Enantioselective Benzylation and Allylation of α-Trifluoromethoxy Indanones under Phase-Transfer Catalysis



Abstract
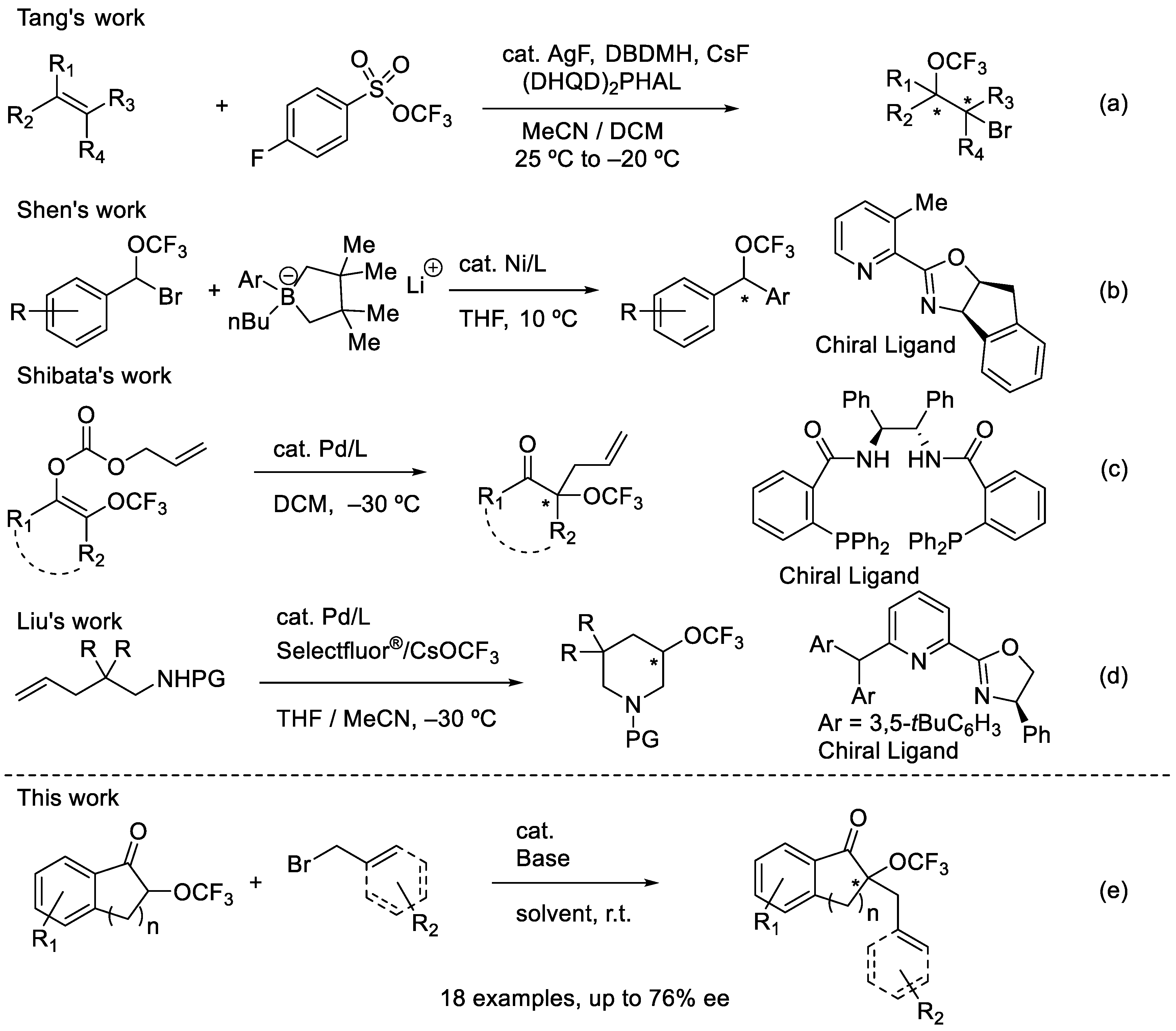
1. Introduction
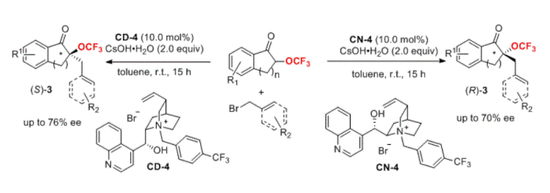
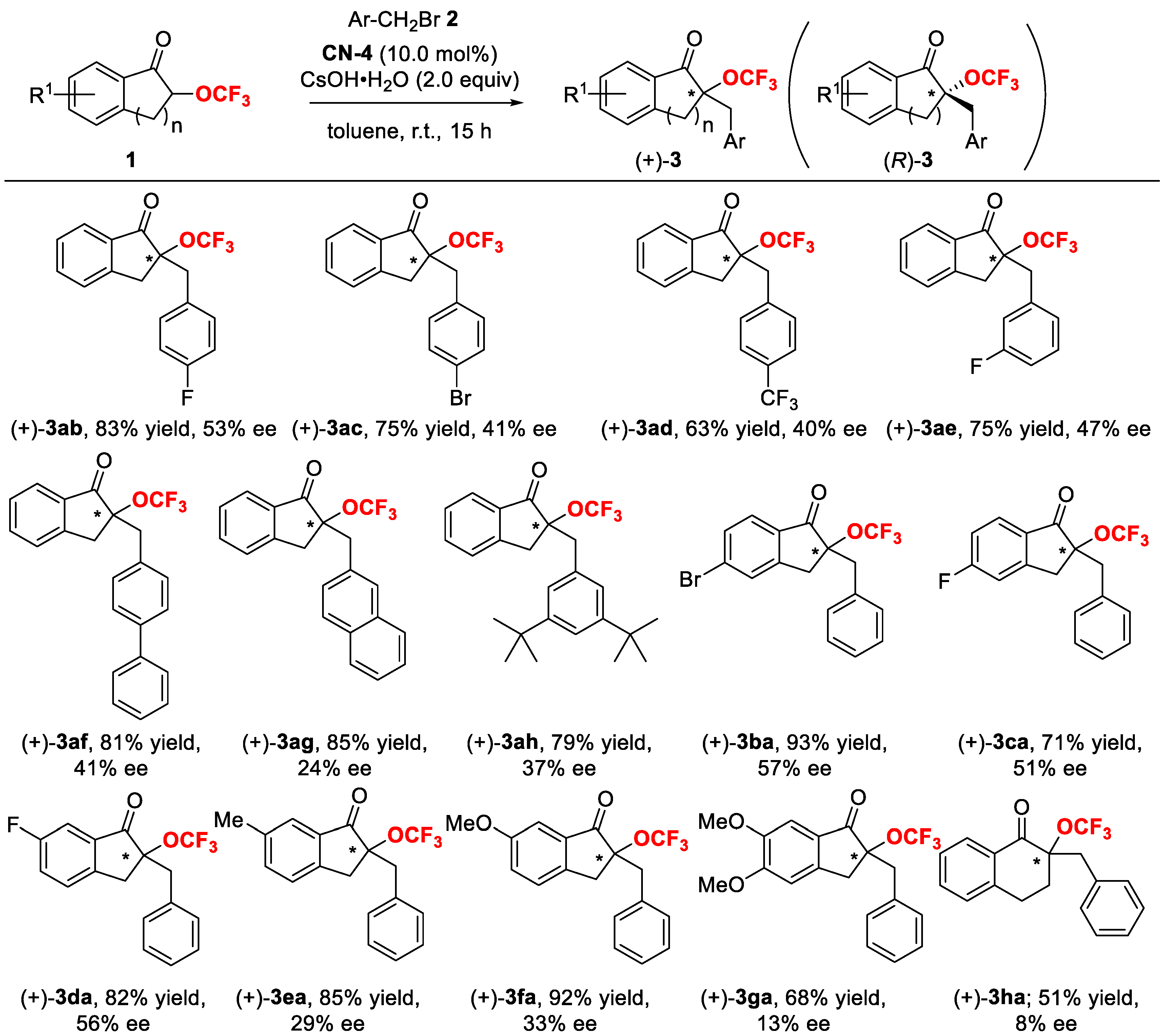
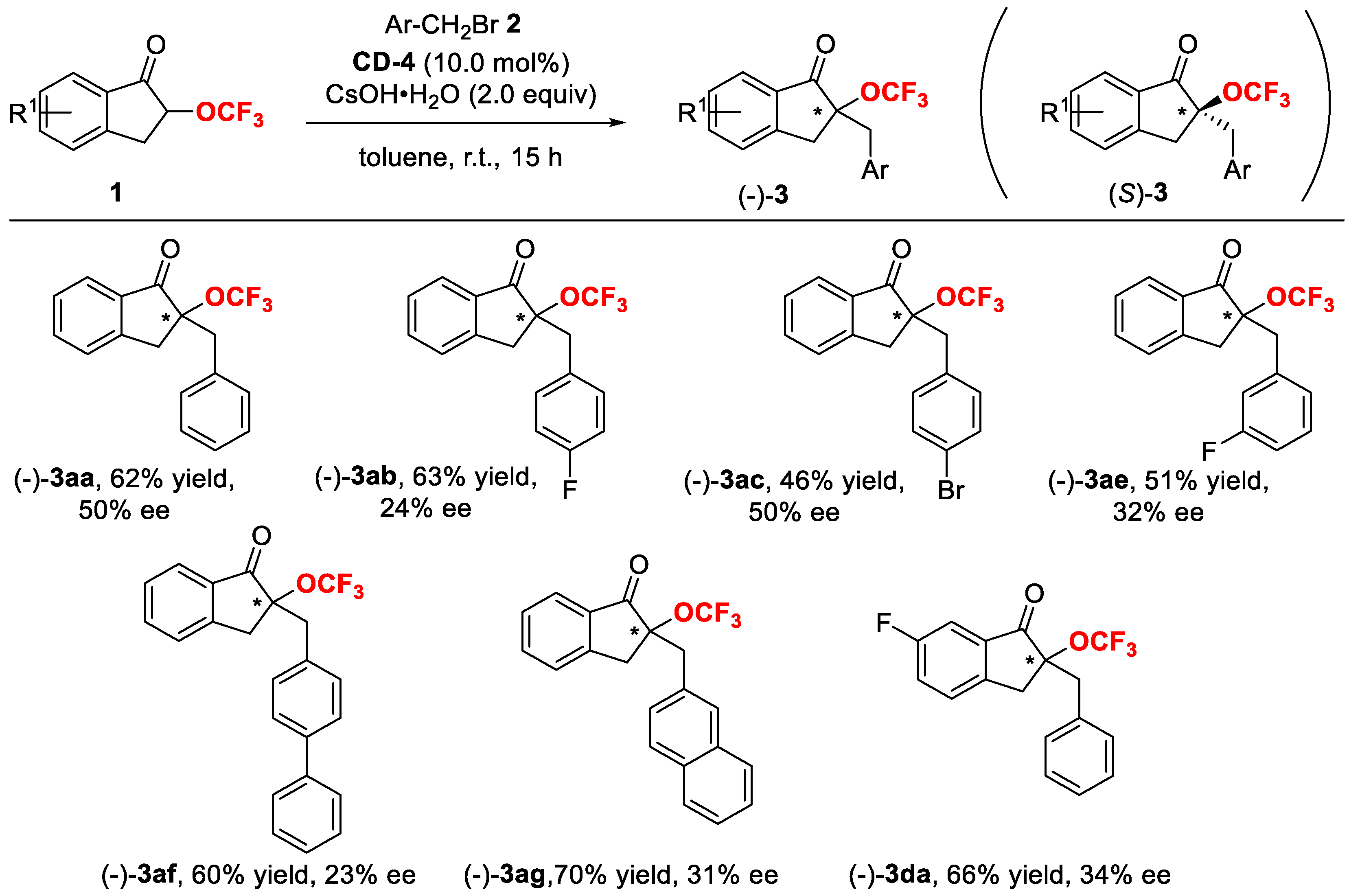
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Preparation of α-OCF3-Substituted Indanones (General Procedure)
3.3. Representative Procedure for the Enantioselective Catalytic Phase Transfer Benzylation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Note
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Organic fluorine compounds: A great opportunity for enhanced materials properties. Chem. Soc. Rev. 2011, 40, 3496–3508. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Isanbor, C.; O’Hagan, D. Fluorine in medicinal chemistry: A review of anti-cancer agents. J. Fluor. Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Kawai, H.; Shibata, N. Asymmetric Synthesis of Agrochemically Attractive Trifluoromethylated Dihydroazoles and Related Compounds under Organocatalysis. Chem. Rec. 2014, 14, 1024–1040. [Google Scholar] [CrossRef]
- Shibata, N.; Mizuta, S.; Kawai, H. Recent advances in enantioselective trifluoromethylation reactions. Tetrahedron Asymmetry 2008, 19, 2633–2644. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V.A.; Coelho, J.A.; Toste, F.D. Modern approaches for asymmetric construction of carbon–fluorine quaternary stereogenic centers: Synthetic challenges and pharmaceutical needs. Chem. Rev. 2018, 118, 3887–3964. [Google Scholar] [CrossRef]
- Mori, S.; Shibata, N. Synthesis and application of trifluoroethoxy-substituted phthalocyanines and subphthalocyanines. Beilstein J. Org. Chem. 2017, 13, 2273–2296. [Google Scholar] [CrossRef]
- Smart, B.E. Fluorine substituent effects (on bioactivity). J. Fluor. Chem. 2001, 109, 3–11. [Google Scholar] [CrossRef]
- Xu, X.H.; Matsuzaki, K.; Shibata, N. Synthetic methods for compounds having CF3–S units on carbon by trifluoromethylation, trifluoromethylthiolation, triflylation, and related reactions. Chem. Rev. 2014, 115, 731–764. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Acenña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next generation of fluorine-containing pharmaceuticals, compounds currently in phase II–III clinical trials of major pharmaceutical companies: New structural trends and therapeutic areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Ishimaru, T.; Nakamura, S.; Toru, T. New approaches to enantioselective fluorination: Cinchona alkaloids combinations and chiral ligands/metal complexes. J. Fluor. Chem. 2007, 128, 469–483. [Google Scholar] [CrossRef]
- Shimizu, M.; Hiyama, T. Modern synthetic methods for fluorine-substituted target molecules. Angew. Chem. Int. Ed. 2005, 44, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Leroux, F.; Jeschke, P.; Schlosser, M. α-Fluorinated ethers, thioethers, and amines: Anomerically biased species. Chem. Rev. 2005, 105, 827–856. [Google Scholar] [CrossRef] [PubMed]
- Landelle, G.; Panossian, A.; R Leroux, F. Trifluoromethyl ethers and–thioethers as tools for Medicinal chemistry and drug discovery. Curr. Top. Med. Chem. 2014, 14, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, P.; Baston, E.; Leroux, F.R. Alpha-fluorinated ethers as “exotic” entity in medicinal chemistry. Mini Rev. Med. Chem. 2007, 7, 1027–1034. [Google Scholar] [CrossRef]
- Kirsch, P.; Bremer, M. Nematic liquid crystals for active matrix displays: Molecular design and synthesis. Angew. Chem. Int. Ed. 2000, 39, 4216–4235. [Google Scholar] [CrossRef]
- Mamada, M.; Shima, H.; Yoneda, Y.; Shimano, T.; Yamada, N.; Kakita, K.; Machita, T.; Tanaka, Y.; Aotsuka, S.; Kumaki, D.; et al. A unique solution-processable n-type semiconductor material design for high-performance organic field-effect transistors. Chem. Mater. 2014, 27, 141–147. [Google Scholar] [CrossRef]
- Coric, V.; Taskiran, S.; Pittenger, C.; Wasylink, S.; Mathalon, D.H.; Valentine, G.; Saksa, J.; Gueorguieva, R.; Sanacora, G.; Malison, R.T.; et al. Riluzole augmentation in treatment-resistant obsessive–compulsive disorder: An open-label trial. Biol. Psychiatry 2005, 58, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Diacon, A.H.; Dawson, R.; von Groote-Bidlingmaier, F.; Symons, G.; Venter, A.; Donald, P.R.; van Niekerk, C.; Everitt, D.; Winter, H.; Becker, P.; et al. 14-day bactericidal activity of PA-824, bedaquiline, pyrazinamide, and moxifloxacin combinations: A randomised trial. Lancet 2012, 380, 986–993. [Google Scholar] [CrossRef]
- Henne, K.R.; Tran, T.B.; VandenBrink, B.M.; Rock, D.A.; Aidasani, D.K.; Subramanian, R.; Mason, A.K.; Stresser, D.M.; Teffera, Y.; Wong, S.G.; et al. Sequential metabolism of AMG 487, a novel CXCR3 antagonist, results in formation of quinone reactive metabolites that covalently modify CYP3A4 Cys239 and cause time-dependent inhibition of the enzyme. Drug Metab. Dispos. 2012, 40, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Leo, A.; Hansch, C.; Elkins, D. Partition coefficients and their uses. Chem. Rev. 1971, 71, 525–616. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Unger, S.H.; Kim, K.H.; Nikaitani, D.; Lien, E.J. Aromatic substituent constants for structure-activity correlations. J. Med. Chem. 1973, 16, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- McClinton, M.A.; McClinton, D.A. Trifluoromethylations and related reactions in organic chemistry. Tetrahedron 1992, 48, 6555–6666. [Google Scholar] [CrossRef]
- Federsel, D.; Herrmann, A.; Christen, D.; Sander, S.; Willner, H.; Oberhammer, H. Structure and conformation of α, α, α-trifluoroanisol, C6H5OCF3. J. Mol. Struct. 2001, 567, 127–136. [Google Scholar] [CrossRef]
- Böhm, H.J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in medicinal chemistry. ChemBioChem 2004, 5, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Farnham, W.B.; Smart, B.E.; Middleton, W.J.; Calabrese, J.C.; Dixon, D.A. Crystal and molecular structure of tris (dimethylamino) sulfonium trifluoromethoxide. Evidence for negative fluorine hyperconjugation. J. Am. Chem. Soc. 1985, 107, 4565–4567. [Google Scholar] [CrossRef]
- Taylor, S.L.; Martin, J.C. Trifluoromethyl triflate: Synthesis and reactions. J. Org. Chem. 1987, 52, 4147–4156. [Google Scholar] [CrossRef]
- Marrec, O.; Billard, T.; Vors, J.P.; Pazenok, S.; Langlois, B.R. A deeper insight into direct trifluoromethoxylation with trifluoromethyl triflate. J. Fluor. Chem. 2010, 131, 200–207. [Google Scholar] [CrossRef]
- Sheppard, W.A. α-Fluorinated Ethers. I. Aryl Fluoroalkyl Ethers1. J. Org. Chem. 1964, 29, 1–11. [Google Scholar] [CrossRef]
- Marrec, O.; Billard, T.; Vors, J.P.; Pazenok, S.; Langlois, B.R. A New and Direct Trifluoromethoxylation of Aliphatic Substrates with 2,4-Dinitro (trifluoromethoxy) benzene. Adv. Synth. Catal. 2010, 352, 2831–2837. [Google Scholar] [CrossRef]
- Huang, C.; Liang, T.; Harada, S.; Lee, E.; Ritter, T. Silver-mediated trifluoromethoxylation of aryl stannanes and arylboronic acids. J. Am. Chem. Soc. 2011, 133, 13308–13310. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, P.; Liu, G. Palladium-catalyzed intramolecular aminotrifluoromethoxylation of alkenes. J. Am. Chem. Soc. 2015, 137, 15648–15651. [Google Scholar] [CrossRef]
- Chen, C.; Luo, Y.; Fu, L.; Chen, P.; Lan, Y.; Liu, G. Palladium-Catalyzed Intermolecular Ditrifluoromethoxylation of Unactivated Alkenes: CF3O-Palladation Initiated by Pd (IV). J. Am. Chem. Soc. 2018, 140, 1207–1210. [Google Scholar] [CrossRef]
- Qi, X.; Chen, P.; Liu, G. Catalytic Oxidative Trifluoromethoxylation of Allylic C−H Bonds Using a Palladium Catalyst. Angew. Chem. Int. Ed. 2017, 56, 9517–9521. [Google Scholar] [CrossRef]
- Zhou, M.; Ni, C.; Zeng, Y.; Hu, J. Trifluoromethyl benzoate: A versatile trifluoromethoxylation reagent. J. Am. Chem. Soc. 2018, 140, 6801–6805. [Google Scholar] [CrossRef]
- Zheng, W.; Morales-Rivera, C.A.; Lee, J.W.; Liu, P.; Ngai, M.Y. Catalytic C−H Trifluoromethoxylation of Arenes and Heteroarenes. Angew. Chem. 2018, 130, 9793–9797. [Google Scholar] [CrossRef]
- Yang, H.; Wang, F.; Jiang, X.; Zhou, Y.; Xu, X.; Tang, P. Silver-Promoted Oxidative Benzylic C−H Trifluoromethoxylation. Angew. Chem. Int. Ed. 2018, 57, 13266–13270. [Google Scholar] [CrossRef] [PubMed]
- Jelier, B.J.; Tripet, P.F.; Pietrasiak, E.; Franzoni, I.; Jeschke, G.; Togni, A. Radical Trifluoromethoxylation of Arenes Triggered by a Visible-Light-Mediated N−O Bond Redox Fragmentation. Angew. Chem. Int. Ed. 2018, 57, 13784–13789. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Cong, F.; Guo, R.; Wang, L.; Tang, P. Asymmetric silver-catalysed intermolecular bromotrifluoromethoxylation of alkenes with a new trifluoromethoxylation reagent. Nat. Chem. 2017, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Wan, X.; Shen, Q. Enantioselective Construction of Trifluoromethoxylated Stereogenic Centers by a Nickel-Catalyzed Asymmetric Suzuki–Miyaura Coupling of Secondary Benzyl Bromides. Angew. Chem. Int. Ed. 2017, 56, 11986–11989. [Google Scholar] [CrossRef]
- Kondo, H.; Maeno, M.; Hirano, K.; Shibata, N. Asymmetric synthesis of α-trifluoromethoxy ketones with a tetrasubstituted α-stereogenic centre via the palladium-catalyzed decarboxylative allylic alkylation of allyl enol carbonates. Chem. Commun. 2018, 54, 5522–5525. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Maeno, M.; Sasaki, K.; Guo, M.; Hashimoto, M.; Shiro, M.; Shibata, N. Synthesis of chiral nonracemic α-difluoromethylthio compounds with tetrasubstituted stereogenic centers via a palladium-catalyzed decarboxylative asymmetric allylic alkylation. Org. Lett. 2018, 20, 7044–7048. [Google Scholar] [CrossRef]
- Chen, C.; Pflüger, P.M.; Chen, P.; Liu, G. Palladium (II)-Catalyzed Enantioselective Aminotrifluoromethoxylation of Unactivated Alkenes using CsOCF3 as a Trifluoromethoxide Source. Angew. Chem. Int. Ed. 2019, 58, 2392–2396. [Google Scholar] [CrossRef]
- We attempted the alkylation reaction using aliphatic halides, but the no reaction was observed.
Sample Availability: Samples of the compounds 1a and 3aa are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Cat. | Base | Solvent | Time | Yield (%) 7 | ee (%) 8 |
|---|---|---|---|---|---|---|
| 1 | CN-1 | KOH | Toluene | 15 | 73 | 1 |
| 2 | CN-2 | KOH | Toluene | 15 | 75 | 3 |
| 3 | CN-3 | KOH | Toluene | 15 | 82 | 14 |
| 4 | CN-4 | KOH | Toluene | 15 | 88 | 26 |
| 5 | CN-5 | KOH | Toluene | 15 | 82 | 14 |
| 6 | CN-6 | KOH | Toluene | 15 | 89 | 0 |
| 7 | CN-7 | KOH | Toluene | 15 | 67 | 0 |
| 8 | CN-8 | KOH | Toluene | 15 | 78 | 0 |
| 9 | CN-4 | KOH | n-hexane | 24 | 38 | 2 |
| 10 | CN-4 | KOH | DCM | 24 | 65 | 5 |
| 11 | CN-4 | KOH | THF | 24 | 68 | 5 |
| 12 | CN-4 | KOH | Et2O | 24 | 40 | 19 |
| 13 | CN-4 | KOH | DMF | 15 | 20 | 1 |
| 14 | CN-4 | K2CO3 | Toluene | 48 | 11 | n.d. |
| 15 | CN-4 | NaOH | Toluene | 48 | 58 | 4 |
| 16 | CN-4 | CsOH·H2O | Toluene | 15 | 83 | 43 |
| 17 | CN-4 | LiOH | Toluene | 48 | NR | - |
| 18 | CN-4 | NaH | Toluene | 48 | 57 | 0 |
| 19 | CN-4 | KOAc | Toluene | 48 | NR | - |
| 20 | CN-4 | K2HPO4 | Toluene | 48 | NR | - |
| 21 | CN-4 | KOtBu | Toluene | 24 | 26 | 17 |
| 22 | CN-4 | KOMe | Toluene | 24 | 6 | n.d. |
| 23 | CN-4 | 50% KOH (aq) | Toluene | 15 | 44 | 6 |
| 24 2 | CN-4 | KOH | Toluene | 15 | 68 | 24 |
| 25 | CN-4 | CsOH·H2O | PhCF3 | 15 | 96 | 9 |
| 26 | CN-4 | CsOH·H2O | Toluene:CHCl3 = 7:3 | 24 | 39 | 35 |
| 27 | CN-9 | CsOH·H2O | Toluene | 15 | 77 | 1 |
| 28 3 | CN-4 | CsOH·H2O | Toluene | 15 | 92 | 49 |
| 29 4 | CN-4 | CsOH·H2O | Toluene | 15 | 83 | 50 |
| 30 5 | CN-4 | CsOH·H2O | Toluene | 15 | 80 | 54 |
| 31 5,6 | CN-4 | CsOH·H2O | Toluene | 72 | 75 | 57 |
| 32 5 | CD-4 | CsOH·H2O | Toluene | 15 | 75 | −50 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Y.; Maeno, M.; Zhao, Z.; Shibata, N. Enantioselective Benzylation and Allylation of α-Trifluoromethoxy Indanones under Phase-Transfer Catalysis. Molecules 2019, 24, 2774. https://doi.org/10.3390/molecules24152774
Liang Y, Maeno M, Zhao Z, Shibata N. Enantioselective Benzylation and Allylation of α-Trifluoromethoxy Indanones under Phase-Transfer Catalysis. Molecules. 2019; 24(15):2774. https://doi.org/10.3390/molecules24152774
Chicago/Turabian StyleLiang, Yumeng, Mayaka Maeno, Zhengyu Zhao, and Norio Shibata. 2019. "Enantioselective Benzylation and Allylation of α-Trifluoromethoxy Indanones under Phase-Transfer Catalysis" Molecules 24, no. 15: 2774. https://doi.org/10.3390/molecules24152774
APA StyleLiang, Y., Maeno, M., Zhao, Z., & Shibata, N. (2019). Enantioselective Benzylation and Allylation of α-Trifluoromethoxy Indanones under Phase-Transfer Catalysis. Molecules, 24(15), 2774. https://doi.org/10.3390/molecules24152774