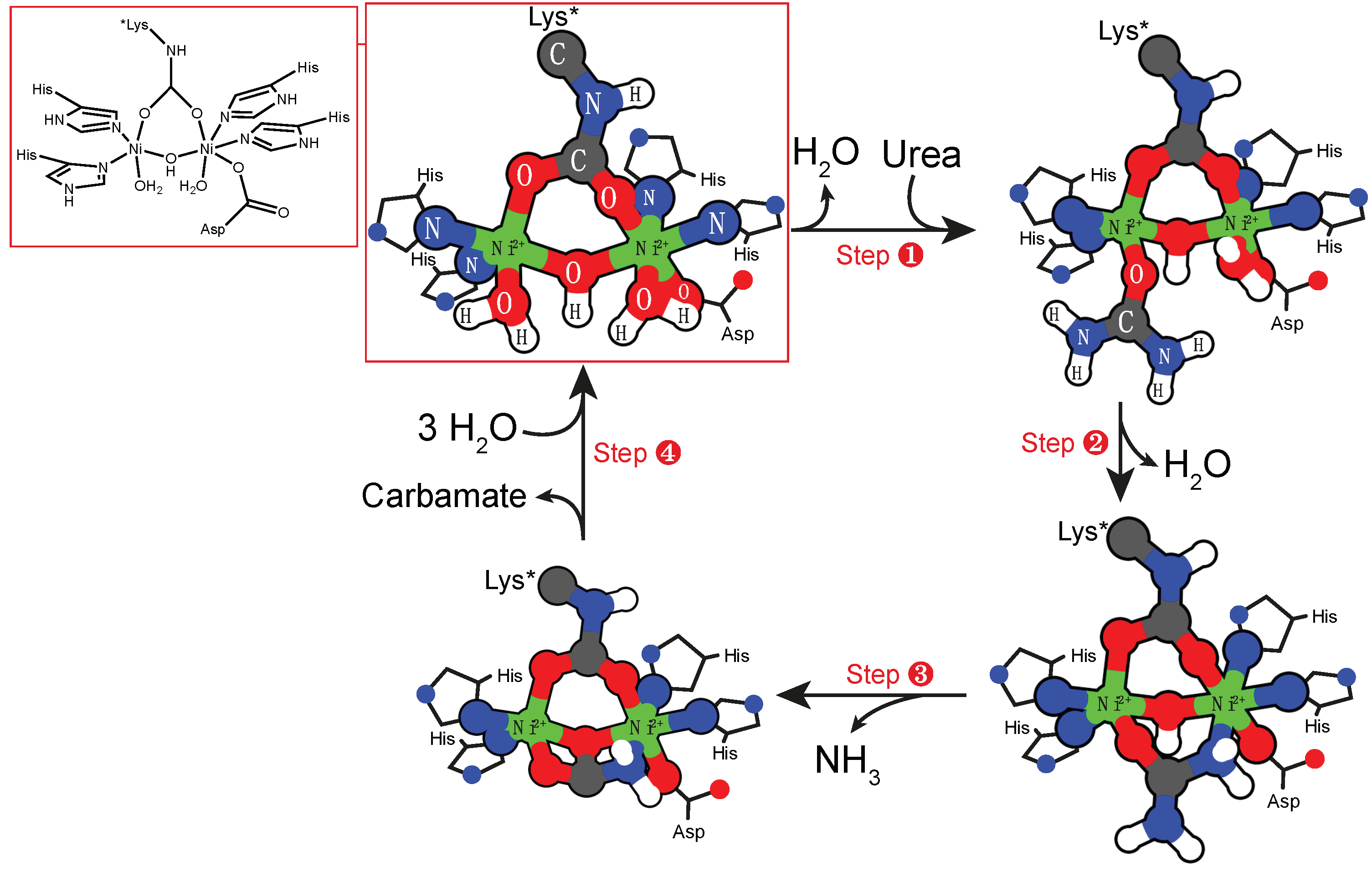
2.3. Binuclear Nickel Cofactor
2.4. Binuclear Zn Cofactor
3. Enzymes that Catalyze the Hydride Transfer Reactions
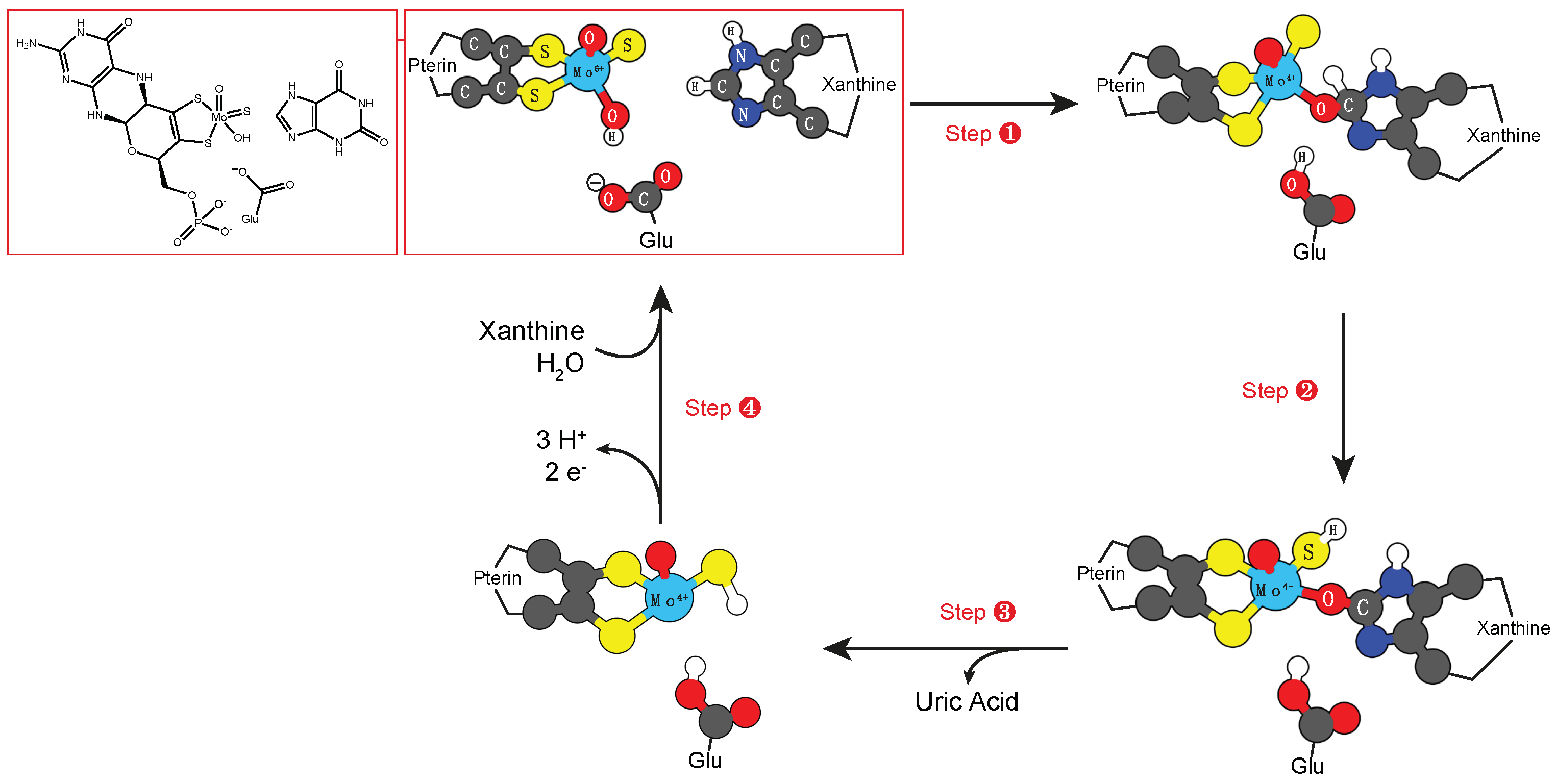
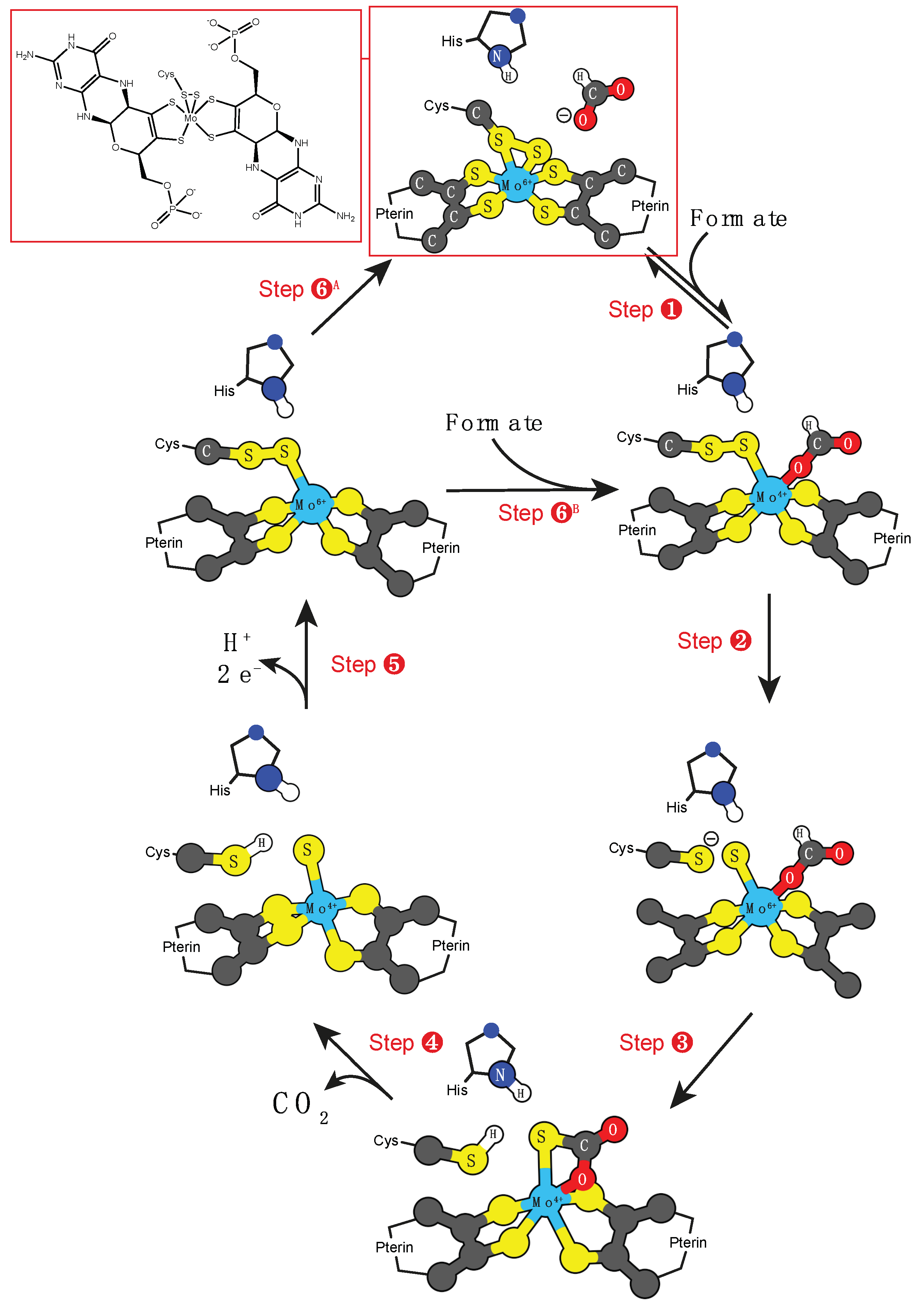
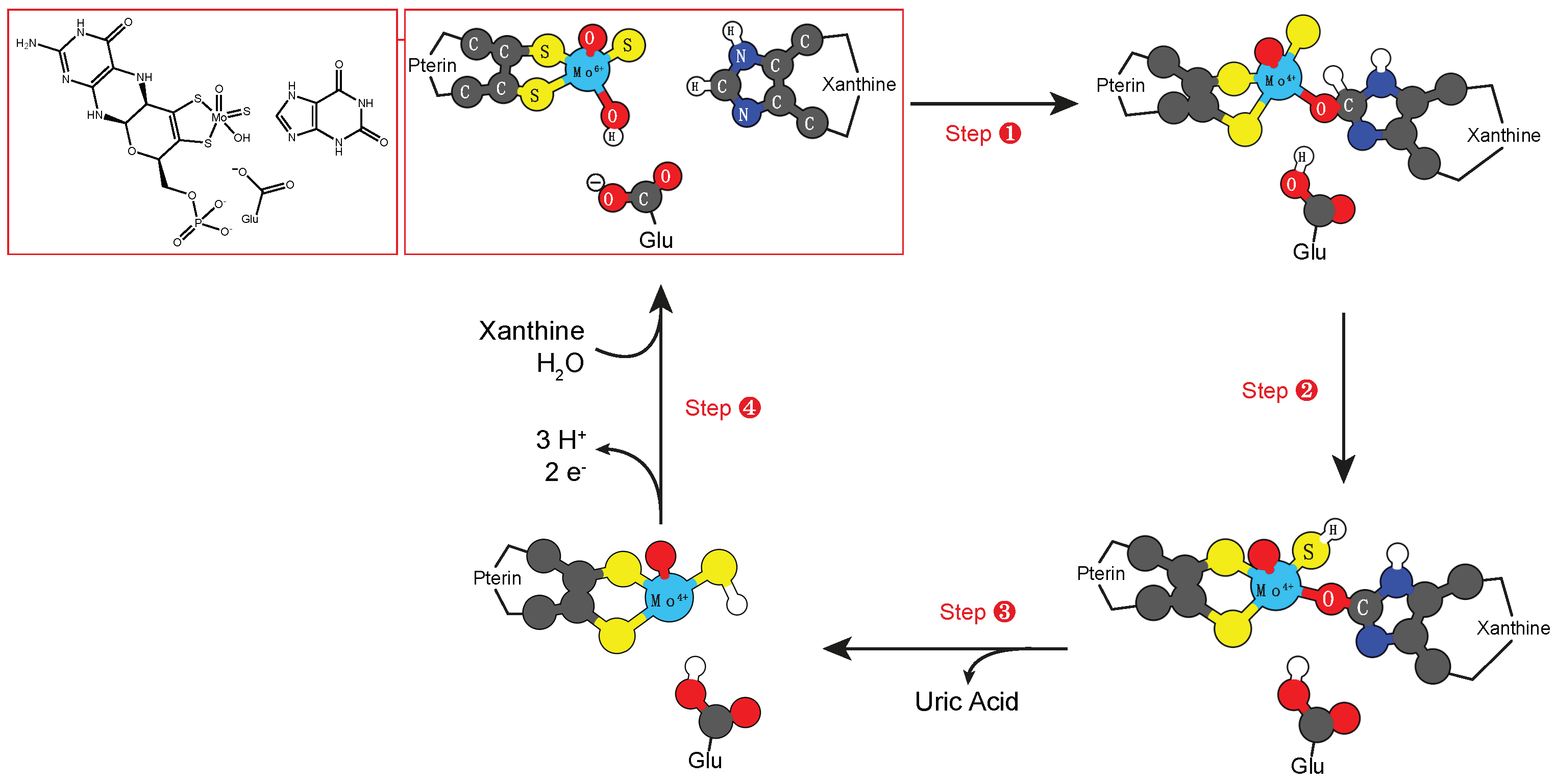
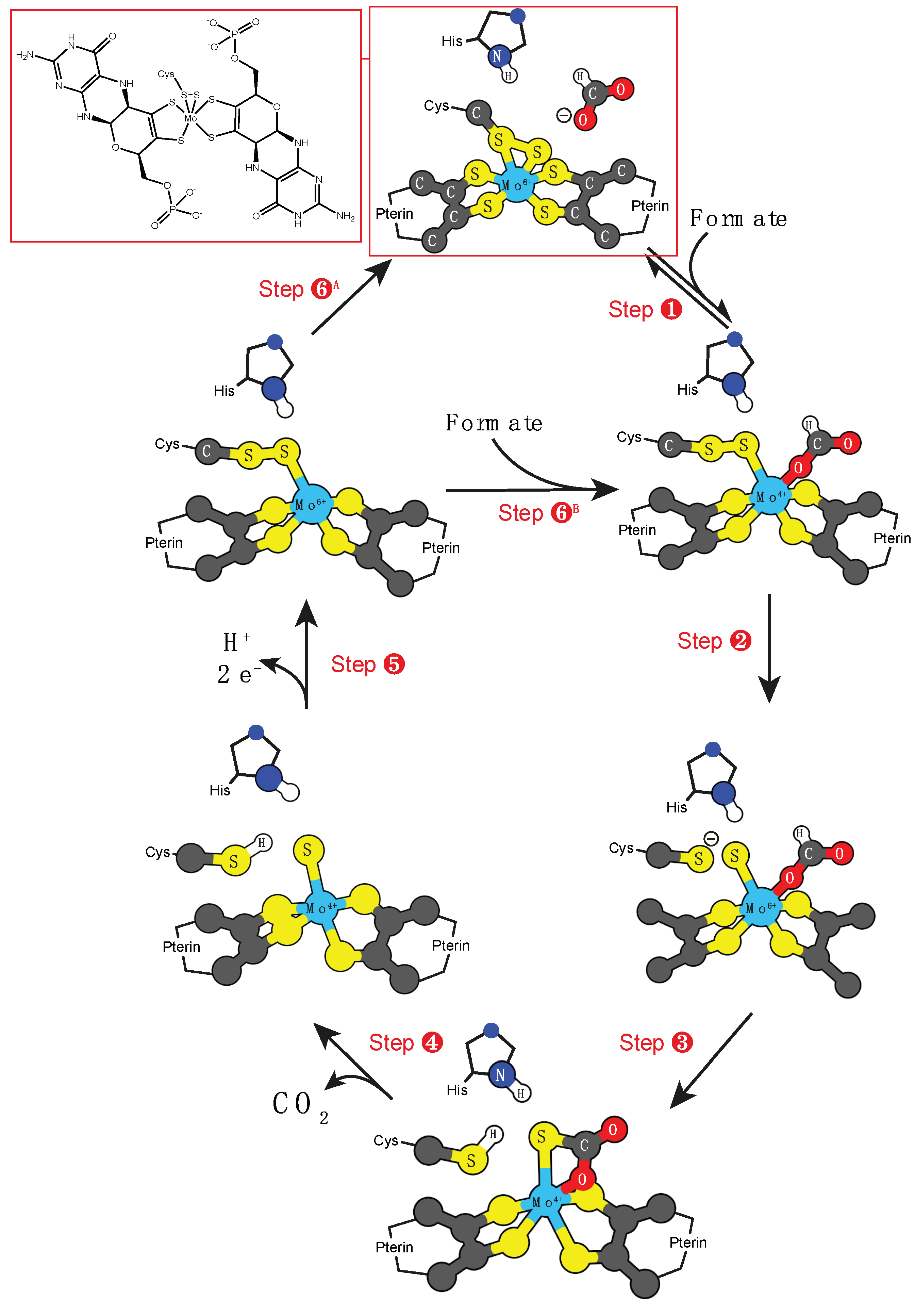
3.1. Molybdenum and Tungsten Cofactors
- Xanthine oxidase: RCHO + H2O → RCO2H + 2 H+ + 2 e−;
- Sulfite oxidase: SO3−2 + H2O → O4−2 + 2 H+ + 2 e−;
- DMSO reductase: HCO2− → O2 + 2 H+ + 2 e−.
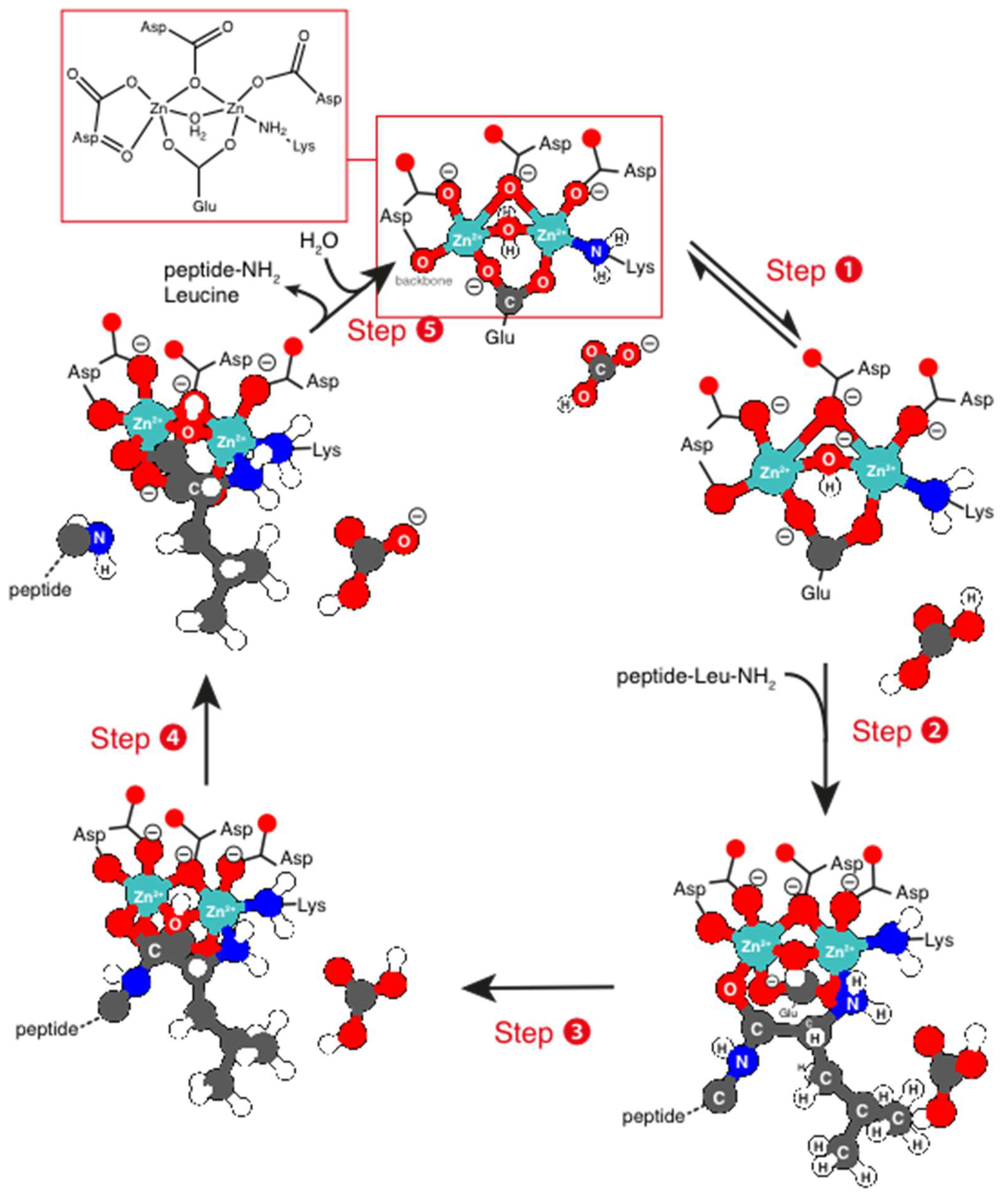
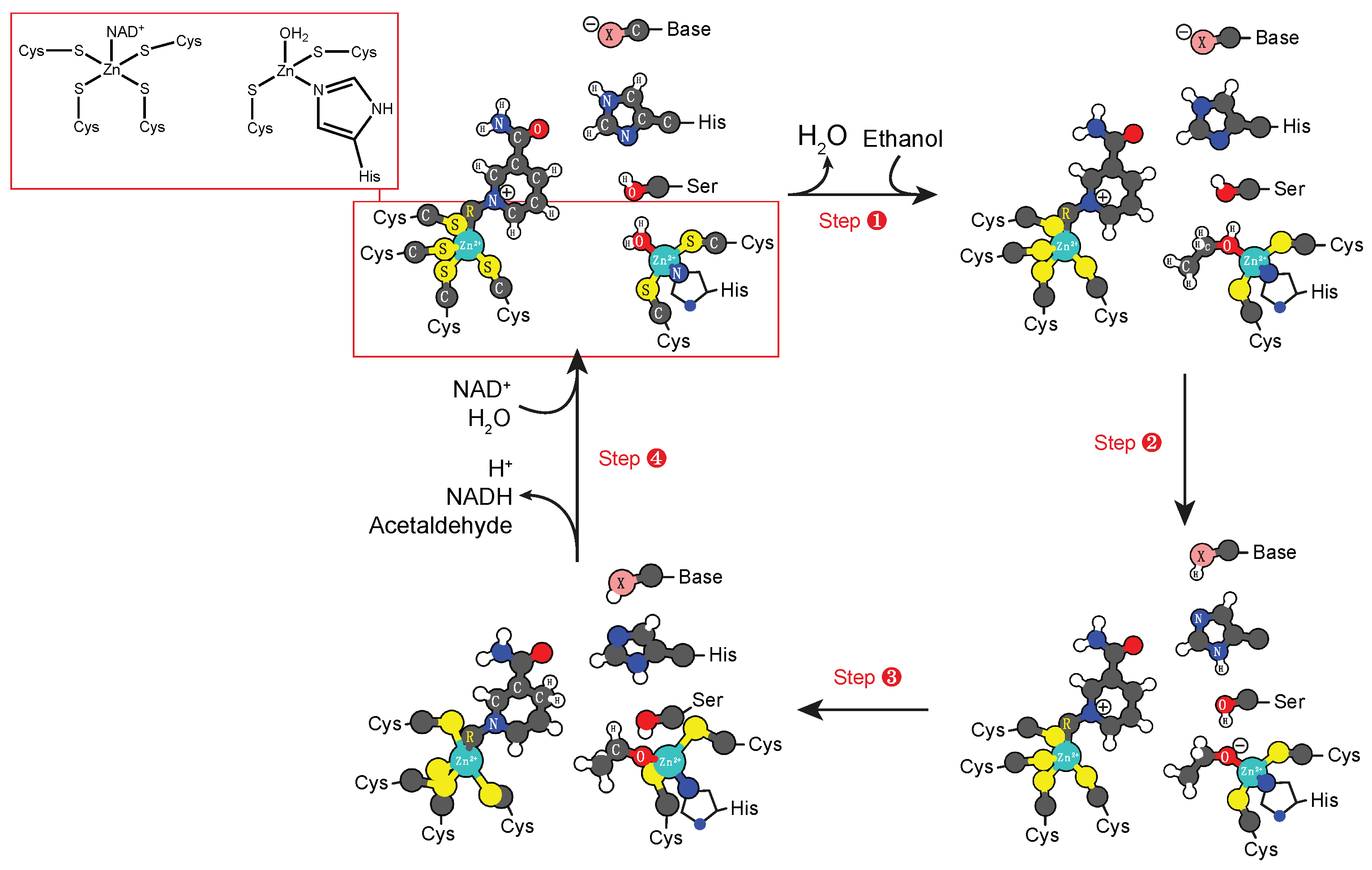
3.2. Zinc Cofactors
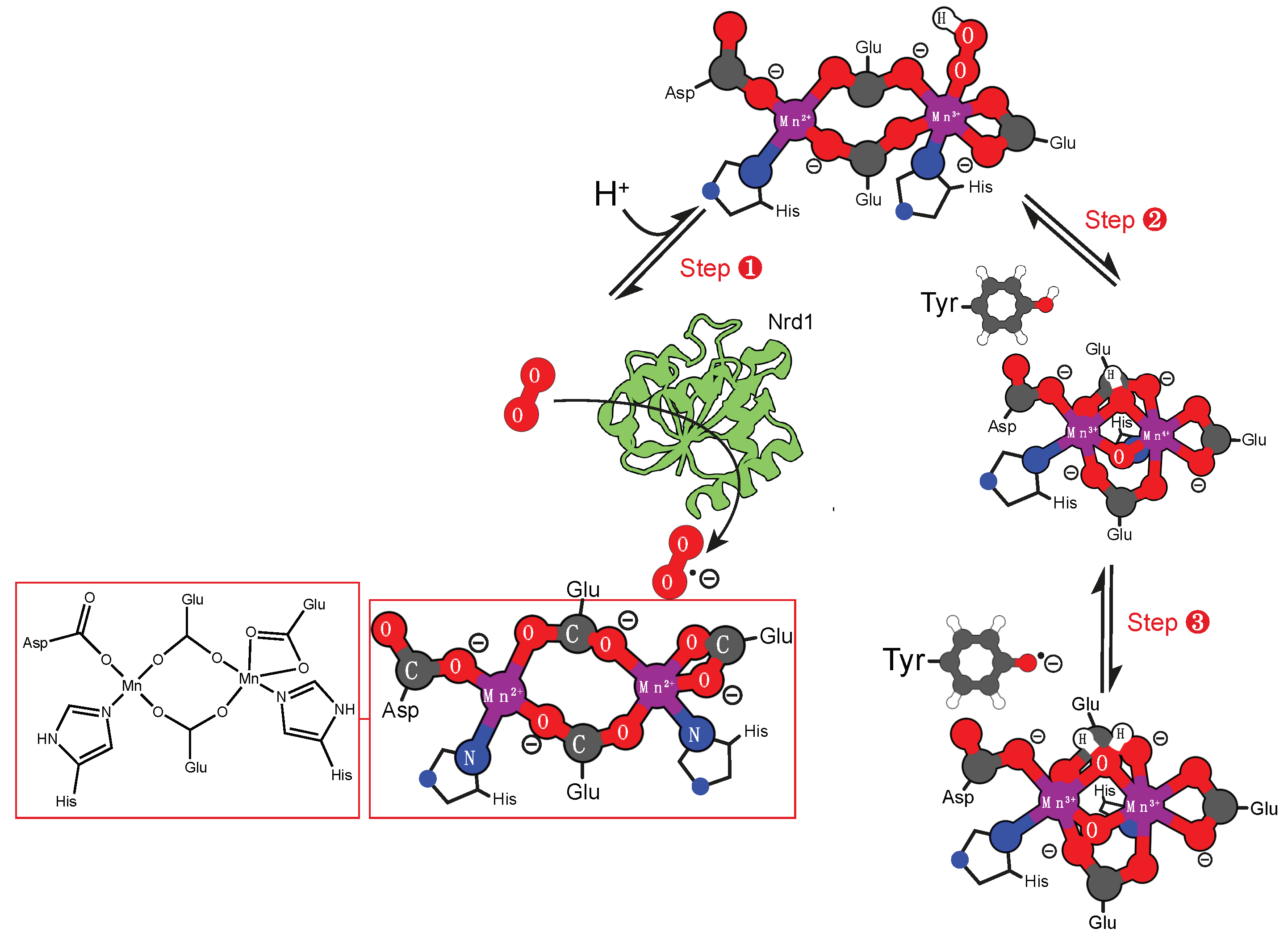
4. Enzymes that Catalyze the Formation of Radicals
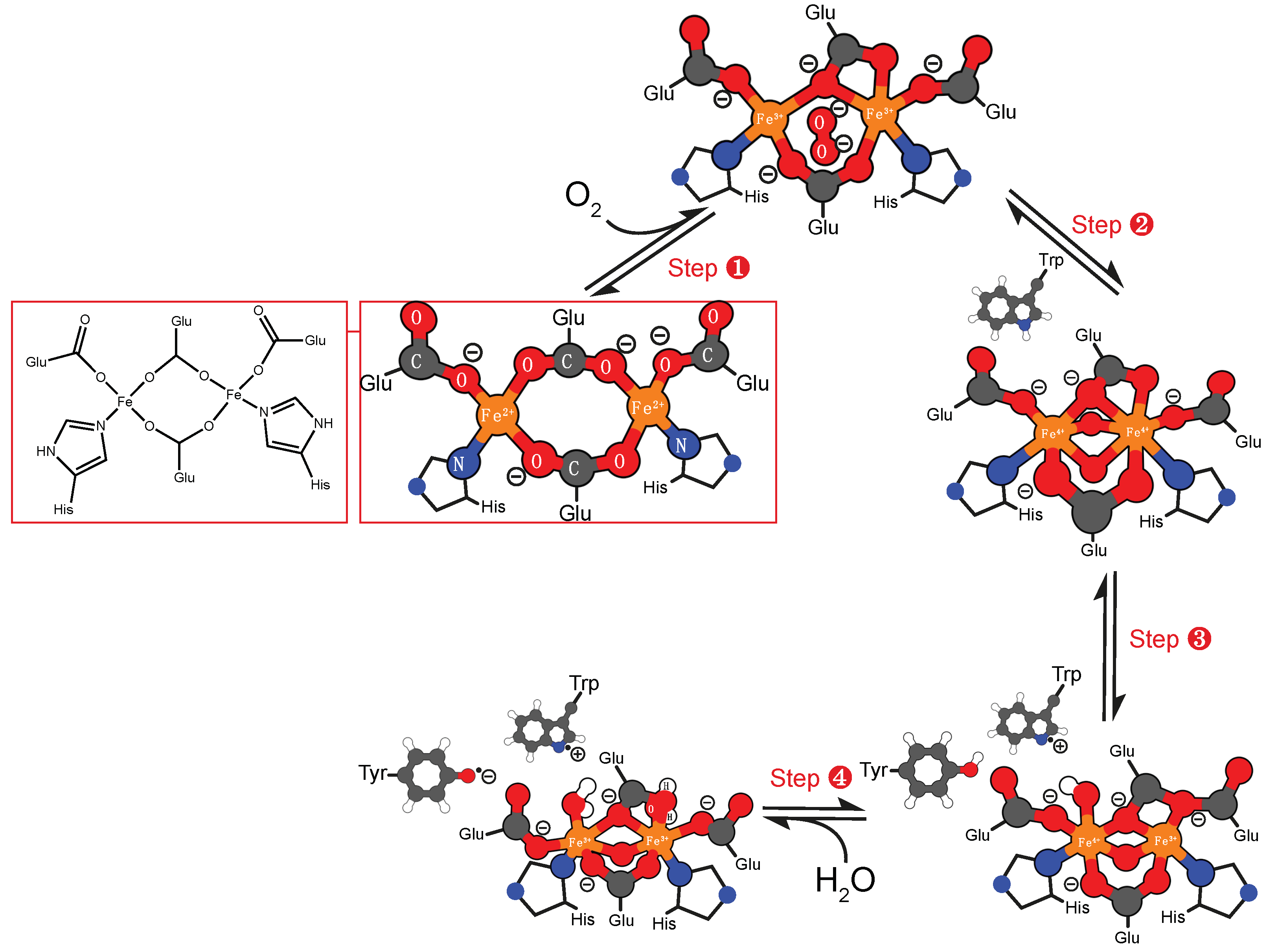
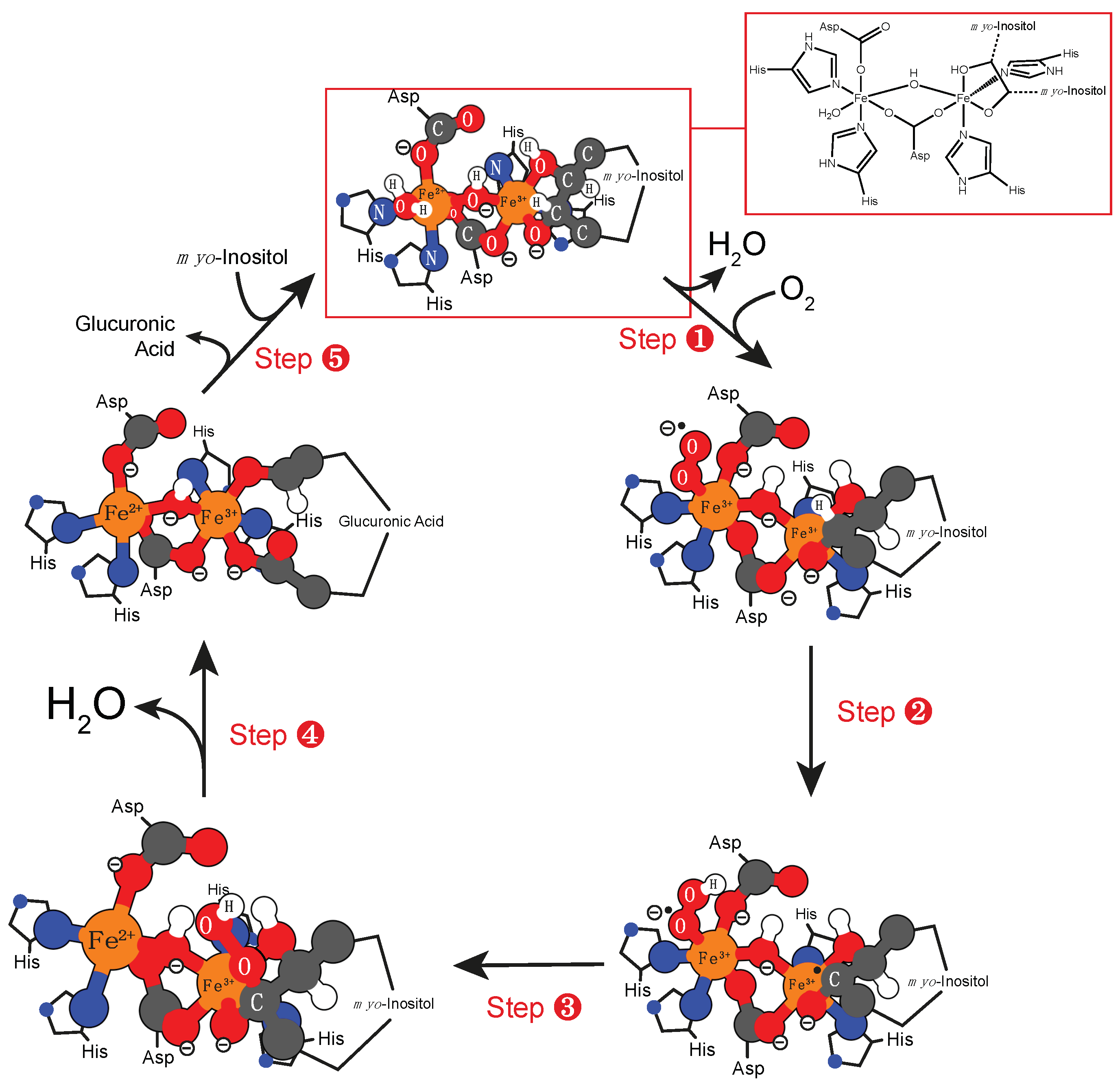
4.1. Iron Cofactor
4.2. Manganese Cofactor
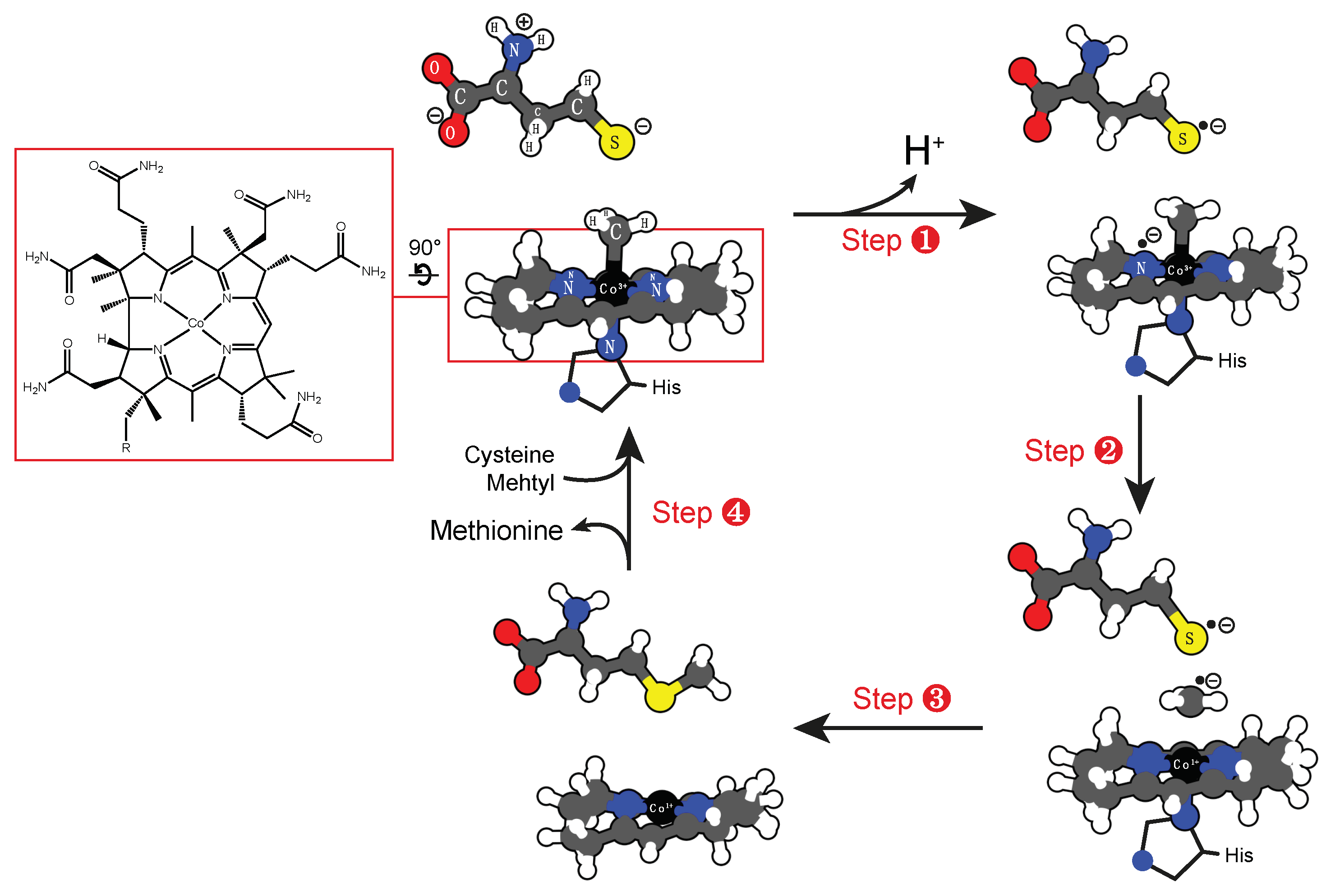
4.3. Cobalt Cofactor
5. Enzymes that Catalyze the Formation of Superoxide
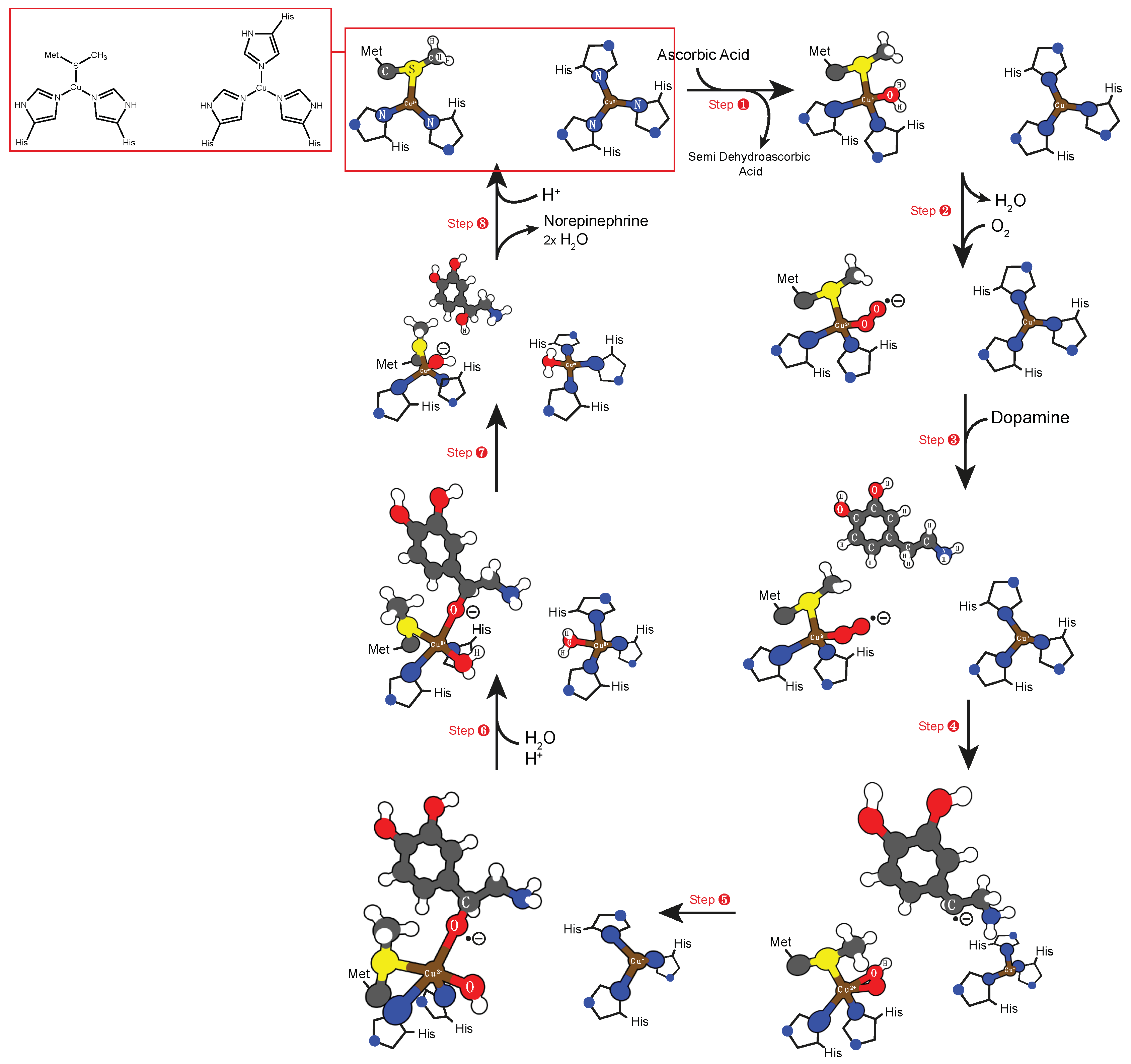
5.1. Copper Cofactor
5.2. Binuclear Iron Cofactor
6. Conclusions
Funding
Conflicts of Interest
References
- Kozlowski, L.P. Proteome-pi: Proteome isoelectric point database. Nucleic Acids Res. 2016, 45, D1112–D1116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ying, H.; Xu, Y. Comparative genomics and metagenomics of the metallomes. Metallomics 2019. [Google Scholar] [CrossRef] [PubMed]
- Pernil, R.; Schleiff, E. Metalloproteins in the biology of heterocysts. Life 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Hausinger, R.P. New metal cofactors and recent metallocofactor insights. Curr. Opin. Struct. Biol. 2019, 59, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mendel, R.R.; Smith, A.G.; Marquet, A.; Warren, M.J. Metal and cofactor insertion. Nat. Prod. Rep. 2007, 24, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Ascone, I.; Fourme, R.; Hasnain, S.S. Introductory overview: X-ray absorption spectroscopy and structural genomics. J. Synchrotron Radiat. 2003, 10, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.D.; Penner-Hahn, J.E.; Stemmler, T.L. Structure and dynamics of metalloproteins in live cells. Methods Cell Biol. 2008, 90, 199–216. [Google Scholar] [PubMed]
- Malmstrom, B.G.; Neilands, J.B. Metalloproteins. Annu. Rev. Biochem 1964, 33, 331–354. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J. Metalloproteins. Nature 2009, 460, 813. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A. Metalloproteins: Simple structure, complex function. Nat. Chem. Biol. 2015, 11, 760. [Google Scholar] [CrossRef] [PubMed]
- Vallee, B.L.; Williams, R. Metalloenzymes: The entatic nature of their active sites. Proc. Natl. Acad. Sci. USA 1968, 59, 498. [Google Scholar] [CrossRef] [PubMed]
- Karlin, K.D. Metalloenzymes, structural motifs, and inorganic models. Science 1993, 261, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Tan, X. Metalloproteins and Metalloenzymes: Roles and Mechanisms of Metals in Functional Proteins, 1st ed.; World Scientific Publishing: Shanghai, China, 2020. [Google Scholar]
- Yu, F.; Cangelosi, V.M.; Zastrow, M.L.; Tegoni, M.; Plegaria, J.S.; Tebo, A.G.; Mocny, C.S.; Ruckthong, L.; Qayyum, H.; Pecoraro, V.L. Protein design: Toward functional metalloenzymes. Chem. Rev. 2014, 114, 3495–3578. [Google Scholar] [CrossRef] [PubMed]
- Yruela, I. Transition metals in plant photosynthesis. Metallomics 2013, 5, 1090–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, J.; Yachandra, V. Mn4ca cluster in photosynthesis: Where and how water is oxidized to dioxygen. Chem. Rev. 2014, 114, 4175–4205. [Google Scholar] [CrossRef] [PubMed]
- de Vries, S.; Dorner, K.; Strampraad, M.J.F.; Friedrich, T. Electron tunneling rates in respiratory complex i are tuned for efficient energy conversion. Angew. Chem. (International ed in English) 2015, 54, 2844–2848. [Google Scholar] [CrossRef] [PubMed]
- Al-Attar, S.; de Vries, S. Energy transduction by respiratory metallo-enzymes: From molecular mechanism to cell physiology. Coord. Chem. Rev. 2013, 257, 64–80. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Shoji, M.; Isobe, H.; Yamanaka, S.; Kawakami, T.; Yamada, S.; Katouda, M.; Nakajima, T. Theory of chemical bonds in metalloenzymes xxi. Possible mechanisms of water oxidation in oxygen evolving complex of photosystem ii. Mol. Phys. 2018, 116, 717–745. [Google Scholar] [CrossRef]
- Augustine, A.J.; Kjaergaard, C.; Qayyum, M.; Ziegler, L.; Kosman, D.J.; Hodgson, K.O.; Hedman, B.; Solomon, E.I. Systematic perturbation of the trinuclear copper cluster in the multicopper oxidases: The role of active site asymmetry in its reduction of O2 to H2O. J. Am. Chem. Soc. 2010, 132, 6057–6067. [Google Scholar] [CrossRef]
- Solomon, E.I.; Augustine, A.J.; Yoon, J. O2 reduction to H2O by the multicopper oxidases. Dalton Trans. 2008, 3921–3932. [Google Scholar] [CrossRef]
- Rees, D.C.; Akif Tezcan, F.; Haynes, C.A.; Walton, M.Y.; Andrade, S.; Einsle, O.; Howard, J.B. Structural basis of biological nitrogen fixation. Philos. Trans. A Math. Phys. Eng. Sci. 2005, 363, 971–984; discussion 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Valdez, C.E.; Smith, Q.A.; Nechay, M.R.; Alexandrova, A.N. Mysteries of metals in metalloenzymes. Acc. Chem. Res. 2014, 47, 3110–3117. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.W.; Osman, D.; Robinson, N.J. Metal preferences and metallation. J. Biol. Chem. 2014, 289, 28095–28103. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.J.; Qian, H.X.; Wang, W.J.; Liao, R.Z. Computational understanding of the selectivities in metalloenzymes. Front. Chem. 2018, 6, 638. [Google Scholar] [CrossRef] [PubMed]
- Christianson, D.W. Structural chemistry and biology of manganese metalloenzymes. Prog. Biophys. Mol. Biol. 1997, 67, 217–252. [Google Scholar] [CrossRef]
- Pain, D.; Dancis, A. Roles of Fe–S proteins: From cofactor synthesis to iron homeostasis to protein synthesis. Curr. Opin. Genet. Dev. 2016, 38, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Shimizu, S. Cobalt proteins. Eur. J. Biochem. 1999, 261, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rubino, J.T.; Franz, K.J. Coordination chemistry of copper proteins: How nature handles a toxic cargo for essential function. J. Inorg. Biochem. 2012, 107, 129–143. [Google Scholar] [CrossRef] [PubMed]
- McCall, K.A.; Huang, C.; Fierke, C.A. Function and mechanism of zinc metalloenzymes. J. Nutr. 2000, 130, 1437S–1446S. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Profiti, G.; Martelli, P.L.; Casadio, R. The human “magnesome”: Detecting magnesium binding sites on human proteins. BMC Bioinformatics 2012, 13 Suppl. 14, S10. [Google Scholar] [CrossRef]
- Mendel, R.R. The molybdenum cofactor. J. Biol. Chem. 2013, 288, 13165–13172. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Schulzke, C.; Kirk, M.L. Molybdenum and tungsten enzymes: Bioinorganic chemistry, 1st ed.; The Royal Society of Chemistry: Cambridge, UK, 2016; Vol. 6. [Google Scholar]
- Sundberg, R.J.; Martin, R.B. Interactions of histidine and other imidazole derivatives with transition metal ions in chemical and biological systems. Chem. Rev. 1974, 74, 471–517. [Google Scholar] [CrossRef]
- Giles, N.M.; Watts, A.B.; Giles, G.I.; Fry, F.H.; Littlechild, J.A.; Jacob, C. Metal and redox modulation of cysteine protein function. Chem. Biol. 2003, 10, 677–693. [Google Scholar] [CrossRef]
- Lamine, W.; Boughdiri, S.; Christ, L.; Morell, C.; Chermette, H. Coordination chemistry of Zn2+ with sal(ph)en ligands: Tetrahedral coordination or penta-coordination? A dft analysis. J. Comput. Chem. 2019, 40, 717–725. [Google Scholar]
- Lamine, W.; Boughdiri, S.; Jeanneau, E.; Sanglar, C.; Morell, C.; Christ, L.; Chermette, H. Unexpected structure of a helical N4-schiff-base Zn(ii) complex and its demetallation: Experimental and theoretical studies. Chemphyschem 2018, 19, 2938–2946. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.S.; Drennan, C.L. Metalloenzymes in natural product biosynthetic pathways. Nat. Prod. Rep. 2018, 35, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Geissler, P.L.; Dellago, C.; Chandler, D.; Hutter, J.; Parrinello, M. Autoionization in liquid water. Science 2001, 291, 2121. [Google Scholar] [CrossRef] [PubMed]
- Crichton, R.R. Chapter 12—zinc—lewis acid and gene regulator. In Biological Inorganic Chemistry (Second Edition); Crichton, R.R., Ed.; Elsevier: Oxford, UK, 2012; pp. 229–246. [Google Scholar]
- Lindskog, S. Structure and mechanism of carbonic anhydrase. Pharmacol. Ther. 1997, 74, 1–20. [Google Scholar] [CrossRef]
- Kannan, K.K.; Petef, M.; Fridborg, K.; Cid-Dresdner, H.; Lovgren, S. Structure and function of carbonic anhydrases. Imidazole binding to human carbonic anhydrase b and the mechanism of action of carbonic anhydrases. FEBS Lett. 1977, 73, 115–119. [Google Scholar] [PubMed]
- Wang, W.; Zhang, Y.; Wang, L.; Jing, Q.; Wang, X.; Xi, X.; Zhao, X.; Wang, H. Molecular structure of thermostable and zinc-ion-binding gamma-class carbonic anhydrases. BioMetals 2019, 32, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Scozzafava, A.; Casini, A. Carbonic anhydrase inhibitors. Med. Res. Rev. 2003, 23, 146–189. [Google Scholar] [CrossRef] [PubMed]
- Piazzetta, P.; Marino, T.; Russo, N. Theoretical investigation on the restoring step of the carbonic anhydrase catalytic cycle for natural and promiscuous substrates. Arch. Biochem. Biophys. 2015, 582, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Miscione, G.P.; Stenta, M.; Spinelli, D.; Anders, E.; Bottoni, A. New computational evidence for the catalytic mechanism of carbonic anhydrase. Theor. Chem. Acc. 2007, 118, 193–201. [Google Scholar] [CrossRef]
- Liang, J.Y.; Lipscomb, W.N. Theoretical study of carbonic anhydrase-catalyzed hydration of CO2: A brief review. Int. J. Quantum Chem 1989, 36, 299–312. [Google Scholar] [CrossRef]
- Liang, J.Y.; Lipscomb, W.N. Binding of substrate CO2 to the active site of human carbonic anhydrase ii: A molecular dynamics study. Proc. Natl. Acad. Sci. USA 1990, 87, 3675–3679. [Google Scholar] [CrossRef] [PubMed]
- Fisher, Z.; Hernandez Prada, J.A.; Tu, C.; Duda, D.; Yoshioka, C.; An, H.; Govindasamy, L.; Silverman, D.N.; McKenna, R. Structural and kinetic characterization of active-site histidine as a proton shuttle in catalysis by human carbonic anhydrase ii. Biochemistry 2005, 44, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Christianson, D.W.; Fierke, C.A. Carbonic anhydrase: Evolution of the zinc binding site by nature and by design. Acc. Chem. Res. 1996, 29, 331–339. [Google Scholar] [CrossRef]
- Silverman, D.N.; Lindskog, S. The catalytic mechanism of carbonic anhydrase: Implications of a rate-limiting protolysis of water. Acc. Chem. Res. 2002, 21, 30–36. [Google Scholar] [CrossRef]
- Liljas, A.; Kannan, K.K.; Bergsten, P.C.; Waara, I.; Fridborg, K.; Strandberg, B.; Carlbom, U.; Jarup, L.; Lovgren, S.; Petef, M. Crystal structure of human carbonic anhydrase c. Nat. New Biol. 1972, 235, 131–137. [Google Scholar] [CrossRef]
- Kannan, K.K.; Liljas, A.; Waara, I.; Bergsten, P.C.; Lovgren, S.; Strandberg, B.; Bengtsson, U.; Carlbom, U.; Fridborg, K.; Jarup, L.; et al. Crystal structure of human erythrocyte carbonic anhydrase c. Vi. The three-dimensional structure at high resolution in relation to other mammalian carbonic anhydrases. Cold Spring Harb. Symp. Quant. Biol. 1972, 36, 221–231. [Google Scholar] [CrossRef]
- Kimura, E. Model studies for molecular recognition of carbonic anhydrase and carboxypeptidase. Acc. Chem. Res. 2001, 34, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Lesburg, C.A.; Kiefer, L.L.; Fierke, C.A.; Christianson, D.W. Reversal of the hydrogen bond to zinc ligand histidine-119 dramatically diminishes catalysis and enhances metal equilibration kinetics in carbonic anhydrase ii. Biochemistry 1996, 35, 3439–3446. [Google Scholar] [CrossRef] [PubMed]
- Lesburg, C.A.; Huang, C.; Christianson, D.W.; Fierke, C.A. Histidine --> carboxamide ligand substitutions in the zinc binding site of carbonic anhydrase ii alter metal coordination geometry but retain catalytic activity. Biochemistry 1997, 36, 15780–15791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hubbard, C.D.; van Eldik, R. Carbonic anhydrase catalysis: A volume profile analysis. J. Phys. Chem. 1996, 100, 9161–9171. [Google Scholar] [CrossRef]
- Tu, C.; Tripp, B.C.; Ferry, J.G.; Silverman, D.N. Bicarbonate as a proton donor in catalysis by Zn(ii)- and Co(ii)-containing carbonic anhydrases. J. Am. Chem. Soc. 2001, 123, 5861–5866. [Google Scholar] [CrossRef]
- Thoms, S. Hydrogen bonds and the catalytic mechanism of human carbonic anhydrase ii. J. Theor. Biol. 2002, 215, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Schröder, D.; Schwarz, H.; Schenk, S.; Anders, E. A gas-phase reaction as a functional model for the activation of carbon dioxide by carbonic anhydrase. Angew. Chem. (International ed in English) 2003, 42, 5087–5090. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.Y.; Lipscomb, W.N. Hydration of carbon dioxide by carbonic anhydrase: Internal protein transfer of Zinc(2+)-bound bicarbonate. Biochemistry 2002, 26, 5293–5301. [Google Scholar] [CrossRef]
- Merz, K.M.; Hoffmann, R.; Dewar, M.J.S. The mode of action of carbonic anhydrase. J. Am. Chem. Soc. 1989, 111, 5636–5649. [Google Scholar] [CrossRef]
- Krauss, M.; Garmer, D.R. Active site ionicity and the mechanism of carbonic anhydrase. J. Am. Chem. Soc. 1991, 113, 6426–6435. [Google Scholar] [CrossRef]
- Zheng, Y.J.; Merz, K.M. Mechanism of the human carbonic anhydrase ii-catalyzed hydration of carbon dioxide. J. Am. Chem. Soc. 1992, 114, 10498–10507. [Google Scholar] [CrossRef]
- Sola, M.; Lledos, A.; Duran, M.; Bertran, J. Ab initio study of the hydration of carbon dioxide by carbonic anhydrase. A comparison between the lipscomb and lindskog mechanisms. J. Am. Chem. Soc. 1992, 114, 869–877. [Google Scholar] [CrossRef]
- Sakurai, M.; Furuki, T.; Inoue, Y. The pka of the zinc-bound water in carbonic anhydrase and its model compounds as studied by the am1 calculation coupled with a reaction field theory. J. Phys. Chem. 1995, 99, 17789–17794. [Google Scholar] [CrossRef]
- Merz, K.M.; Banci, L. Binding of bicarbonate to human carbonic anhydrase ii: A continuum of binding states. J. Am. Chem. Soc. 1997, 119, 863–871. [Google Scholar] [CrossRef]
- Lu, D.; Voth, G.A. Proton transfer in the enzyme carbonic anhydrase: Anab initiostudy. J. Am. Chem. Soc. 1998, 120, 4006–4014. [Google Scholar] [CrossRef]
- Toba, S.; Colombo, G.; Merz, K.M. Solvent dynamics and mechanism of proton transfer in human carbonic anhydrase ii. J. Am. Chem. Soc. 1999, 121, 2290–2302. [Google Scholar] [CrossRef]
- Denisov, V.P.; Jonsson, B.-H.; Halle, B. Dynamics of functional water in the active site of native carbonic anhydrase from17o magnetic relaxation dispersion. J. Am. Chem. Soc. 1999, 121, 2327–2328. [Google Scholar] [CrossRef]
- Mauksch, M.; Bräuer, M.; Weston, J.; Anders, E. New insights into the mechanistic details of the carbonic anhydrase cycle as derived from the model system [(NH3)3Zn(OH)]+/CO2: How does the H2O/HCO3− replacement step occur? ChemBioChem 2001, 2, 190–198. [Google Scholar] [CrossRef]
- Bräuer, M.; Pérez-Lustres, J.L.; Weston, J.; Anders, E. Quantitative reactivity model for the hydration of carbon dioxide by biomimetic zinc complexes. Inorg. Chem. 2002, 41, 1454–1463. [Google Scholar] [CrossRef]
- Smedarchina, Z.; Siebrand, W.; Fernández-Ramos, A.; Cui, Q. Kinetic isotope effects for concerted multiple proton transfer: A direct dynamics study of an active-site model of carbonic anhydrase ii. J. Am. Chem. Soc. 2003, 125, 243–251. [Google Scholar] [CrossRef]
- Garmer, D.R. Carbonic anhydrase reactivity, mutation, and inhibition probed with a model of ab initioquantum chemistry within a protein. J. Phys. Chem. B 1997, 101, 2945–2953. [Google Scholar] [CrossRef]
- Cui, Q.; Karplus, M. Is a “proton wire” concerted or stepwise? A model study of proton transfer in carbonic anhydrase. J. Phys. Chem. B 2003, 107, 1071–1078. [Google Scholar] [CrossRef]
- Muguruma, C. Ab initio Mo study on the catalytic mechanism in the active site of carbonic anhydrase. J. Mol. Struct. THEOCHEM 1999, 461-462, 439–452. [Google Scholar] [CrossRef]
- Bottoni, A.; Lanza, C.Z.; Miscione, G.P.; Spinelli, D. New model for a theoretical density functional theory investigation of the mechanism of the carbonic anhydrase: How does the internal bicarbonate rearrangement occur? J. Am. Chem. Soc. 2004, 126, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Jeske, L.; Placzek, S.; Schomburg, I.; Chang, A.; Schomburg, D. Brenda in 2019: A european elixir core data resource. Nucleic Acids Res. 2019, 47, D542–D549. [Google Scholar] [CrossRef] [PubMed]
- Amata, O.; Marino, T.; Russo, N.; Toscano, M. Catalytic activity of a zeta-class zinc and cadmium containing carbonic anhydrase. Compared work mechanisms. PCCP 2011, 13, 3468–3477. [Google Scholar] [CrossRef] [PubMed]
- Nechay, M.R.; Gallup, N.M.; Morgenstern, A.; Smith, Q.A.; Eberhart, M.E.; Alexandrova, A.N. Histone deacetylase 8: Characterization of physiological divalent metal catalysis. J. Phys. Chem. B 2016, 120, 5884–5895. [Google Scholar] [CrossRef]
- Banerjee, S.; Adhikari, N.; Amin, S.A.; Jha, T. Histone deacetylase 8 (HDAC8) and its inhibitors with selectivity to other isoforms: An overview. Eur. J. Med. Chem. 2019, 164, 214–240. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, X.X.; Wu, Y.D.; Wiest, O. Inhibition and mechanism of HDAC8 revisited. J. Am. Chem. Soc. 2014, 136, 11636–11643. [Google Scholar] [CrossRef]
- Gantt, S.M.; Decroos, C.; Lee, M.S.; Gullett, L.E.; Bowman, C.M.; Christianson, D.W.; Fierke, C.A. General base-general acid catalysis in human histone deacetylase 8. Biochemistry 2016, 55, 820–832. [Google Scholar] [CrossRef]
- Wu, R.; Wang, S.; Zhou, N.; Cao, Z.; Zhang, Y. A proton-shuttle reaction mechanism for histone deacetylase 8 and the catalytic role of metal ions. J. Am. Chem. Soc. 2010, 132, 9471–9479. [Google Scholar] [CrossRef] [PubMed]
- Gantt, S.L.; Gattis, S.G.; Fierke, C.A. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry 2006, 45, 6170–6178. [Google Scholar] [CrossRef] [PubMed]
- Laitaoja, M.; Valjakka, J.; Jänis, J. Zinc coordination spheres in protein structures. Inorg. Chem. 2013, 52, 10983–10991. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. New perspectives of zinc coordination environments in proteins. J. Inorg. Biochem. 2012, 111, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Maret, W.; Li, Y. Coordination dynamics of zinc in proteins. Chem. Rev. 2009, 109, 4682–4707. [Google Scholar] [CrossRef] [PubMed]
- Alberto, M.E.; Leopoldini, M.; Russo, N. Can human prolidase enzyme use different metals for full catalytic activity? Inorg. Chem. 2011, 50, 3394–3403. [Google Scholar] [CrossRef] [PubMed]
- Kitchener, R.L.; Grunden, A.M. Prolidase function in proline metabolism and its medical and biotechnological applications. J. Appl. Microbiol. 2012, 113, 233–247. [Google Scholar] [CrossRef]
- Vanhoof, G.; Goossens, F.; De Meester, I.; Hendriks, D.; Scharpe, S. Proline motifs in peptides and their biological processing. FASEB J. 1995, 9, 736–744. [Google Scholar] [CrossRef]
- Fujii, M.; Nagaoka, Y.; Imamura, S.; Shimizu, T. Purification and characterization of a prolidase from aureobacterium esteraromaticum. Biosci. Biotechnol. Biochem. 1996, 60, 1118–1122. [Google Scholar]
- Wilk, P.; Uehlein, M.; Kalms, J.; Dobbek, H.; Mueller, U.; Weiss, M.S. Substrate specificity and reaction mechanism of human prolidase. FEBS J. 2017, 284, 2870–2885. [Google Scholar] [CrossRef] [Green Version]
- Wilk, P.; Uehlein, M.; Piwowarczyk, R.; Dobbek, H.; Mueller, U.; Weiss, M.S. Structural basis for prolidase deficiency disease mechanisms. FEBS J. 2018, 285, 3422–3441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metz, S.; Thiel, W. A combined qm/mm study on the reductive half-reaction of xanthine oxidase: Substrate orientation and mechanism. J. Am. Chem. Soc. 2009, 131, 14885–14902. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.M.; Lancaster, G.L.; Choy, F.Y.; Hechtman, P. Purification and characterization of activated human erythrocyte prolidase. Biochem. Cell Biol. 1989, 67, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.P.; Miller, B.R.; Roitberg, A.E.; Merz, K.M. Wide-open flaps are key to urease activity. J. Am. Chem. Soc. 2012, 134, 9934–9937. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, B.; van Eldik, R.; Brindell, M. Temperature- and pressure-dependent stopped-flow kinetic studies of jack bean urease. Implications for the catalytic mechanism. J. Biol. Inorg. Chem. 2012, 17, 1123–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hameed, A.; Al-Rashida, M.; Uroos, M.; Qazi, S.U.; Naz, S.; Ishtiaq, M.; Khan, K.M. A patent update on therapeutic applications of urease inhibitors (2012–2018). Expert Opin. Ther. Pat. 2019, 29, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, H.; Nordlander, E. Computational modeling of the mechanism of urease. Bioinorg. Chem. Appl. 2010. [Google Scholar] [CrossRef]
- Mazzei, L.; Cianci, M.; Benini, S.; Ciurli, S. The structure of the elusive urease-urea complex unveils the mechanism of a paradigmatic nickel-dependent enzyme. Angew. Chem. (International ed. in English) 2019. [Google Scholar]
- Pearson, M.A.; Park, I.S.; Schaller, R.A.; Michel, L.O.; Karplus, P.A.; Hausinger, R.P. Kinetic and structural characterization of urease active site variants. Biochemistry 2000, 39, 8575–8584. [Google Scholar] [CrossRef]
- Suarez, D.; Diaz, N.; Merz, K.M., Jr. Ureases: Quantum chemical calculations on cluster models. J. Am. Chem. Soc. 2003, 125, 15324–15337. [Google Scholar] [CrossRef]
- Estiu, G.; Merz, K.M., Jr. The hydrolysis of urea and the proficiency of urease. J. Am. Chem. Soc. 2004, 126, 6932–6944. [Google Scholar] [CrossRef]
- Sanderink, G.J.; Artur, Y.; Siest, G. Human aminopeptidases: A review of the literature. J. Clin. Chem. Clin. Biochem. 1988, 26, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Fowler, J.H.; Walling, L.L. Leucine aminopeptidases: Diversity in structure and function. Biol. Chem. 2006, 387, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A. Aminopeptidases: Structure and function. FASEB J. 1993, 7, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Lowther, W.T.; Matthews, B.W. Metalloaminopeptidases: Common functional themes in disparate structural surroundings. Chem. Rev. 2002, 102, 4581–4608. [Google Scholar] [CrossRef] [PubMed]
- Liew, S.M.; Tay, S.T.; Puthucheary, S.D. Enzymatic and molecular characterisation of leucine aminopeptidase of burkholderia pseudomallei. BMC Microbiol. 2013, 13, 110. [Google Scholar]
- Sträter, N.; Sun, L.; Kantrowitz, E.R.; Lipscomb, W.N. A bicarbonate ion as a general base in the mechanism of peptide hydrolysis by dizinc leucine aminopeptidase. Proc. Natl. Acad. Sci. USA 1999, 96, 11151–11155. [Google Scholar] [CrossRef] [Green Version]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef]
- Sousa, S.F.; Lopes, A.B.; Fernandes, P.A.; Ramos, M.J. The zinc proteome: A tale of stability and functionality. Dalton Trans. 2009, 7946–7956. [Google Scholar] [CrossRef]
- Zastrow, M.L.; Pecoraro, V.L. Designing hydrolytic zinc metalloenzymes. Biochemistry 2014, 53, 957–978. [Google Scholar] [CrossRef]
- Zhu, X.; Barman, A.; Ozbil, M.; Zhang, T.; Li, S.; Prabhakar, R. Mechanism of peptide hydrolysis by co-catalytic metal centers containing leucine aminopeptidase enzyme: A DFT approach. JBIC J. Biol. Inorg. Chem. 2012, 17, 209–222. [Google Scholar] [CrossRef]
- Zhang, T.; Ozbil, M.; Barman, A.; Paul, T.J.; Bora, R.P.; Prabhakar, R. Theoretical insights into the functioning of metallopeptidases and their synthetic analogues. Acc. Chem. Res. 2015, 48, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.P.; Yamada, A.H.; Carpenter, F.H. Kinetic parameters of metal-substituted leucine aminopeptidase from bovine lens. Biochemistry 1983, 22, 3778–3783. [Google Scholar] [CrossRef] [PubMed]
- Cappiello, M.; Alterio, V.; Amodeo, P.; Del Corso, A.; Scaloni, A.; Pedone, C.; Moschini, R.; De Donatis, G.M.; De Simone, G.; Mura, U. Metal ion substitution in the catalytic site greatly affects the binding of sulfhydryl-containing compounds to leucyl aminopeptidase. Biochemistry 2006, 45, 3226–3234. [Google Scholar] [CrossRef] [PubMed]
- Kostić, D.A.; Dimitrijević, D.S.; Stojanović, G.S.; Palić, I.R.; Đorđević, A.S.; Ickovski, J.D. Xanthine oxidase: Isolation, assays of activity, and inhibition. J. Chem. 2015, 2015, 1–8. [Google Scholar] [CrossRef]
- Cherak, S.J.; Turner, R.J. Assembly pathway of a bacterial complex iron sulfur molybdoenzyme. Biomol. Concepts 2017, 8, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, M.J.; Johnson, J.L.; Rajagopalan, K.V.; Kisker, C. The 1.2 Å structure of the human sulfite oxidase cytochrome b5 domain. Acta crystallographica. Section D 2003, 59, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Hall, J.; Basu, P. The mononuclear molybdenum enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef]
- Gonzalez, P.J.; Rivas, M.G.; Mota, C.S.; Brondino, C.D.; Moura, I.; Moura, J.J.G. Periplasmic nitrate reductases and formate dehydrogenases: Biological control of the chemical properties of mo and w for fine tuning of reactivity, substrate specificity and metabolic role. Coord. Chem. Rev. 2013, 257, 315–331. [Google Scholar] [CrossRef]
- Maia, L.B.; Moura, J.J.; Moura, I. Molybdenum and tungsten-dependent formate dehydrogenases. J. Biol. Inorg. Chem. 2015, 20, 287–309. [Google Scholar] [CrossRef]
- Metz, S.; Thiel, W. Qm/mm studies of xanthine oxidase: Variations of cofactor, substrate, and active-site glu802. J. Phys. Chem. B 2010, 114, 1506–1517. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Matsumura, T.; Ichida, K.; Okamoto, K.; Nishino, T. Human xanthine oxidase changes its substrate specificity to aldehyde oxidase type upon mutation of amino acid residues in the active site: Roles of active site residues in binding and activation of purine substrate. J. Biochem. 2007, 141, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Winters, K.; Tollin, G.; Enemark, J.H. Elucidating the catalytic mechanism of sulfite oxidizing enzymes using structural, spectroscopic, and kinetic analyses. Biochemistry 2010, 49, 7242–7254. [Google Scholar] [CrossRef]
- Cerqueira, N.M.F.S.A.; Pakhira, B.; Sarkar, S. Theoretical studies on mechanisms of some mo enzymes. J. Biol. Inorg. Chem. 2015, 20, 323–335. [Google Scholar] [CrossRef]
- Cerqueira, N.M.F.S.A.; Gonzalez, P.J.; Fernandes, P.A.; Moura, J.J.G.; Ramos, M.J. Periplasmic nitrate reductase and formate dehydrogenase: Similar molecular architectures with very different enzymatic activities. Acc. Chem. Res. 2015, 48, 2875–2884. [Google Scholar] [CrossRef]
- Hartmann, T.; Schrapers, P.; Utesch, T.; Nimtz, M.; Rippers, Y.; Dau, H.; Mroginski, M.A.; Haumann, M.; Leimkuhler, S. The molybdenum active site of formate dehydrogenase is capable of catalyzing c-h bond cleavage and oxygen atom transfer reactions. Biochemistry 2016, 55, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Niks, D.; Hille, R. Molybdenum- and tungsten-containing formate dehydrogenases and formylmethanofuran dehydrogenases: Structure, mechanism, and cofactor insertion. Protein Sci. 2019, 28, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Mota, C.S.; Rivas, M.G.; Brondino, C.D.; Moura, I.; Moura, J.J.G.; Gonzalez, P.J.; Cerqueira, N.M.F.S.A. The mechanism of formate oxidation by metal-dependent formate dehydrogenases. J. Biol. Inorg. Chem. 2011, 16, 1255–1268. [Google Scholar] [CrossRef] [Green Version]
- Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Gonzalez, P.J.; Moura, J.J.G.; Ramos, M.J. The sulfur shift: An activation mechanism for periplasmic nitrate reductase and formate dehydrogenase. Inorg. Chem. 2013, 52, 10766–10772. [Google Scholar] [CrossRef]
- Coelho, C.; Gonzalez, P.J.; Moura, J.J.G.; Moura, I.; Trincao, J.; Romao, M.J. The crystal structure of cupriavidus necator nitrate reductase in oxidized and partially reduced states. J. Mol. Biol. 2011, 408, 932–948. [Google Scholar] [CrossRef]
- Schrapers, P.; Hartmann, T.; Kositzki, R.; Dau, H.; Reschke, S.; Schulzke, C.; Leimkuhler, S.; Haumann, M. ‘Sulfido and cysteine ligation changes at the molybdenum cofactor during substrate conversion by formate dehydrogenase (FDH) from rhodobacter capsulatus. Inorg. Chem. 2015, 54, 3260–3271. [Google Scholar] [CrossRef]
- Robinson, W.E.; Bassegoda, A.; Reisner, E.; Hirst, J. Oxidation-state-dependent binding properties of the active site in a mo-containing formate dehydrogenase. J. Am. Chem. Soc. 2017, 139, 9927–9936. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, H.C.A.; Romao, M.J. Formate-reduced e-coli formate dehydrogenase h: The reinterpretation of the crystal structure suggests a new reaction mechanism. J. Biol. Inorg. Chem. 2006, 11, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Ryde, U. Reaction mechanism of formate dehydrogenase studied by computational methods. J. Biol. Inorg. Chem. 2018, 23, 1243–1254. [Google Scholar] [CrossRef]
- Axley, M.J.; Grahame, D.A. Kinetics for formate dehydrogenase of escherichia coli formate-hydrogenlyase. J. Biol. Chem. 1991, 266, 13731–13736. [Google Scholar] [PubMed]
- Bergquist, C.; Storrie, H.; Koutcher, L.; Bridgewater, B.M.; Friesner, R.A.; Parkin, G. Factors influencing the thermodynamics of zinc alkoxide formation by alcoholysis of the terminal hydroxide complex, [tp(but,me)]znoh: An experimental and theoretical study relevant to the mechanism of action of liver alcohol dehydrogenase. J. Am. Chem. Soc. 2000, 122, 12651–12658. [Google Scholar] [CrossRef]
- Kvassman, J.; Pettersson, G. Unified mechanism for proton-transfer reactions affecting the catalytic activity of liver alcohol-dehydrogenase. Eur. J. Biochem. 1980, 103, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.H.; Liu, C.Y.; Cai, S.; Guo, L.B.; Kim, I.W.; Kalia, V.C.; Lee, J.K.; Zhang, Y.W. Cloning, expression and characterization of a highly active alcohol dehydrogenase for production of ethyl (S)-4-chloro-3-hydroxybutyrate. Indian J. Med. Microbiol. 2019, 59, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Plapp, B.V. Substitution of cysteine-153 ligated to the catalytic zinc in yeast alcohol dehydrogenase with aspartic acid and analysis of mechanisms of related medium chain dehydrogenases. Chem. Biol. Interact. 2019, 302, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Moa, S.; Himo, F. Quantum chemical study of mechanism and stereoselectivity of secondary alcohol dehydrogenase. J. Inorg. Biochem. 2017, 175, 259–266. [Google Scholar] [CrossRef]
- Agarwal, P.K.; Webb, S.P.; Hammes-Schiffer, S. Computational studies of the mechanism for proton and hydride transfer in liver alcohol dehydrogenase. J. Am. Chem. Soc. 2000, 122, 4803–4812. [Google Scholar] [CrossRef]
- Hammes-Schiffer, S.; Benkovic, S.J. Relating protein motion to catalysis. Annu. Rev. Biochem 2006, 75, 519–541. [Google Scholar] [CrossRef] [PubMed]
- Bogin, O.; Peretz, M.; Burstein, Y. Thermoanaerobacter brockii alcohol dehydrogenase: Characterization of the active site metal and its ligand amino acids. Protein Sci. 1997, 6, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Sjoberg, B.M. Biochemistry. A never-ending story. Science 2010, 329, 1475–1476. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.M.; Fernandes, P.A.; Ramos, M.J. Ribonucleotide reductase: A critical enzyme for cancer chemotherapy and antiviral agents. Recent Pat. Anticancer. Drug Discov. 2007, 2, 11–29. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.M.F.S.A.; Pereira, S.; Fernandes, P.A.; Ramos, M.J. Overview of ribonucleotide reductase inhibitors: An appealing target in anti-tumour therapy. Curr. Med. Chem. 2005, 12, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.M.F.S.A.; Femandes, P.A.; Eriksson, L.A.; Ramos, M.J. New insights into a critical biological control step of the mechanism of ribonucleotide reductase. J. Mol. Struct. 2004, 709, 53–65. [Google Scholar] [CrossRef]
- Perez, M.A.S.; Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Ramos, M.J. Ribonucleotide reductase: A mechanistic portrait of substrate analogues inhibitors. Curr. Med. Chem. 2010, 17, 2854–2872. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, G.; Huang, M. Ribonucleotide reductase metallocofactor: Assembly, maintenance and inhibition. Front. Biol. (Beijing) 2014, 9, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Bollinger, J.M., Jr.; Jiang, W.; Green, M.T.; Krebs, C. The manganese(iv)/iron(iii) cofactor of chlamydia trachomatis ribonucleotide reductase: Structure, assembly, radical initiation, and evolution. Curr. Opin. Struct. Biol. 2008, 18, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Cotruvo, J.A.; Stubbe, J. Escherichia coli class ib ribonucleotide reductase contains a dimanganese(iii)-tyrosyl radical cofactor in vivo. Biochemistry 2011, 50, 1672–1681. [Google Scholar] [CrossRef] [PubMed]
- Rose, H.R.; Maggiolo, A.O.; McBride, M.J.; Palowitch, G.M.; Pandelia, M.E.; Davis, K.M.; Yennawar, N.H.; Boal, A.K. Structures of class id ribonucleotide reductase catalytic subunits reveal a minimal architecture for deoxynucleotide biosynthesis. Biochemistry 2019, 58, 1845–1860. [Google Scholar] [CrossRef] [PubMed]
- Kolberg, M.; Strand, K.R.; Graff, P.; Andersson, K.K. Structure, function, and mechanism of ribonucleotide reductases. Biochim. Biophys. Acta 2004, 1699, 1–34. [Google Scholar] [CrossRef]
- Eklund, H.; Uhlin, U.; Farnegardh, M.; Logan, D.T.; Nordlund, P. Structure and function of the radical enzyme ribonucleotide reductase. Prog. Biophys. Mol. Biol. 2001, 77, 177–268. [Google Scholar] [CrossRef]
- Minnihan, E.C.; Nocera, D.G.; Stubbe, J. Reversible, long-range radical transfer in E. Coli class ia ribonucleotide reductase. Acc. Chem. Res. 2013, 46, 2524–2535. [Google Scholar] [CrossRef] [PubMed]
- Hogbom, M.; Huque, Y.; Sjoberg, B.M.; Nordlund, P. Crystal structure of the di-iron/radical protein of ribonucleotide reductase from corynebacterium ammoniagenes. Biochemistry 2002, 41, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Merkx, M.; Kopp, D.A.; Sazinsky, M.H.; Blazyk, J.L.; Muller, J.; Lippard, S.J. Dioxygen activation and methane hydroxylation by soluble methane monooxygenase: A tale of two irons and three proteins a list of abbreviations can be found in section 7. Angew. Chem. (International ed. in English) 2001, 40, 2782–2807. [Google Scholar] [CrossRef]
- Buckel, W. Enzymatic reactions involving ketyls: From a chemical curiosity to a general biochemical mechanism. Biochemistry 2019. [Google Scholar] [CrossRef] [PubMed]
- Kashlan, O.B.; Scott, C.P.; Lear, J.D.; Cooperman, B.S. A comprehensive model for the allosteric regulation of mammalian ribonucleotide reductase. Functional consequences of ATP- and dATP-induced oligomerization of the large subunit. Biochemistry 2002, 41, 462–474. [Google Scholar] [CrossRef]
- Ge, J.; Yu, G.; Ator, M.A.; Stubbe, J. Pre-steady-state and steady-state kinetic analysis of E. Coliclass i ribonucleotide reductase. Biochemistry 2003, 42, 10071–10083. [Google Scholar] [CrossRef]
- Cotruvo, J.A.; Stich, T.A.; Britt, R.D.; Stubbe, J. Mechanism of assembly of the dimanganese-tyrosyl radical cofactor of class ib ribonucleotide reductase: Enzymatic generation of superoxide is required for tyrosine oxidation via a Mn(iii)Mn(iv) intermediate. J. Am. Chem. Soc. 2013, 135, 4027–4039. [Google Scholar] [CrossRef]
- Boal, A.K.; Cotruvo, J.A., Jr.; Stubbe, J.; Rosenzweig, A.C. The dimanganese(ii) site of bacillus subtilis class ib ribonucleotide reductase. Biochemistry 2012, 51, 3861–3871. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Ragsdale, S.W. The many faces of vitamin b12: Catalysis by cobalamin-dependent enzymes. Annu. Rev. Biochem. 2003, 72, 209–247. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.S.; Ramos, M.J.; Cerqueira, N.M.F.S.A. Catalytic mechanism of the serine hydroxymethyltransferase: A computational oniom qm/mm study. ACS Catal. 2018, 8, 10096–10110. [Google Scholar] [CrossRef]
- Bandarian, V.; Pattridge, K.A.; Lennon, B.W.; Huddler, D.P.; Matthews, R.G.; Ludwig, M.L. Domain alternation switches b(12)-dependent methionine synthase to the activation conformation. Nat. Struct. Mol. Biol. 2002, 9, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Drennan, C.L.; Huang, S.; Drummond, J.T.; Matthews, R.G.; Ludwig, M.L. How a protein binds b 12: A 3.0 å x-ray structure of b 12 -binding domains of methionine synthase. Science 1994, 266, 1669–1674. [Google Scholar] [CrossRef]
- Drennan, C.L.; Matthews, R.G.; Ludwig, M.L. Cobalamin-dependent methionine synthase: The structure of a methylcobalamin-binding fragment and implications for other b12-dependent enzymes. Curr. Opin. Struct. Biol. 1994, 4, 919–929. [Google Scholar] [CrossRef]
- Kumar, N.; Kozlowski, P.M. Mechanistic insights for formation of an organometallic Co-C bond in the methyl transfer reaction catalyzed by methionine synthase. J. Phys. Chem. B 2013, 117, 16044–16057. [Google Scholar] [CrossRef]
- Smith, A.E.; Matthews, R.G. Protonation state of methyltetrahydrofolate in a binary complex with cobalamin-dependent methionine synthase. Biochemistry 2000, 39, 13880–13890. [Google Scholar] [CrossRef]
- Jensen, K.P.; Ryde, U. Conversion of homocysteine to methionine by methionine synthase: A density functional study. J. Am. Chem. Soc. 2003, 125, 13970–13971. [Google Scholar] [CrossRef]
- Kozlowski, P.M.; Kuta, J.; Galezowski, W. Reductive cleavage mechanism of methylcobalamin: Elementary steps of Co-C bond breaking. J. Phys. Chem. B 2007, 111, 7638–7645. [Google Scholar] [CrossRef]
- Alfonso-Prieto, M.; Biarnes, X.; Kumar, M.; Rovira, C.; Kozlowski, P.M. Reductive cleavage mechanism of Co-C bond in cobalamin-dependent methionine synthase. J. Phys. Chem. B 2010, 114, 12965–12971. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.P.; Mamun, A.A.; Lodowski, P.; Jaworska, M.; Kozlowski, P.M. Mechanism of the photo-induced activation of CoC bond in methylcobalamin-dependent methionine synthase. J. Photochem. Photobiol. B Biol. 2018, 189, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Amaratunga, M.; Drennan, C.L.; Scholten, J.D.; Sands, R.H.; Ludwig, M.L.; Matthews, R.G. Mutations in the b12-binding region of methionine synthase: How the protein controls methylcobalamin reactivity. Biochemistry 1996, 35, 2464–2475. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Shandilya, M.; Kundu, S. Structural insight of dopamine beta-hydroxylase, a drug target for complex traits, and functional significance of exonic single nucleotide polymorphisms. PLoS ONE 2011, 6, e26509. [Google Scholar] [CrossRef] [PubMed]
- Vendelboe, T.V.; Harris, P.; Zhao, Y.; Walter, T.S.; Harlos, K.; El Omari, K.; Christensen, H.E. The crystal structure of human dopamine beta-hydroxylase at 2.9 a resolution. Sci. Adv. 2016, 2, e1500980. [Google Scholar] [CrossRef] [PubMed]
- Cowley, R.E.; Tian, L.; Solomon, E.I. Mechanism of O2 activation and substrate hydroxylation in noncoupled binuclear copper monooxygenases. Proc. Natl. Acad. Sci. USA 2016, 113, 12035–12040. [Google Scholar] [CrossRef]
- Wimalasena, K.; Dharmasena, S.; Wimalasena, D.S.; Hughbanks-Wheaton, D.K. Reduction of dopamine β-monooxygenase: A unified model for apparent negative cooperativity and fumarate activation. J. Biol. Chem. 1996, 271, 26032–26043. [Google Scholar] [CrossRef] [PubMed]
- Klinman, J.P.; Krueger, M. Dopamine beta-hydroxylase: Activity and inhibition in the presence of beta-substituted phenethylamines. Biochemistry 1982, 21, 67–75. [Google Scholar] [CrossRef]
- Sirimanne, S.R.; Herman, H.H.; May, S.W. Interaction of dopamine β-mono-oxygenase with substituted imidazoles and pyrazoles. Catalysis and inhibition. Biochem. J. 1987, 242, 227–233. [Google Scholar] [CrossRef]
- Bollinger, J.M., Jr.; Diao, Y.; Matthews, M.L.; Xing, G.; Krebs, C. Myo-inositol oxygenase: A radical new pathway for O2 and C-H activation at a nonheme diiron cluster. Dalton Trans. 2009, 905–914. [Google Scholar] [CrossRef]
- Hirao, H.; Morokuma, K. Insights into the (superoxo)Fe(iii)Fe(iii) intermediate and reaction mechanism of myo-inositol oxygenase: Dft and ONIOM(DFT:MM) study. J. Am. Chem. Soc. 2009, 131, 17206–17214. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.M.; Caradoc-Davies, T.T.; Dickson, J.M.J.; Cooper, G.J.S.; Loomes, K.M.; Baker, E.N. Crystal structure of a substrate complex of myo-inositol oxygenase, a di-iron oxygenase with a key role in inositol metabolism. Proc. Natl. Acad. Sci. USA 2006, 103, 15032–15037. [Google Scholar] [CrossRef] [PubMed]
- Arner, R.J.; Prabhu, K.S.T.; Thompson, J.R.; Hildenbrandt, G.D.; Liken, A.; Reddy, C.C. Myo-inositol oxygenase: Molecular cloning and expression of a unique enzyme that oxidizes myo-inositol and D-chiro-inositol. Biochem. J. 2001, 360. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysed Reaction | ||||
|---|---|---|---|---|
| Metal | Hydroxide | Hydride | Radical | Superoxide |
 | Prolidase | RNR-Ib | ||
| (Asp)Mn-(Asp)(Glu)(OH2)-Mn(Glu)(His) | (His)(Asp)Mn-(Glu)2-Mn(His)(Glu) | |||
 | RNR-Ia | MIOX | ||
| (His)(Glu)Fe-(Glu)2-Fe(His)(Glu) | (His)2(OH2)(Asp)Fe-(OH)(Asp)-Fe(His)2 | |||
 | MetH | |||
| Co(Cobalamin)(His) | ||||
 | Urease | |||
| (His)2(OH2)Ni-(Lys*)(OH2)-Ni(His)2(OH2)(Asp) *carbamylated | ||||
 | DBH | |||
| (His)2(Met)Cu--Cu(His)3 | ||||
 | CA | |||
| Zn(His)3(OH2) | ||||
| HDAC8 | ||||
| Zn(Asp)2(His)(OH2) | ||||
| LeuAP | ADH | |||
| (Asp)Zn-(Asp)(Glu)(OH2)-Zn(Lys)(Asp) | (Cys)4Zn✸--Zn(His)(Cys)2(OH2) | |||
 | XO | |||
| MoOS(OH)(Pterin) | ||||
| FdH | ||||
| MoS(Pterin)2(Cys) | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, H.S.; Teixeira, C.S.S.; Sousa, S.F.; Cerqueira, N.M.F.S.A. Formation of Unstable and very Reactive Chemical Species Catalyzed by Metalloenzymes: A Mechanistic Overview. Molecules 2019, 24, 2462. https://doi.org/10.3390/molecules24132462
Fernandes HS, Teixeira CSS, Sousa SF, Cerqueira NMFSA. Formation of Unstable and very Reactive Chemical Species Catalyzed by Metalloenzymes: A Mechanistic Overview. Molecules. 2019; 24(13):2462. https://doi.org/10.3390/molecules24132462
Chicago/Turabian StyleFernandes, Henrique S., Carla S. Silva Teixeira, Sérgio F. Sousa, and Nuno M. F. S. A. Cerqueira. 2019. "Formation of Unstable and very Reactive Chemical Species Catalyzed by Metalloenzymes: A Mechanistic Overview" Molecules 24, no. 13: 2462. https://doi.org/10.3390/molecules24132462