Synthesis and Biological Evaluation of Disubstituted Pyrimidines as Selective 5-HT2C Agonists
by
 ,
,
Juhyeon Kim
1,2,† ,
,
Yoon Jung Kim
3,†,
Ashwini M. Londhe
4,5,
Ae Nim Pae
4,5,
Hyunah Choo
1,4,
Hak Joong Kim
2 and
Sun-Joon Min
3,6,* 1
Center for Neuro-Medicine, Korea Institute of Science and Technology (KIST), Seoul 02792, Korea
2
Department of Chemistry, Korea University, Seoul 02841, Korea
3
Department of Applied Chemistry, Hanyang University, Ansan, Gyeonggi-do 15588, Korea
4
Division of Bio-Medical Science & Technology, KIST School, Korea University of Science and Technology, Seoul 02792, Korea
5
Convergence Research Center for Diagnosis, Treatment and Care System of Dementia, Korea Institute of Science and Technology, Seoul 02792, Korea
6
Department of Chemical & Molecular Engineering, Hanyang University, Ansan, Gyeonggi-do 15588, Korea
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2019, 24(18), 3234; https://doi.org/10.3390/molecules24183234
Submission received: 1 August 2019
/
Revised: 31 August 2019
/
Accepted: 3 September 2019
/
Published: 5 September 2019
(This article belongs to the Special Issue Fabulous Fluorine in Organic and Medicinal Chemistry)
Abstract
:Here, we describe the synthesis of disubstituted pyrimidine derivatives and their biological evaluation as selective 5-HT2C agonists. To improve selectivity for 5-HT2C over other subtypes, we synthesized two series of disubstituted pyrimidines with fluorophenylalkoxy groups at either the 5-position or 4-position and varying cyclic amines at the 2-position. The in vitro cell-based assay and binding assay identified compounds 10a and 10f as potent 5-HT2C agonists. Further studies on selectivity to 5-HT subtypes and drug-like properties indicated that 2,4-disubstituted pyrimidine 10a showed a highly agonistic effect on the 5-HT2C receptor, with excellent selectivity, as well as exceptional drug-like properties, including high plasma and microsomal stability, along with low CYP inhibition. Thus, pyrimidine 10a could be considered a viable lead compound as a 5-HT2C selective agonist.
1. Introduction
Serotonin (5-hydroxytryptamine, 5-HT), a monoamine neurotransmitter, plays a critical role in the regulation of various neurological functions, including mood, sleep, cognition, anxiety, sexual behavior, and appetite [1,2]. There are 14 variants of serotonin receptors (5-HT receptors), which mostly belong to G-protein-coupled receptors (GPCRs), except for the 5-HT3 receptor, a ligand-gated ion channel. Activation of the 5-HT receptors induces both inhibitory and excitatory signal transduction by modulating the release of many neurotransmitters, such as glutamate, dopamine, epinephrine, and acetylcholine. Therefore, they have been considered crucial therapeutic targets for a variety of neurological diseases, including depression, psychiatric disorders, sexual dysfunction, obesity, and urinary incontinence [3,4,5].
The 5-HT2 receptors comprise three subtypes: 5-HT2A, 5-HT2B, and 5-HT2C. Recent studies revealed that activation of 5-HT2C receptors in the central nerve system (CNS) is a potential drug target for effective treatment of schizophrenia, as well as obesity [6,7,8,9,10]. One of the crucial problems faced during the development of 5-HT2C agonists is selectivity over the structurally related 5-HT2A and 5-HT2B receptors. 5-HT2A agonism in humans is implicated in acute adverse effects, such as hallucination and cardiovascular effects. Stimulation of the 5-HT2B receptor is related to other side effects, including chronic cardiac valvulopathy and pulmonary hypertension.
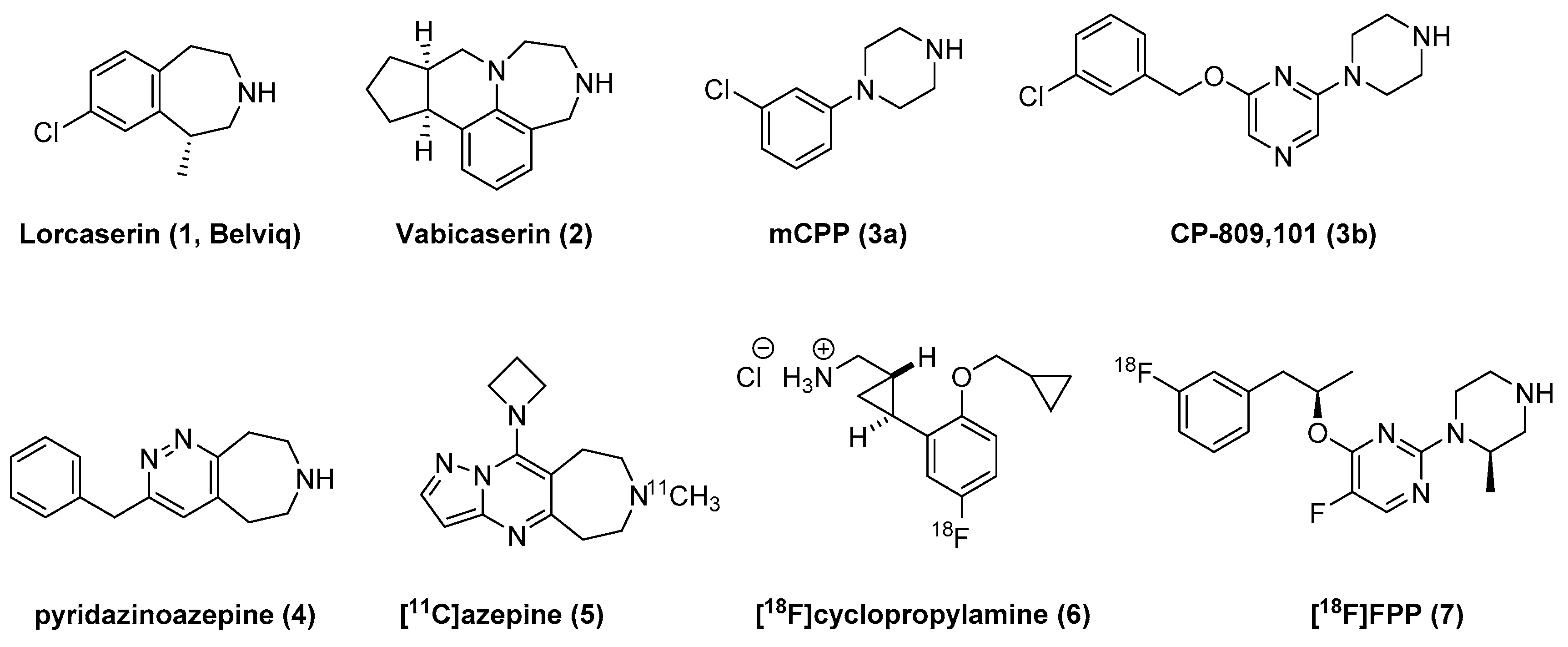
Potent and selective 5-HT2C agonists have been developed, as shown in Figure 1. Lorcaserin 1 was approved by the FDA (Food and Drug Administration) in 2012 for treating obesity [11], and vabicaserin 2 was clinically evaluated as a potential antipsychotic [12]. Numerous 5-HT2C agonists that have undergone preclinical or early clinical studies have been reported [13,14,15,16,17,18,19]. In addition, several 5-HT2C-selective positron emission tomography (PET) radioligands (5 and 6) have been developed as chemical tools for in vivo imaging of the 5-HT2C receptor [20,21].
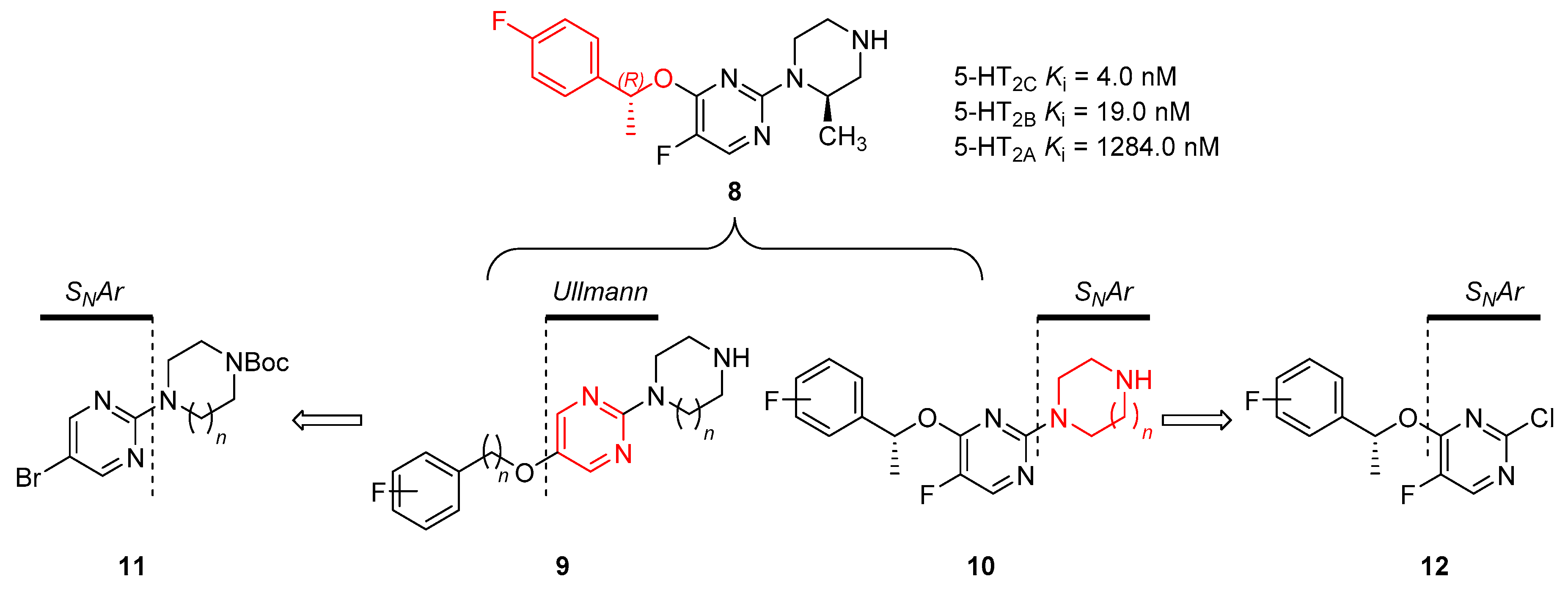
Despite the discovery of potential 5-HT2C agonists, the selectivity of these compounds over other 5-HT2 subtypes is still moderate; therefore, the search for highly selective 5-HT2C agonists remains challenging. In this regard, our group has previously reported optically active pyrimidine derivative 8 as a potent and selective 5-HT2C agonist (Figure 2) [22]. The structure activity relationship (SAR) between related pyrimidine derivatives and 5-HT2 subtypes revealed that a subtle change in the fluorophenylalkoxy moiety attached to pyrimidine 8 was essential to control the activation of selectivity for 5-HT2C over 5-HT2A and 5-HT2B. Although compound 8 was discovered as a lead compound with excellent potency and good selectivity for the 5-HT2 subtypes, further structural modification of these pyrimidine derivatives is required to improve selectivity over other 5-HT subtypes. Furthermore, selective 5-HT2C agonists could be applied in the development of chemical probes such as PET radiotracers [23,24,25,26].
We first planned to alter the substitution pattern of pyrimidines for the structural adjustment of compound 8 toward the 5-HT2C binding pocket. Recent studies demonstrated that pyridazine-fused azepine 4 containing a remote phenyl group (Figure 1) showed high potency and selectivity to 5-HT2C over 5-HT2B with a good stability profile [19]. Accordingly, we hypothesized that the linear form of 2,5-disubstituted pyrimidine 9, possessing a flexible fluorophenylalkoxy group at the 5-position, might be suitable for exploring the relationship between structural change and selectivity (Figure 2). Based on the molecular docking study of several 5-HT2C agonists published in literature [27,28], we also assumed that the terminal-free amine of the piperazine moiety could be a key functional group binding to a conserved residue, D134, in 5-HT2C protein. Taken together with the fact that compound 8 derived from (R)-isomer of secondary benzyl alcohol showed better binding affinity and selectivity to 5-HT2C than its diastereomer [22], we designed an alternative series of pyrimidine derivatives 10 possessing a variety of cyclic amines to examine their agonistic effect on 5-HT receptor activation. Herein, we report the synthesis of disubstituted pyrimidine derivatives with different substitution patterns and their biological evaluation as selective 5-HT2C receptor agonists.
2. Results and Discussion
2.1. Synthesis of 2,5-Disubstituted Pyrimidine Derivatives
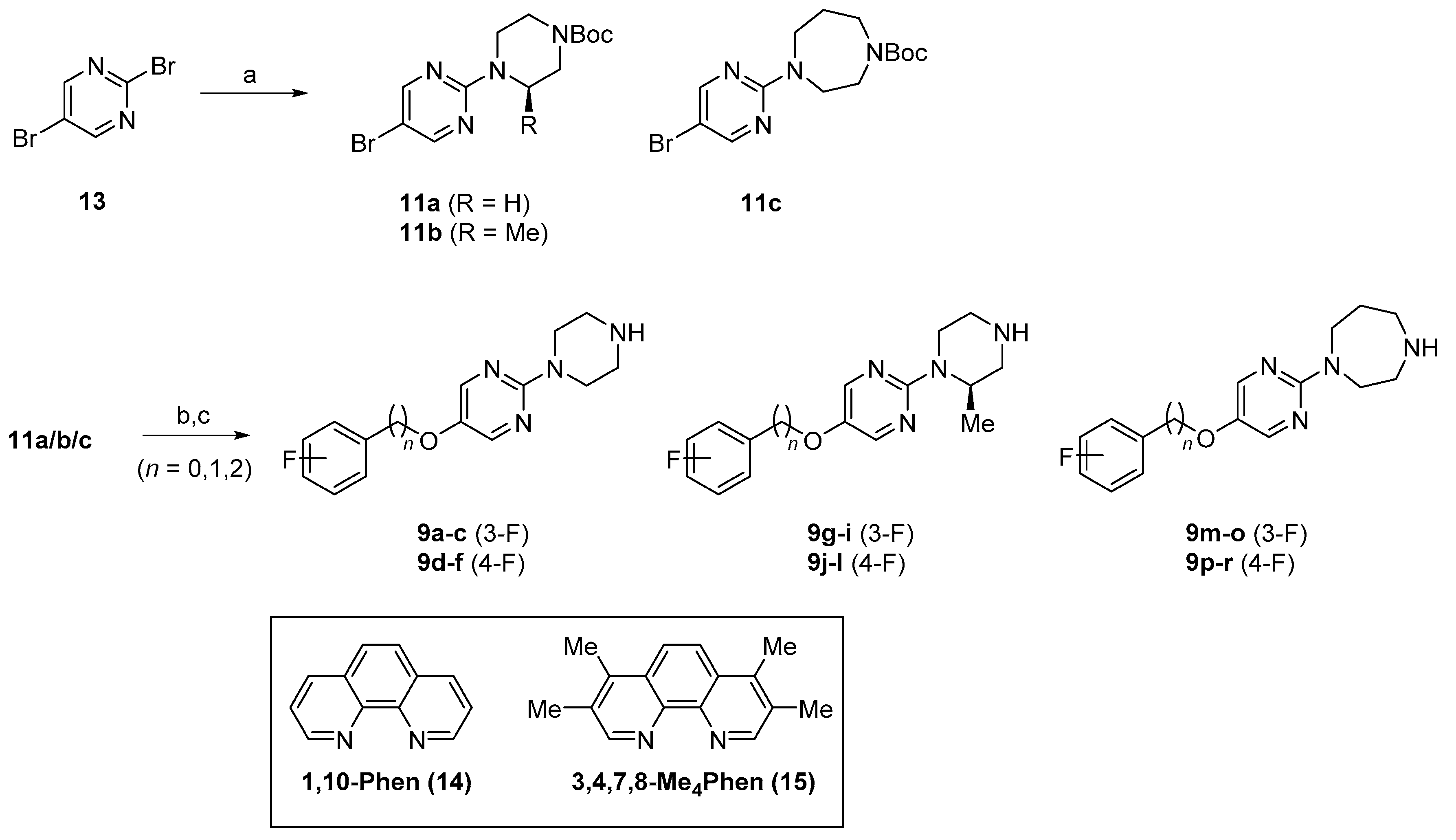
Initially, we synthesized 2,5-disubstituted pyrimidines with flexible phenylalkoxy groups at the 5-position, as shown in Scheme 1. Starting from 2,5-dibromopyrimidine 13, three kinds of 2-aminopyrimidines 11a–c were prepared through SNAr type amination. Pyrimidine derivatives 11a, 11b, and 11c were reacted with 3- or 4-fluoro-phenols, benzyl alcohols, and phenethyl alcohols in the presence of copper catalyst and 1,10-phenanthroline ligands 14 or 15 [29,30,31] to afford 2,5-disubstituted pyrimidines possessing an N-Boc-protecting group at the terminal amine. It is noted that the use of 15 as a ligand in this transformation led to formation of the desired products in higher yields. Finally, the 2-amino-5-alkoxypyrimidines 9a–r were obtained after removal of the Boc group under acidic conditions.
2.2. Synthesis of 2,4-Disubstituted Pyrimidine Derivatives
The synthesis of a series of 2,4-disubstituted pyrimidines 10, possessing different cyclic amines, is described in Scheme 2. According to our protocol [22], we efficiently synthesized optically active 1-(3- or 4-fluorophenyl)ethan-1-ol (R)-16a and (R)-16b using an enzymatic kinetic resolution method. As per the procedure previously reported in literature [32], nucleophilic aromatic substitution (SNAr) of 2,4-dichloropyrimidine with alcohols (R)-16a and (R)-16b selectively produced 4-alkoxypyrimidines 17a and 17b, which were subjected to an additional SNAr reaction to afford 2-alkoxy-4-aminopyrimidines as free or N-Boc-protected amine forms. The latter were further treated with a strong acid, such as HCl or TFA, to give the desired disubstituted pyrimidines. Besides pyrimidine analogues 10, we synthesized compounds 20a and 20b possessing a direct connection between the fluorophenyl group and pyrimidine via ether linkage using a similar experimental process. In this case, these compounds 20a/b might allow us to examine the optimal size of the fluorophenylalkoxy group through a comparison of their in vitro activity with that of 8.
2.3. In Vitro Evaluation of Pyrimidine Derivatives
In vitro agonistic activities of pyrimidines against 5-HT2C were evaluated using a fluorescence-based receptor functional assay, and the results are summarized in Table 1. Among the first series of pyrimidines possessing different sizes of phenylalkoxy groups at the 5-position, we found that the cellular agonistic effect of only compound 9b was higher than 50% at a concentration of 10 μM. Regarding the effect of cyclic amines at the 2-position of pyrimidine, only compounds 9g–l with methyl piperazine showed low to moderate activities, with up to 48% activation. Based on this result, we concluded that the substitution pattern of cyclic amines and the fluorophenylalkoxy group on the pyrimidine ring is important for 5-HT2C agonism. Thus, we decided not to proceed with further biological evaluation of this first series of compounds because an activation value higher than 50% is considered to represent significant agonistic effects of the compounds.
Next, the in vitro biological activities of 2,4-disubstituted pyrimidines 10a–j and 20a–b against the 5-HT2C receptor were evaluated, as shown in Table 2. At this point, the ligand binding assay, as well as the cell-based functional assay, were performed to identify more potent and selective 5-HT2C agonists. Among the tested compounds, four pyrimidine derivatives showed greater than 50% activation against 5-HT2C in the cell-based assay. Studies on the binding affinity of this series to 5-HT2C indicated that compounds 10a and 10f, with 1,4-diazepane at the 2-position of pyrimidine, exhibited excellent Ki values of 7.9 nM and 19.0 nM, respectively. Although the result of the cell-based assay is important to explore the cellular function of the 5-HT2C receptor, binding affinity plays a key role in determining whether the compound is suitable for the development of a 5-HT2C-specific chemical probe. Thus, we selected compounds 10a and 10f for further examination of the 5-HT subtype selectivity and drug-like properties.
The evaluation of compounds 10a and 10f for binding affinity to the 5-HT receptor subtypes is illustrated in Table 3. We calculated the selectivity index (SI) of binding affinities between 5-HT2C and other subtypes by dividing the given subtype Ki value with the 5-HT2C Ki value. The results indicate that compound 10a displayed higher selectivity for most 5-HT subtypes compared to compound 10f, except for 5-HT1B and 5-HT1E, to which the binding affinities of both compounds were negligible. Most importantly, compound 10a showed good selectivity for 5-HT2C over 5-HT2A and 5-HT2B, which are associated with the primary side effects of non-specific 5-HT2C agonists.
Considering the 5-HT2C cellular agonistic effect, binding affinity, and subtype selectivity of 10a, we further investigated its drug-like properties, such as plasma stability, microsomal stability, and CYP inhibition, as shown in Table 4. For plasma stability, 10 μM of compound 10a was incubated in human plasma; the remaining amount of 10a was measured after incubation for 30 min and 2 h. For microsomal stability, human liver microsomes were treated with 10a at a concentration of 1 μM; the % remaining amount of 10a was analyzed at 30 min after treatment. CYP inhibition values against five human CYP450 isozymes were recorded as % remaining activity values of the given isozymes. We found that compound 10a was stable in both the plasma and liver, indicating that compound 10a would be less affected by hepatic metabolism or plasma decomposition throughout systemic circulation. Furthermore, the inhibitory activity of 10a against all the tested CYP isozymes was significantly low (%remaining activity of isozymes >60%), suggesting that compound 10a might not exert toxic effects due to drug–drug interaction (DDI). Overall, compound 10a exhibited considerably excellent plasma and microsomal stability with low CYP inhibitory activity.
2.4. Molecular Docking Study
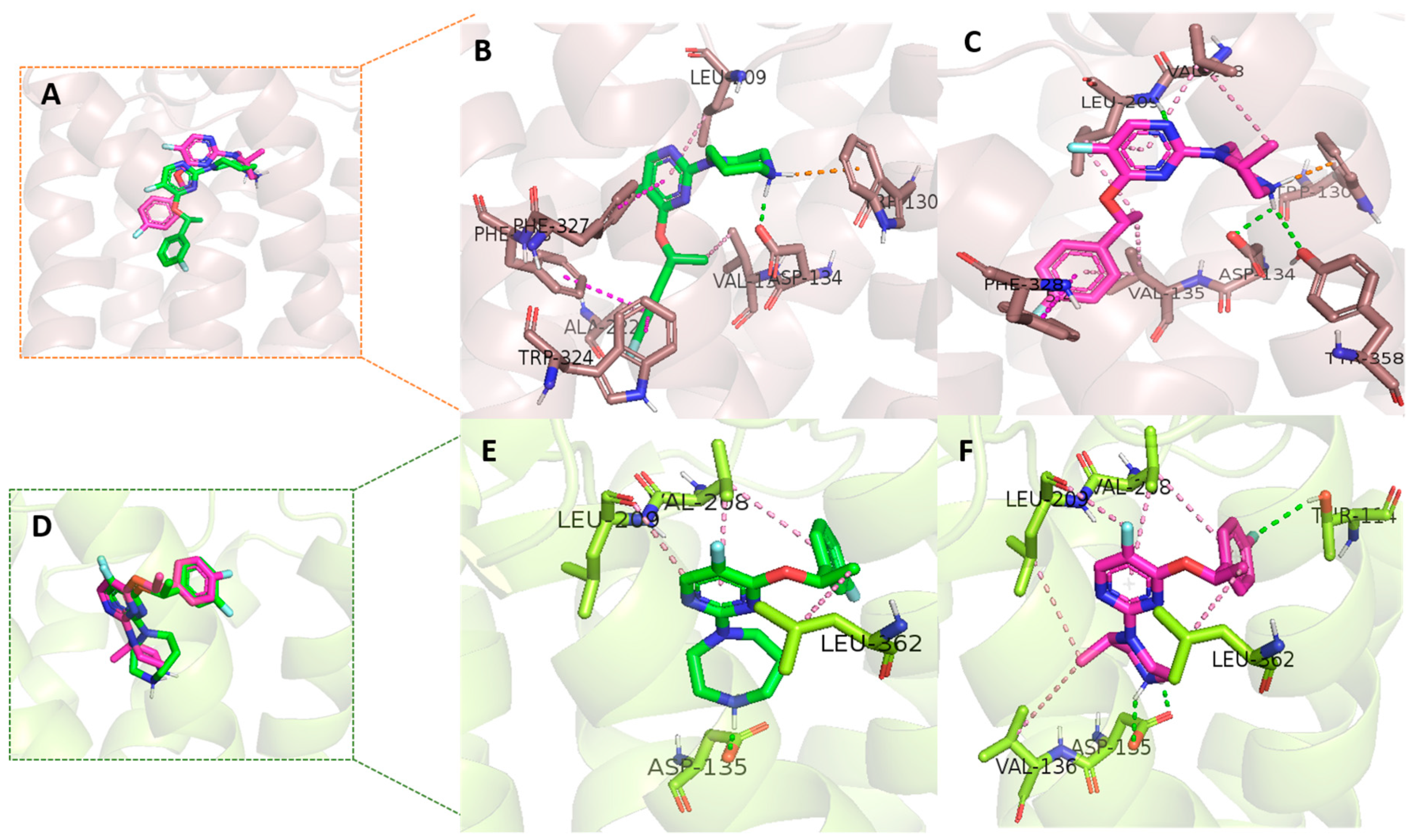
In order to investigate the good selectivity of 10a to 5-HT2C over 5-HT2B, the molecular docking study was performed using Discovery Studio software. Thus, the interaction of 10a and 8 with those receptors were evaluated using the crystal structures of 5-HT2C (PDB ID = 6BQH) and 5-HT2B (PDB ID = 4IB4), which were recently reported in the literatures [28,33].
In the case of 5-HT2C receptor docking study, the binding orientation of both ligands 10a (-CDOCKER score 36.31) and 8 (-CDOCKER score 36.96) are similar but not precisely overlapping over each other (Figure 3A). Both of these ligands have shown hydrogen bonds with crucial aspartic acids Asp134 (Figure 3B,C). NH groups of piperazine (8) and 1,4-diazepane ring (10a) are involved in hydrogen bonds and π-cationic interactions with Trp130 and Asp134. Additionally, 8 has shown two more hydrogen bonds with Tyr358 and Leu209. The hydrogen of the NH group of the piperazine ring has shown a hydrogen bond with Tyr358 and nitrogen of the pyrimidine ring has shown one hydrogen bond with Leu209. In total, we recognized four hydrogen bond interactions between 5-HT2C and 8. Fluorophenyl rings of 8 were involved in π–π stacking interactions with Phe328 residue. In the case of 10a, in addition to hydrogen bonds, strong π–π stacking contacts were observed with Phe327, phe328, and Trp324. Several hydrophobic contacts in both ligands were identified, mediated by Val135 and Leu209 in 10a and Val135, Val208, and Leu209 in 8.
In the case of 5-HT2B receptor docking study, overlap of ligand 10a (-CDOCKER score 37.40) and 8 (-CDOCKER score 39.53) inside 5-HT2B binding pocket (Figure 3D) shows that ligand bind at the same place but differ in pyrimidine, fluorophenyl ring, and diazepane ring/piperidine orientation. Compound 8 has shown three hydrogen bonds with binding site residues. The hydrogen (NH group) of the piperazine ring showed two hydrogen bonds with Asp135 (Figure 3F). The fluorine group on the phenyl ring in 8 has shown an additional hydrogen bond with Thr114. Beside these hydrogen bonding interactions, several hydrophobic residues, Val136, Val208, Leu209, and Leu362, hold the molecule by several hydrophobic interactions. Compound 10a has only one hydrogen bond with Asp135 (Figure 3E). Compared to 8, 10a has fewer hydrophobic contacts with the 5-HT2B receptor binding pocket (Val2018, Leu209, and Leu362).
Although both 10a and 8 bind in a similar way inside the 5-HT2B binding pocket, a minor difference in aromatic ring orientation affects the hydrophobic interactions of 10a. The difference in fluorophenyl ring orientation is responsible for the additional hydrogen bond between Thr114 and 8. These extra hydrogen bonds and hydrophobic contacts inside the 5-HT2B receptors make the potency of compound 8 superior to that of compound 10a in the 5-HT2B binding assay. Compared to the 5-HT2B receptor, 10a has stronger interactions (hydrogen bond and hydrophobic) with the 5-HT2C receptor. Overall, the decrease in hydrophobic contacts inside the 5-HT2B receptor makes our molecule 10a more selective towards the 5-HT2C receptor.
3. Materials and Methods
3.1. General Methods
All reactions were conducted under oven-dried glassware, under an atmosphere of nitrogen. All commercially available reagents were purchased and used without further purification. Solvents and gases were dried according to standard procedures. Organic solvents were evaporated with reduced pressure using a rotary evaporator. Reactions were followed by analytical thin layer chromatography (TLC) analysis using glass plates precoated with silica gel (0.25 mm). TLC plates were visualized by exposure to UV light (UV), and then were visualized with a KMnO4 or p-anisaldehyde stain followed by brief heating on a hot plate. Flash column chromatography was performed using silica gel 60 (230–400 mesh, Merck) with the indicated solvents. 1H-NMR spectra were measured with 400 MHz and 13C-NMR spectra were measured with 100 MHz using CDCl3 and CD3OD. 1H NMR spectra are represented as follows: Chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), integration, and coupling constant (J) in Hertz (Hz). 1H NMR chemical shifts are reported relative to CDCl3 (7.26 ppm). 13C NMR was recorded relative to the central line of CDCl3 (77.0 ppm). High resolution mass spectra (LR-MS) were obtained using positive electrospray ionization and mass/charge (m/z) ratios are reported as values in atomic mass units.
3.2. Synthesis of 2,5-Disubstituted Pyrimidines
3.2.1. General Procedure for Preparing Compounds 11a–c
In a dry sealed tube under argon were placed, to a solution of 1-Boc-piperazine (5.04 mmol), potassium carbonate (13.1 mmol) in acetonitrile (12.6 mL) was added 2,5-dibromo pyrimidine 13 (5.04 mmol). The mixture was allowed to stir at 80 °C for 12 h. After completion of the reaction (monitored by TLC), the mixture was then cooled at room temperature, then it was quenched with saturated aqueous NH4Cl (10 mL) and extracted with EtOAc. The organic layers were dried over anhydrous MgSO4 and concentrated in vacuo. The resulting residue was purified by flash column chromatography on silica gel (EtOAc:n-hexane = 1:8) to afford piperazine 11a.
tert-Butyl 4-(5-bromopyrimidin-2-yl)piperazine-1-carboxylate (11a): Yield: 88%; 1H-NMR (400 MHz, CDCl3) δ 8.30 (s, J = 2.0 Hz, 2H), 3.78-–3.75 (t, J = 5.2 Hz, 4H), 3.49–3.47 (t, J = 5.2 Hz, 4H), 1.48 (s, 9H). 13C-NMR (100 MHz, CD3OD) δ 161.2, 159.2, 156.46, 107.15, 81.5, 44.8, 28.6.
tert-Butyl (R)-4-(5-bromopyrimidin-2-yl)-3-methylpiperazine-1-carboxylate (11b): Yield: 92%; 1H-NMR (400 MHz, CDCl3) δ 8.28 (s, J = 2.0 Hz, 1H), 4.81–4.70 (m, 1H), 4.39–4.36 (m, 1H), 4.15–3.90 (m, 2H), 3.19–3.08 (m, 1H), 3.08–2.90 (m, 2H), 1.47 (s, 9H), 1.16 (d, J = 6.7 Hz, 3H). 13C-NMR (100 MHz, CD3OD) δ 160.9, 159.2, 156.9, 107.0, 81.4, 45.1, 44.0, 39.4, 14.5.
tert-Butyl 4-(5-bromopyrimidin-2-yl)-1,4-diazepane-1-carboxylate (11c): Yield: 97%; 1H-NMR (400 MHz, CDCl3) δ 8.28 (s, 2H), 3.86–3.80 (m, 2H), 3.73–3.70 (t, J = 6.1 Hz, 2H), 3.55–3.52 (t, J = 5.4 Hz, 2H), 3.38–3.35 (t, J = 5.9 Hz, 1H), 3.28–3.25 (t, J = 6.1 Hz, 1H), 1.96–1.90 (m, 2H), 1.43 (d, J = 8.4 Hz, 9H). 13C-NMR (100 MHz, CDCl3) δ 159.5, 158.1, 155.3, 105.8, 79.6, 49.0, 48.5, 47.4, 46.9, 46.3, 46.1, 28.5, 25.4.
3.2.2. General Procedure for Preparing Compounds 9a–r
In a dry sealed tube under argon were placed 11b (0.56 mmol), 3-fluorobenzyl alcohol (1.68 mmol), cesium carbonate (0.84 mmol), copper iodide (0.056 mmol) (or copper(I) oxide), 1,10-phenanthroline (0.11 mmol) (or 3,4,7,8-tetramethyl-1,10-phenanthroline) in toluene (1.1 mL) and the mixture was heated at 130 °C for 20 h. After completion of the reaction (monitored by TLC), the mixture was then cooled at room temperature and it was quenched with saturated aqueous NH4Cl (3 × 50 mL), extracted with EtOAc. The organic layers were dried over anhydrous MgSO4 and concentrated in vacuo. The resulting residue was purified by flash column chromatography on silica gel (EtOAc: Acetone: Hexane = 1:1:15) to afford intermediate pre-9h.
To a solution of methyl piperazine carboxylate pre-9h (0.136 mmol) in dichloromethane (1.3 mL) was added trifluoroacetic acid (0.21 mL). The mixture was allowed to stir at 0 °C for 3 h. After completion of the reaction (monitored by TLC), then neutralized by sodium hydrogen carbonate until the mixture to be pH 8 and extracted with EtOAc. The organic layers were dried over anhydrous MgSO4 and concentrated in vacuo. The resulting residue was purified by flash column chromatography on silica gel (MeOH:DCM = 1:15) to afford pyrimidine 9h, which was isolated as HCl salt form after treating with 4 M HCl in diethyl ether.
tert-Butyl 4-(5-((3-fluorophenoxy)pyrimidin-2-yl)piperazine-1-carboxylate (pre-9a): Yield: 21%; 1H-NMR (400 MHz, CDCl3) δ 8.17 (s, 2H), 7.27–7.21 (m, 1H), 6.77–6.73 (m, 1H), 6.71–6.62 (m, 2H), 3.80 (t, J = 5.1 Hz, 4H), 3.52 (t, J = 5.0 Hz, 4H), 1.49 (s, 9H).
4-(5-((3-fluorophenoxy)pyrimidin-2-yl)piperazin-1-ium∙hydrochloride (9a): Yield: 21%; 1H-NMR (400MHz, CDCl3) δ 8.17 (s, 2H), 7.24 (dd, J = 15.4, 7.5 Hz, 1H), 6.75 (m, 1H), 6.70 (d, J = 8.4 Hz, 1H), 6.64 (d, J = 10.2 Hz, 1H), 3.89 (t, J = 4.5 Hz, 4H), 3.07 (t, J = 4.7 Hz, 4H). 13C-NMR (100 MHz, CD3OD) δ 162.4 (d, 1J = 259 Hz), 159.7, 152.3, 152.0, 144.5, 132.2 (d, 3J = 9 Hz), 113.4 (d, 4J = 3 Hz), 110.9 (d, 2J = 21 Hz), 105.4 (d, 2J = 25 Hz), 44.4, 42.5; LRMS-EI (m/z): [M–Cl]+ calcd for C14H15FN4O: 274.30, found: 274.30
tert-Butyl 4-(5-((3-fluorobenzyl)oxy)pyrimidin-2-yl)piperazine-1-carboxylate(pre-9b): Yield: 79%; 1H-NMR (400 MHz, CDCl3) δ 8.11 (s, 2H), 7.37–7.32 (m, 1H), 7.17–7.11 (m, 2H), 7.05–6.99 (m, 1H), 5.01 (s, 2H), 3.71 (t, J = 5.2 Hz, 4H), 3.49 (t, J = 5. 2 Hz, 4H), 1.48 (s, 9H).
4-(5-((3-fluorobenzyl)oxy)pyrimidin-2-yl)piperazin-1-ium∙hydrochloride (9b): Yield: 58%; 1H-NMR (400 MHz, CD3OD) δ 8.23 (s, 1H), 7.42–7.37 (m, 1H), 7.25–7.17 (m, 2H), 7.06 (td, J = 8.5, 2.4 Hz, 1H), 7.07–7.02 (m, 1H), 5.12 (s, 2H), 3.95 (t, J = 5.3 Hz, 4H), 3.23 (t, J = 5.3 Hz, 4H). 13C-NMR (100 MHz, CD3OD) δ 164.3 (d, 1J = 243 Hz), 158.2, 147.7, 147.4, 140.8 (d, 3J = 7 Hz), 131.5 (d, 3J = 8 Hz), 124.4 (d, 4J = 3 Hz), 115.8 (d, 2J = 21 Hz), 115.3 (d, 2J = 22 Hz), 71.9, 44.3, 42.6; LRMS-EI (m/z): [M–Cl]+ calcd for C15H17FN4O: 288.33, found: 288.33
tert-Butyl 4-(5-((3-fluorophenethoxy)pyrimidin-2-yl)piperazine-1-carboxylate (pre-9c): Yield: 46%; 1H-NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.24–7.20 (m, 2H), 7.02–6.98 (m, 2H), 4.12 (t, J = 6.8 Hz, 2H), 3.69 (t, J = 5.2 Hz, 2H), 3.48 (t, J = 5.2 Hz, 2H), 3.03 (t, J = 6.8 Hz, 2H), 1.48 (s, 9H).
4-(5-((3-fluorophenethoxy)pyrimidin-2-yl)piperazin-1-ium∙hydrochloride (9c): Two-step yield: 46%; 1H-NMR (400 MHz, CD3OD) δ 8.17 (s, 2H), 7.34–7.29 (m, 1H), 7.13 (d, J = 8 Hz, 1H), 7.07 (d, J = 10.1 Hz, 1H), 6.96 (td, J = 8.7, 2.3 Hz, 1H), 4.25 (t, J = 6.5 Hz, 2H), 3.97 (t, J = 5.2 Hz, 4H), 3.27 (t, J = 5.2 Hz, 4H), 3.09 (t, J = 6.5 Hz, 2H). 13C-NMR (100 MHz, CD3OD) δ 164.3 (d, 1J = 242 Hz), 158.1, 148.2, 146.8, 142.5 (d, 3J = 7 Hz), 131.1 (d, 3J = 8 Hz), 125.9 (d, 4J = 3 Hz), 116.7 (d, 2J = 21 Hz), 114.3 (d, 2J = 21 Hz), 71.28, 44.3, 42.7, 36.4, 36.4; LRMS-EI (m/z): [M–Cl]+ calcd for C16H19FN4O: 302.35, found: 302.35
tert-Butyl 4-(5-((4-fluorophenoxy)pyrimidin-2-yl)piperazine-1-carboxylate (pre-9d): Yield: 21%; 1H-NMR (400 MHz, CDCl3) δ 8.17 (s, 2H), 7.03–6.99 (m, 2H), 6.92–6.89 (m, 2H), 3.80 (t, J = 5.2 Hz, 4H), 3.52 (t, J = 5.2 Hz, 4H), 1.49 (s, 9H).
4-(5-((4-fluorophenoxy)pyrimidin-2-yl)piperazin-1-ium∙hydrochloride (9d): Yield: 60%; 1H-NMR (400 MHz, CDCl3) δ 8.15 (s, 2H), 7.02-6.98 (m, 2H), 6.91–6.88 (m, 2H), 3.77 (t, J = 5.2 Hz, 4H), 3.51 (t, J = 5.1 Hz, 4H). 13C-NMR (100 MHz, CD3OD) δ 158.9 (d, 1J = 241 Hz), 157.9, 153.8, 150.1, 144.4, 118.6 (d, 3J = 8 Hz), 116.6 (d, 2J = 23 Hz), 43.4, 41.5; LRMS-EI (m/z): [M–Cl]+ calcd for C14H15FN4O: 274.30, found: 274.30.
tert-Butyl 4-(5-((4-fluorobenzyl)oxy)pyrimidin-2-yl)piperazine-1-carboxylate (pre-9e): Yield: 76%; 1H-NMR (400 MHz, CDCl3) δ 8.10 (s, 1H), 7.37 (dd, J = 8.7, 5.3 Hz, 2H), 7.09–7.05 (m, 2H), 4.98 (s, 2H), 3.71 (t, J = 5.2 Hz, 4H), 3.49 (t, J = 5.2 Hz, 4H), 1.48 (s, 9H).
4-(5-((4-fluorobenzyl)oxy)pyrimidin-2-yl)piperazin-1-ium∙hydrochloride (9e): Two-step yield: 67%; 1H-NMR (400 MHz, CD3OD) δ 8.22 (s, 2H), 7.47–7.43 (m, 2H), 7.13–7.08 (m, 2H), 5.08 (s, J = 2.5 Hz, 2H), 3.95 (t, J = 5.3 Hz, 4H), 3.24 (t, J = 5.2 Hz, 4H). 13C-NMR (100 MHz, CD3OD) δ 164.0 (d, 1J = 243 Hz), 158.4, 147.4, 134.0 (d, 4J = 3 Hz), 132.3, 131.0 (d, 3J = 8 Hz), 116.3 (d, 2J = 22 Hz), 72.1, 44.7, 43.3; LRMS-EI (m/z): [M–Cl]+ calcd for C15H17FN4O: 288.33, found: 288.33.
tert-Butyl 4-(5-((4-fluorophenethoxy)pyrimidin-2-yl)piperazine-1-carboxylate (pre-9f): Yield: 54%; 1H-NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.24–7.20 (m, 2H), 7.02–6.98 (m, 2H), 4.12 (t, J = 6.8 Hz, 2H), 3.70 (t, J = 5.2 Hz, 2H), 3.48 (t, J = 5.2 Hz, 2H), 3.03 (t, J = 6.8 Hz, 2H), 1.48 (s, 9H).
4-(5-((4-fluorophenethoxy)pyrimidin-2-yl)piperazin-1-ium∙hydrochloride (9f): Two-step yield: 53%; 1H-NMR (400 MHz, CD3OD) δ 8.16 (s, 2H), 7.32–7.28 (m, 2H), 7.04–6.99 (m, 2H), 4.21 (t, J = 6.6 Hz, 2H), 3.95 (t, J = 5.1 Hz, 4H), 3.27 (t, J = 5.1 Hz, 4H), 3.05 (t, J = 6.6 Hz, 2H). 13C-NMR (100 MHz, CD3OD) δ 163.1 (d, 1J = 241 Hz), 158.6, 147.7, 146.8, 135.51, 131.7 (d, 3J = 8 Hz), 116.0 (d, 2J = 21 Hz), 71.7, 45.3, 44.5, 35.9; LRMS-EI (m/z): [M–Cl]+ calcd for C16H19FN4O: 302.35, found: 302.35.
tert-Butyl (R)-4-(5-((3-fluorophenoxy)pyrimidin-2-yl)-3-methylpiperazine-1-carboxylate (pre-9g): Yield: 17%; 1H-NMR (400 MHz, CDCl3) δ 8.18 (s, 2H), 7.27–7.22 (m, 1H), 6.78–6.70 (m, 2H), 6.65–6.63 (m, 1H), 4.90–4.84 (m, 1H), 4.43–4.40 (m, 1H), 4.18–3.93 (m, 2H), 3.24–3.18 (m, 1H), 3.13–2.94 (m, 2H), 1.49 (s, 9H), 1.20 (d, J = 6.8 Hz, 3H).
(R)-4-(5-((3-fluorophenoxy)pyrimidin-2-yl)-3-methylpiperazine-1-ium∙hydrochloride (9g): Yield: 21%; 1H-NMR (400 MHz, CDCl3) δ 8.18 (s, 2H), 7.27–7.21 (m, 1H), 6.77–6.65 (m, 2H), 6.63–6.62 (m, 1H), 4.78–4.75 (m, 1H), 4.41–4.38 (m, 1H), 3.16–3.08 (m, 2H), 3.04–3.00 (m, 1H), 2.92–2.89 (m, 1H), 2.83–2.76 (m, 1H), 1.28 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CD3OD) δ 159.3, 152.1, 146.9, 144.3, 132.2 (d, 3J = 9 Hz), 131.8, 113.4, 110.8 (d, 2J = 21 Hz), 105.4 (d, 2J = 25 Hz), 45.8, 44.8, 36.9, 35.9, 13.7; LRMS-EI (m/z): [M–Cl]+ calcd for C15H17FN4O: 288.33, found: 288.33.
tert-Butyl (R)-4-(5-((3-fluorobenzyl)oxy)pyrimidin-2-yl)-3-methylpiperazine-1-carboxylate (pre-9h): Yield: 43%; 1H-NMR (400 MHz, CDCl3) δ 8.11 (s, 2H), 7.37–7.32 (m, 1H), 7.15–7.11 (m, 2H), 7.04–6.99 (m, 1H), 5.00 (s, 2H), 4.75 (s, 1H), 4.31 (d, J = 12.5 Hz, 1H), 4.10 (d, J = 12.5 Hz, 1H, broad), 3.17–3.10 (m, 1H), 3.10–2.91 (brs, 1H), 1.48 (s, 9H), 1.14 (d, J = 6.7 Hz, 3H).
(R)-4-(5-((3-fluorobenzyl)oxy)pyrimidin-2-yl)-3-methylpiperazine-1-ium∙hydrochloride (9h): Yield: 70%; 1H-NMR (400 MHz, CDCl3) δ 8.12 (s, 2H), 7.37–7.33 (m, 1H), 7.17–7.11 (m, 2H), 7.05–7.00 (td, J = 8.4, 2.3 Hz, 1H), 5.01 (s, 1H), 4.85–4.82 (m, 1H), 4.45–4.40 (m, 1H), 3.23–3.17 (m, 2H), 3.10–3.01 (m, 2H), 2.89–2.82(m, 1H), 1.30 (d, J = 6.9 Hz, 3H). 13C-NMR (100 MHz, CD3OD) δ 165.6 (d, 1J = 244 Hz), 157.8, 147.4, 140.8, 131.4 (d, 3J = 9 Hz), 124.3, 115.8 (d, 2J = 20 Hz), 115.3 (d, 2J = 22 Hz), 71.9, 45.8, 44.8, 37.2, 13.4; LRMS-EI (m/z): [M–Cl]+ calcd for C16H19FN4O: 302.35, found: 302.35
tert-Butyl (R)-4-(5-((3-fluorophenethoxy)pyrimidin-2-yl)-3-methylpiperazine-1-carboxylate (pre-9i): Yield: 24%; 1H-NMR (400 MHz, CDCl3) δ 8.06 (s, 2H), 7.30–7.26 (m, 1H), 7.04–7.02 (m, 1H), 6.98–6.91 (m, 2H), 4.98 (s, 2H), 4.80–4.70 (m, 1H), 4.31–4.28 (m, 1H), 4.14 (t, J = 6.7 Hz, 2H), 3.97–3.90 (m, 2H), 3.17–3.10 (m, 2H), 3.06 (t, J = 6.7 Hz, 2H), 3.00–2.80 (m. 2H), 1.48 (s, 9H), 1.14 (d, J = 6.7 Hz, 3H).
(R)-4-(5-((3-fluorophenethoxy)pyrimidin-2-yl)-3-methylpiperazin-1-ium∙hydrochloride (9i): Two-step yield: 37%; 1H-NMR (400 MHz, CDCl3) δ 8.06 (s, 2H), 7.28 (dd, J = 13.9, 7.9 Hz, 1H), 7.03 (d, J = 7.6 Hz, 1H), 6.98–6.91 (m, 2H), 4.86 (s, 2H), 4.45 (d, J = 12.4 Hz, 1H), 4.15 (t, J = 6.7 Hz, 2H), 3.28–3.14 (m, 2H), 3.11–3.08 (m, 1H), 3.06 (t, J = 6.7 Hz, 2H), 2.87 (td, J = 12.6, 4.1 Hz, 2H), 1.26 (d, J = 6.9 Hz, 2H). 13C-NMR (100 MHz, CD3OD) δ 163.10 (d, 1J = 245 Hz), 157.6, 147.9, 146.1, 142.5 (d, 3J = 7 Hz), 131.1 (d, 3J = 8 Hz), 125.9 (d, 4J = 3 Hz), 116.7 (d, 2J = 21 Hz), 114.2 (d, 2J = 21 Hz), 71.3, 45.6, 44.6, 36.7, 36.4, 30.9, 13.3; LRMS-EI (m/z): [M–Cl]+ calcd for C17H21FN4O: 316.38, found: 316.38.
tert-Butyl (R)-4-(5-((4-fluorophenoxy)pyrimidin-2-yl)-3-methylpiperazine-1-carboxylate (pre-9j): Yield: 27%; 1H-NMR (400 MHz, CDCl3) δ 8.17 (s, 2H), 7.02–6.98 (m, 2H), 6.91–6.89 (m, 2H), 4.90–4.83 (m, 1H), 4.43–4.39 (m, 1H), 4.18–3.92 (m, 3H), 3.25–3.18 (m, 1H), 3.15–2.93 (m, 2H), 1.49 (s, 9H), 1.20 (d, J = 6.7 Hz, 3H).
(R)-4-(5-((4-fluorophenoxy)pyrimidin-2-yl)-3-methylpiperazine-1-ium∙hydrochloride (9j): Yield: 39%; 1H-NMR (400 MHz, CDCl3) δ 8.15 (s, 2H), 7.01–6.96 (m, 2H), 6.91–6.87 (m, 2H), 4.79–4.76 (m, 1H), 4.42–4.38 (m, 1H), 3.16–3.09 (m, 2H), 3.05–3.01 (m, 1H), 2.94–2.91 (m, 1H), 2.84–2.77 (m, 1H), 2.77–2.70 (m, 1H), 1.28 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CD3OD) δ 160.1 (d, 1J = 239 Hz), 159.0, 155.5, 155.5 (d, 4J = 2 Hz), 145.5, 119.8 (d, 3J = 8 Hz), 117.4 (d, 2J = 24 Hz), 45.9, 44.9, 37.2, 30.9, 30.7, 13.5; LRMS-EI (m/z): [M–Cl]+ calcd for C15H17FN4O: 288.33, found: 288.33.
tert-Butyl (R)-4-(5-((4-fluorobenzyl)oxy)pyrimidin-2-yl)-3-methylpiperazine-1-carboxylate (pre-9k): Yield: 41%; 1H-NMR (400 MHz, CDCl3): δ 8.10 (s, 2H), 7.37 (dd, J = 8.5, 5.4 Hz, 2H), 7.09–7.05 (m, 2H), 4.97 (s, 2H), 4.80–4.74 (m, 1H), 4.32–4.28 (m, 1H), 4.15–3.89 (m, 3H), 3.17–3.10 (m, 1H), 3.10–2.90 (m, 2H), 1.48 (s, 9H), 1.14 (d, J = 6.7 Hz, 3H).
(R)-4-(5-((4-fluorobenzyl)oxy)pyrimidin-2-yl)-3-methylpiperazin-1-ium∙hydrochloride (9k): Yield: 73%; 1H-NMR (400 MHz, CDCl3) δ 8.11 (s, 2H), 7.39–7.35 (m, 2H), 7.09–7.05 (m, 2H), 4.98 (s, 2H), 4.94–4.89 (m, 1H), 4.64–4.61 (m, 1H), 3.48–3.39 (m, 2H), 3.28–3.25 (m, 1H), 3.20–3.12 (m, 1H), 3.01–2.94 (m, 1H), 1.42 (d, J = 7.0 Hz, 3H). 13C-NMR (100 MHz, CD3OD) δ 162.8 (d, 1J = 245 Hz), 156.4, 146.4, 146.1, 131.9, 129.7 (d, 3J = 8 Hz), 115.8 (d, 2J = 21 Hz), 71.5, 47.2, 44.3, 43.3, 35.7, 14.0; LRMS-EI (m/z): [M–Cl]+ calcd for C16H19FN4O: 302.35, found: 302.35.
tert-Butyl (R)-4-(5-((4-fluorophenethoxy)pyrimidin-2-yl)-3-methylpiperazine-1-carboxylate (pre-9l): Yield: 39%; 1H-NMR (400MHz, CDCl3) δ 8.05 (s, 2H), 7.23–7.20 (m, 2H), 7.02–6.98 (m, 2H), 4.80–4.70 (m, 1H), 4.30–4.27 (m, 1H), 4.12 (t, J = 6.9 Hz, 2H), 3.98–3.87 (m, 2H), 3.17–3.09 (m, 1H), 3.03 (t, J = 6.8 Hz, 2H), 3.01–2.89 (m, 2H), 1.48 (s, 9H), 1.14 (d, J = 6.7 Hz, 3H).
(R)-4-(5-((4-fluorophenethoxy)pyrimidin-2-yl)-3-methylpiperazin-1-ium∙hydrochloride (9l): Yield: 85%; 1H-NMR (400 MHz, CDCl3): δ 8.06 (s, 2H), 7.22 (dd, J = 8.4, 5.5 Hz, 2H), 7.00 (t, J = 8.7 Hz, 2H), 4.87–4.85 (m, 1H), 4.46–4.42 (m, 1H), 4.12 (t, J = 6.8 Hz, 2H), 3.27–3.16 (m, 2H), 3.09–3.05 (m, 2H), 3.03 (t, J = 6.7 Hz, 2H), 2.90–2.84 (m, 1H), 1.37 (d, J = 7.0 Hz, 3H). 13C-NMR (100 MHz, CD3OD): δ 163.08 (d, 1J = 241 Hz), 157.7, 147.8, 146.9, 135.5 (d, 4J = 3 Hz), 131.7 (d, 3J = 8 Hz), 116.0 (d, 2J = 21 Hz), 71.7, 45.8, 44.8, 37.2, 35.9, 30.9, 30.7, 13.3; LRMS-EI (m/z): [M–Cl]+ calcd for C17H21FN4: 316.38, found: 316.38.
tert-Butyl 4-(5-((3-fluorophenoxy)pyrimidin-2-yl)-1,4-diazepane-1-carboxylate (pre-9m): Yield: 19%; 1H-NMR (400 MHz, CDCl3) δ 8.17 (s, 2H), 7.28–7.22 (m, 1H), 6.78–6.70 (m, 2H), 6.71 (m, 1H), 6.65–6.60 (m, 1H), 3.90–3.88 (m, 2H), 3.80–3.78 (m, 2H), 3.59–3.58 (m, 2H), 3.40–3.39 (m, 1H), 3.33–3.30 (m, 1H), 1.99–1.96 (m, 2H), 1.44 (s, 9H).
4-(5-((3-fluorophenoxy)pyrimidin-2-yl)-1,4-diazepan-1-ium∙hydrochloride (9m): Yield: 40%; 1H-NMR (400 MHz, CDCl3) δ 8.17 (s, 2H), 7.26–7.22 (m, 1H), 6.77–6.73 (m, 1H), 6.70–6.67 (m, 1H), 6.65–6.62 (m, 1H), 4.08–4.01 (s, 2H), 3.90 (t, J = 6.3 Hz, 2H), 3.36–3.27 (m, 2H), 3.16–3.15 (m, 2H), 2.16–2.14 (m, 2H). 13C-NMR (100 MHz, CD3OD) δ 165.0 (d, 1J = 244 Hz), 159.6, 152.3, 143.9, 132.1 (d, 3J = 10 Hz), 131.6 (d, 3J = 8 Hz), 113.3, 110.7 (d, 2J = 21 Hz), 105.2 (d, 2J = 25 Hz), 46.8, 46.5, 44.7, 26.8; LRMS-EI (m/z): [M–Cl]+ calcd for C15H17FN4O: 288.33, found: 288.33.
tert-Butyl 4-(5-((3-fluorobenzyl)oxy)pyrimidin-2-yl)-1,4-diazepane-1-carboxylate (pre-9n): Yield: 79%; 1H-NMR (400 MHz, CDCl3): δ 8.09 (s, 2H), 7.37–7.32 (m, 1H), 7.17–7.11 (m, 2H), 7.04–6.99 (m, 1H), 4.99 (s, 2H), 3.84–3.78 (m, 2H), 3.71–3.68 (m, 2H), 3.53 (s, 2H), 3.35 (t, J = 5.8 Hz, 1H), 3.25 (t, J = 5.9 Hz, 1H), 1.96–1.90 (m, 2H), 1.42 (s, 9H, a mixture of rotamers).
4-(5-((3-fluorobenzyl)oxy)pyrimidin-2-yl)-1,4-diazepan-1-ium∙hydrochloride (9n): Two-step yield: 53%; 1H-NMR (400 MHz, CDCl3) δ 8.10 (s, 2H), 7.38–7.32 (m, 1H), 7.17–7.11 (m, 2H), 7.05–7.01 (m, 1H), 5.01 (s, 2H), 3.92–3.90 (m, 2H), 3.81–3.77 (m, 2H), 3.76–3.71 (m, 2H), 3.58 (t, J = 6.0 Hz, 1H), 3.52 (t, J = 6.0 Hz, 1H), 2.07–1.97 (m, 2H). 13C-NMR (100 MHz, CD3OD) δ 164.4 (d, 1J = 243 Hz), 157.9, 147.8, 147.1, 140.9 (d, 3J = 7 Hz), 131.4 (d, 3J = 8 Hz), 124.3 (d, 4J = 3 Hz), 115.8 (d, 2J = 21 Hz), 115.3 (d, 2J = 22 Hz), 72.1, 72.1, 47.0, 46.6, 44.6, 26.9; LRMS-EI (m/z): [M–Cl]+ calcd for C16H19FN4O: 302.35, found: 302.35.
tert-Butyl 4-(5-((4-fluorophenoxy)pyrimidin-2-yl)-1,4-diazepane-1-carboxylate (pre-9p): Yield: 31%; 1H-NMR (400 MHz, CDCl3) δ 8.14 (s, 2H), 7.01–6.96 (m, 2H), 6.87–6.75 (m, 2H), 3.89–3.84 (m, 2H), 3.76–3.73 (m, 2H), 3.58–5.55 (m, 2H), 3.39 (t, J = 6.0 Hz, 1H), 3.31 (d, J = 6.0 Hz, 1H), 3.31 (t, J = 6.0 Hz, 1H), 1.98–1.92 (m, 2H), 1.44 (s, 9H).
4-(5-((4-fluorophenoxy)pyrimidin-2-yl)-1,4-diazepan-1-ium∙hydrochloride (9p): Yield: 34%; 1H-NMR (400 MHz, CDCl3) δ 8.14 (s, 2H), 7.00–6.96 (m, 2H), 6.90–6.87 (m, 2H), 4.10–4.01 (m, 2H), 3.98–3.88 (m, 1H), 3.38–3.30 (m, 2H), 3.27–3.20 (m, 2H), 2.20–2.15 (m, 2H). 13C-NMR (100 MHz, CD3OD) δ 163.9 (d, 1J = 244 Hz), 157.9, 147.9, 147.1, 134.1, 131.0 (d, 3J = 8 Hz), 116.3 (d, 2J = 21 Hz), 72.4, 47.1, 46.6, 46.5, 44.5, 26.8; LRMS-EI (m/z): [M–Cl]+ calcd for C15H17FN4O: 288.33, found: 288.33.
4-(5-((3-fluorophenethoxy)pyrimidin-2-yl)-1,4-diazepan-1-ium∙hydrochloride (9o): Yield: 6.5% (2 steps); 1H-NMR (400 MHz, CDCl3) δ 8.04 (s, 2H), 7.30–7.28 (m, 2H), 7.04–7.03 (m, 1H), 6.99–6.94 (m, 2H), 4.15 (t, J = 6.6 Hz, 2H), 3.91–3.89 (m, 2H), 3.79–3.77 (m, 2H), 3.75–3.72 (m, 2H), 3.57–3.55(m, 1H), 3,50–3.49 (m, 1H), 3.06 (t, J = 6.5 Hz, 2H), 2.05–1.98 (m, 2H). 13C-NMR (100 MHz, CD3OD) δ 147.4, 147.3, 147.2, 131.1, 131.0, 125.9, 125.9, 116.8, 116.6, 114.2, 114.0, 71.59, 47.7, 46.66, 46.5, 36.5, 25.9; LRMS-EI (m/z): [M–Cl]+ calcd for C17H21FN4O: 316.38, found: 316.38.
4-(5-((4-fluorobenzyl)oxy)pyrimidin-2-yl)-1,4-diazepan-1-ium∙hydrochloride (9q): Two-step yield: 7%; 1H-NMR (400 MHz, CDCl3) δ 8.11 (s, 2H), 7.37 (dd, J = 8.4, 5.4 Hz, 2H), 7.08 (t, J = 8.6 Hz, 2H), 4.97 (s, 2H), 4.07–4.03 (s, 2H), 3.89–3.86 (m, 2H), 3.35–3.29 (m, 2H), 3.20–3.17 (m, 2H), 2.23–2.22 (m, 2H). 13C-NMR (100 MHz, CD3OD) δ 165.4 (d, 1J = 243 Hz), 155.0, 147.0, 146.8, 133.5, 131.2 (d, 3J = 8 Hz), 116.4 (d, 2J = 21 Hz), 72.5, 47.2, 46.9, 46.6, 27.0; LRMS-EI (m/z): [M–Cl]+ calcd for C16H19FN4O: 302.35, found: 302.35.
4-(5-((4-fluorophenethoxy)pyrimidin-2-yl)-1,4-diazepan-1-ium∙hydrochloride (9r): Two-step yield: 3.5%; 1H-NMR (400 MHz, CDCl3) δ 8.04 (s, 2H), 7.22 (dd, J = 8.4, 5.5 Hz, 2H), 7.01 (t, J = 8.6 Hz, 2H), 4.12 (t, J = 6.8 Hz, 2H), 3.92–3.89 (m, 2H), 3.81–3.77 (m, 2H), 3.75–3.71 (m, 2H), 3.57 (t, J = 6.0 Hz, 1H), 3.51 (t, J = 6.0 Hz, 1H), 3.03 (t, J = 6.7 Hz, 2H), 2.07–1.96 (m, 2H). 13C-NMR (100 MHz, CD3OD) δ 156.4, 146.2, 145.8, 134.2, 130.3 (d, 3J = 8 Hz), 114.6 (d, 2J = 21 Hz), 70.5, 60.1, 45.6, 45.2, 43.2, 34.6, 29.5, 25.5; LRMS-EI (m/z): [M–Cl]+ calcd for C17H21FN4O: 316.38, found: 316.38.
3.3. Synthesis of 2,4-Disubstituted Pyrimidines 10a–j
3.3.1. General Procedure for Preparing Compound (R)-(+)-16
To a solution of 1-(3 or 4-fluorophenyl)ethanol (6.67 mmol) in n-hexane (22.2 mL) was added CAL-B (147 mg), vinyl acetate (3.34 mmol), and triethylamine (0.667 mmol). The reaction mixture was allowed to stir at the room temperature for 1 h. After completion of the reaction (monitored by TLC), the mixture was filtered and concentrated in vacuo. The resulting residue was purified by flash column chromatography on silica gel (EtOAc:n-hexane = 1:8) to afford acetate intermediate (315 mg) as a colorless oil. To a solution acetate (1.73 mmol) in MeOH (3.45 mL) was added 1M NaOH (2.59 mmol). The reaction mixture was allowed to stir at the room temperature for 1 h. After completion of the reaction (monitored by TLC), it was quenched with distilled water and extracted with EtOAc. The organic layers were dried over anhydrous MgSO4 and concentrated in vacuo. The resulting residue was purified by flash column chromatography on silica gel (EtOAc:n-hexane = 1:8) to afford alcohol (R)-(+)-16.
(R)-1-(3-Fluorophenyl)ethan-1-ol ((R)-(+)-16a): Yield: 17%; 1H-NMR (400 MHz, CDCl3) δ 7.32–7.26 (m, 1H), 7.12–7.07 (m, 2H), 6.97–6.92 (m, 1H), 4.87 (q, J = 6.4 Hz, 1H), 2.18 (s, 1H), 1.47 (d, J = 6.4 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 163.0 (d, 1J = 244 Hz), 148.5 (d, 3J = 6 Hz), 130.0 (d, 3J = 8 Hz), 121.0 (d, 4J = 3 Hz), 114.2 (d, 2J = 21 Hz), 112.3 (d, 2J = 21 Hz), 69.8, 25.2; optical rotation for (R)-(+)-16a: [α]D26 + 43.7 ° (c 0.7, CHCl3).
(R)-1-(4-Fluorophenyl)ethan-1-ol ((R)-(+)-16b): Yield: 32%; 1H-NMR (400 MHz, CDCl3) δ 7.32–7.26 (m, 2H), 7.03–6.97 (m, 2H), 4.84 (q, J = 6.1 Hz, 1H), 2.34 (s, 1H), 1.44 (d, J = 6.4 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 162.1 (d, 1J = 244 Hz), 141.6 (d, 4J = 3 Hz), 127.1 (d, 3J = 8 Hz), 115.2 (d, 2J = 21 Hz), 69.7, 25.3; optical rotation for (R)-(+)-16b: [α]D27 + 51.9 ° (c 0.5, CHCl3).
3.3.2. General Procedure for Preparing Compound 17
A solution of sodium tert-butoxide (1.43 mmol) in toluene (7.10 mL) was treated with primary or secondary alcohol 16 (0.713 mmol) dropwise at 0 °C. After 5 min, 2,4-dichloro-5-fluoropyrimidine (0.713 mmol) was added to the mixture. The reaction mixture was allowed to stir at the room temperature for 1 h. After completion of the reaction (monitored by TLC), it was quenched with saturated aqueous NH4Cl, extracted with EtOAc, and washed with brine. The organic layers were dried over anhydrous MgSO4 and concentrated in vacuo. The resulting residue was purified by flash column chromatography on silica gel (EtOAc:n-hexane = 1:8) to afford pyrimidine 17.
(R)-2-Chloro-5-fluoro-4-(1-(3-fluorophenyl)ethoxy)pyrimidine ((R)-17a): Yield: 72%; 1H-NMR (400 MHz, CDCl3) δ 8.19 (d, J = 2.2 Hz, 1H), 7.37–7.31 (m, 1H), 7.23 (d, J = 7.7 Hz, 1H), 7.18–7.15 (m, 1H), 7.03–6.99 (m, 1H), 6.30 (q, J = 6.5 Hz, 1H), 1.71 (d, J = 6.6 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 162.9 (d, 1J = 245 Hz), 158.6 (d, 2J = 11 Hz), 153.2 (d, 4J = 5 Hz), 146.0 (d, 1J = 263 Hz), 144.4 (d, 2J = 20 Hz), 142.9 (d, 3J = 7 Hz), 130.3 (d, 3J = 8 Hz), 122.0 (d, 4J = 3 Hz), 115.3 (d, 2J = 21 Hz), 113.3 (d, 2J = 22 Hz), 75.6 (d, 4J = 2 Hz), 22.2; optical rotation for (R)-6d: [α]D27 +178.3 ° (c 0.7, CHCl3).
(R)-2-Chloro-5-fluoro-4-(1-(4-fluorophenyl)ethoxy)pyrimidine ((R)-17b): Yield: 68%; 1H-NMR (400 MHz, CDCl3) δ 8.16 (d, J = 2.2 Hz, 1H), 7.47–7.42 (m, 2H), 7.08–7.02 (m, 2H), 6.30 (q, J = 6.6 Hz, 1H), 1.71 (d, J = 6.6 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 162.6 (d, 1J = 245 Hz), 158.7 (d, 2J = 11 Hz), 153.1 (d, 4J = 4 Hz), 146.0 (d, 1J = 263 Hz), 144.3 (d, 2J = 20 Hz), 136.1 (d, 4J = 4 Hz), 128.4 (d, 3J = 9 Hz), 115.6 (d, 2J = 22 Hz), 75.9, 22.2; optical rotation for (R)-6e: [α]D27 +197.3 ° (c 0.8, CHCl3).
2-Chloro-5-fluoro-4-(3-fluorophenoxy)pyrimidine (19a): Yield: 51%; 1H-NMR (400MHz, CDCl3) δ 8.38 (d, J = 2.0 Hz, 1H), 7.45–7.39 (m, 1H), 7.06–7.01 (m, 2H), 6.99–6.96 (m, 1H). 13C-NMR (100 MHz, CD3OD) δ 164.5 (d, 1J = 245 Hz), 159.8 (d, 2J = 11 Hz), 154.2 (d, 4J = 4 Hz), 153.7 (d, 3J = 11 Hz), 147.4 (d, 1J = 262 Hz), 147.4 (d, 2J = 20 Hz), 132.0 (d, 3J = 9 Hz), 118.5 (d, 4J = 3 Hz), 114.4 (d, 2J = 21 Hz), 110.5 (d, 2J = 25 Hz).
2-Chloro-5-fluoro-4-(4-fluorophenoxy)pyrimidine (19b): Yield: 52%; 1H-NMR (400MHz, CDCl3) δ 8.36 (d, J = 2.0 Hz, 1H), 7.19–7.11 (m, 4H). 13C-NMR (100 MHz, CD3OD) δ 162.0 (d, 1J = 242 Hz), 160.2 (d, 2J = 11 Hz), 154.2 (d, 4J = 5 Hz), 148.7 (d, 4J = 2 Hz), 147.2 (d, 2J = 20 Hz), 146.10, 124.3 (d, 3J = 9 Hz), 117.4 (d, 2J = 24 Hz).
3.3.3. General Procedure for Preparing Compounds pre-10b–e and pre-10g–j
To a solution of pyrimidine 17 (0.154 mmol) in toluene (0.300 mL) was added cyclic amine (0.231 mmol) and triethylamine (0.231 mmol). The reaction mixture was allowed to stir at 90 °C for 12 h. After completion of the reaction (monitored by TLC), it was quenched with saturated aqueous NH4Cl, extracted with EtOAc, and washed with brine. The organic layers were dried over anhydrous MgSO4 and concentrated in vacuo. The resulting residue was purified by flash column chromatography on silica gel (EtOAc/n-hexane = 1:3) to afford carboxylate pre-10b–e and pre-10g–j.
tert-Butyl 7-(5-fluoro-4-((R)-1-(3-fluorophenyl)ethoxy)pyrimidin-2-yl)-2,7-diazaspiro[4.4]nonane-2- carboxylate (pre-10b): Yield: 92%; 1H-NMR (400 MHz, CDCl3, a mixture of rotamers) δ 7.94 (s, 1H), 7.31–7.26 (m, 1H), 7.17 (d, J = 7.6 Hz, 1H), 7.11 (d, J = 9.6 Hz, 1H) 6.95 (t, J = 8.3 Hz, 1H), 6.15–6.10 (m, 1H), 3.59–3.23 (m, 8H), 1.94–1.83 (m, 4H), 1.66 (d, J = 6.6 Hz, 3H), 1.45 (s, 9H). 13C-NMR (100 MHz, CDCl3, a mixture of rotamers) δ 162.9 (d, 1J = 244 Hz), 157.1 (d, 2J = 11 Hz), 156.0, 154.6, 145.0 (d, 3J = 7 Hz), 143.4 (d, 2J = 20 Hz), 140.0 (d, 1J = 245 Hz), 130.0 (d, 3J = 8 Hz), 121.6, 114.6 (d, 2J = 21 Hz), 113.1 (d, 2J = 22 Hz), 79.4, 73.7, 55.9, 55.3, 54.7, 54.6, 48.6, 47.8, 46.0, 45.2, 35.3, 34.9, 34.5, 28.5, 22.7.
tert-Butyl (3aR,6aS)-5-(5-fluoro-4-((R)-1-(3-fluorophenyl)ethoxy)pyrimidin-2-yl) hexahydropyrrolo [3,4-c]pyrrole-2(1H)-carboxylate (pre-10c): Yield: 82%; 1H-NMR (400 MHz, CDCl3, a mixture of rotamers) δ 7.94 (d, J = 3.1 Hz, 1H), 7.32–7.28 (m, 1H), 7.17 (d, J = 7.7 Hz, 1H), 7.11 (dd, J = 2.0, 9.7 Hz, 1H), 6.95 (td, J = 2.0, 8.4 Hz, 1H), 6.14 (q, J = 6.6 Hz, 1H), 3.69–3.59 (m, 4H), 3.40–3.21 (m, 4H), 2.94 (bs, 2H), 1.66 (d, J = 6.6 Hz, 3H), 1.47 (s, 9H). 13C-NMR (100 MHz, CDCl3, a mixture of rotamers) δ 162.9 (d, 1J = 244 Hz), 157.1 (d, 2J = 11 Hz), 156.0 (d, 4J = 3 Hz), 154.5, 145.0 (d, 3J = 10 Hz), 143.4 (d, 2J = 20 Hz), 140.1 (d, 1J = 246 Hz), 130.0 (d, 3J = 8 Hz), 121.6, 114.6 (d, 2J = 21 Hz), 113.0 (d, 2J = 21 Hz), 79.5, 73.6, 50.8, 50.1, 49.8, 42.2, 41.2, 28.5, 22.7.
tert-Butyl (R)-3-((5-fluoro-4-((R)-1-(3-fluorophenyl)ethoxy)pyrimidin-2-yl)aminopyrrolidine-1- carboxylate (pre-10d): Yield: 68%; 1H-NMR (400 MHz, CDCl3, a mixture of rotamers) δ 7.89 (s, 1H), 7.31–7.28 (m, 1H), 7.14 (d, J = 7.7 Hz, 1H), 7.08 (d, J = 9.6 Hz, 1H) 6.94 (t, J = 7.9 Hz, 1H), 6.08 (d, J = 4.4 Hz, 1H), 5.12 (bs, 1H), 4.24–4.16 (m, 1H), 3.71–3.65 (m, 1H), 3.43–3.37 (m, 2H), 3.28–3.12 (m, 1H), 2.01–1.98 (m, 1 H), 1.65 (d, J = 6.6 Hz, 3H), 1.45 (s, 9H). 13C-NMR (100 MHz, CDCl3, a mixture of rotamers) δ 162.9 (d, 1J = 245 Hz), 157.6 (d, 2J = 11 Hz), 157.3, 154.6, 144.7 (d, 3J = 6 Hz), 143.7 (d, 2J = 20 Hz), 140.6 (d, 1J = 248 Hz), 130.1 (d, 3J = 8 Hz), 121.3, 114.6 (d, 2J = 22 Hz), 112.8 (d, 2J = 22 Hz), 79.5, 73.9, 51.8, 51.7, 51.4, 51.0, 44.1, 43.7, 31.8, 31.1, 28.5, 22.8.
tert-Butyl (S)-3-((5-fluoro-4-((R)-1-(3-fluorophenyl)ethoxy)pyrimidin-2-yl)aminopyrrolidine-1- carboxylate (pre-10e): Yield: 52%; 1H-NMR (400 MHz, CDCl3, a mixture of rotamers) δ 7.90 (d, J = 2.9 Hz, 1H), 7.33–7.26 (m, 1H), 7.15 (d, J = 7.7 Hz, 1H), 7.10 (d, J = 9.6 Hz, 1H) 6.98–6.94 (m, 1H), 6.11 (q, J = 6.5 Hz, 1H), 5.12 (bs, 1H), 4.28 (bs, 1H), 3.47–3.42 (m, 3H), 3.21–3.04 (m, 1H), 2.20–2.12 (m, 1H), 1.86 (bs, 1H), 1.65 (d, J = 6.6 Hz, 3H), 1.45 (s, 9H). 13C-NMR (100 MHz, CDCl3, a mixture of rotamers) δ 162.9 (d, 1J = 245 Hz), 157.6 (d, 2J = 11 Hz), 157.2, 154.6, 143.6 (d, 2J = 20 Hz), 143.3 (d, 1J = 271 Hz), 139.4, 130.1 (d, 3J = 8 Hz), 121.4, 114.7 (d, 2J = 21 Hz), 112.8 (d, 2J = 22 Hz), 79.5, 73.8, 51.9, 51.6, 51.4, 51.0, 44.1, 43.8, 31.9, 31.1, 28.5, 22.8.
tert-Butyl 7-(5-fluoro-4-((R)-1-(4-fluorophenyl)ethoxy)pyrimidin-2-yl)-2,7-diazaspiro[4.4]nonane-2- carboxylate (pre-10g): Yield: 98%; 1H-NMR (400 MHz, CDCl3, a mixture of rotamers) δ 7.93 (s, 1H), 7.40–7.37 (m, 2H), 7.03–6.99 (m, 2H), 6.18–6.13 (m, 1H), 3.61–3.22 (m, 8H), 1.95–1.82 (m, 4H), 1.66 (d, J = 6.6 Hz, 3H), 1.45 (s, 9H). 13C-NMR (100 MHz, CDCl3, a mixture of rotamers) δ 162.2 (d, 1J = 244 Hz), 157.2 (d, 2J = 11 Hz), 156.0, 154.6, 143.3 (d, 2J = 20 Hz), 140.1 (d, 1J = 246 Hz), 138.0, 127.9 (d, 3J = 8 Hz), 115.3 (d, 2J = 21 Hz), 79.4, 73.6, 55.9, 55.4, 54.8, 54.6, 48.6, 47.8, 46.0, 45.2, 44.9, 35.3, 34.9, 34.5, 28.5, 22.8.
tert-Butyl (3aR,6aS)-5-(5-fluoro-4-((R)-1-(4-fluorophenyl)ethoxy)pyrimidin-2-yl)hexahydropyrrolo [3,4-c]pyrrole-2(1H)-carboxylate (pre-10h): Yield: 93%; 1H-NMR (400 MHz, CDCl3, a mixture of rotamers) δ 7.92 (d, J = 3.1 Hz, 1H), 7.40–7.36 (m, 2H), 7.00 (t, J = 8.6 Hz, 2H), 6.15 (d, J = 5.7 Hz, 1H), 3.73–3.60 (m, 4H), 3.41–3.21 (m, 4H), 2.91 (bs, 2H), 1.64 (d, J = 6.6 Hz, 3H), 1.44 (s, 9H). 13C-NMR (100 MHz, CDCl3, a mixture of rotamers) δ 162.2 (d, 1J = 245 Hz), 157.2 (d, 2J = 11 Hz), 156.0 (d, 4J = 3 Hz), 154.5, 143.3 (d, 2J = 20 Hz), 140.1 (d, 1J = 245 Hz), 138.0, 127.8 (d, 3J = 8 Hz), 115.3 (d, 2J = 21 Hz), 79.5, 73.6, 50.8, 50.1, 49.8, 42.2, 41.2, 28.5, 22.8.
tert-Butyl (R)-3-((5-fluoro-4-((R)-1-(4-fluorophenyl)ethoxy)pyrimidin-2-yl)amino)pyrrolidine-1- carboxylate (pre-10i): Yield: 52%; 1H-NMR (400 MHz, CDCl3, a mixture of rotamers) δ 7.88 (d, J = 2.0 Hz, 1H), 7.38–7.34 (m, 2H), 7.01 (t, J = 8.5 Hz, 2H), 6.14–6.09 (m, 1H), 5.13 (bs, 1H), 4.26–4.19 (m, 1H), 3.69–3.66 (m, 1H), 3.40 (bs, 2H), 3.29–3.16 (m, 1H), 2.09–2.01 (m, 1H), 1.74–1.73 (m, 1H), 1.64 (d, J = 6.6 Hz, 3H), 1.46 (s, 9H). 13C-NMR (100 MHz, CDCl3, a mixture of rotamers) δ 162.2 (d, 1J = 245 Hz), 157.7 (d, 2J = 11 Hz), 157.3, 154.6, 143.5 (d, 2J = 21 Hz), 140.6 (d, 1J = 246 Hz), 137.8 (d, 2J = 20 Hz), 127.6 (d, 3J = 12 Hz), 115.4 (d, 2J = 21 Hz), 79.5, 73.9, 51.8, 51.7, 51.5, 51.0, 44.1, 43.8, 31.8, 31.1, 28.5, 22.8.
tert-Butyl (S)-3-((5-fluoro-4-((R)-1-(4-fluorophenyl)ethoxy)pyrimidin-2-yl)aminopyrrolidine-1- carboxylate (pre-10j): Yield: 44%; 1H-NMR (400 MHz, CDCl3, a mixture of rotamers) δ 7.89 (d, J = 3.0 Hz, 1H), 7.39–7.36 (m, 2H), 7.03 (t, J = 8.6 Hz, 2H), 6.14 (q, J = 6.5 Hz, 1H), 4.98 (bs, 1H), 4.31 (m, 1H), 3.62–3.44 (m, 3H), 3.24–3.06 (m, 1H), 2.28-2.13 (m, 1H), 1.86 (bs, 1H), 1.64 (d, J = 6.6 Hz, 3H), 1.45 (s, 9H). 13C-NMR (100 MHz, CDCl3, a mixture of rotamers) δ 162.3 (d, 1J = 244 Hz), 157.7 (d, 2J = 11 Hz), 157.2, 154.6, 143.5 (d, 2J = 19 Hz), 140.8 (d, 1J = 247 Hz), 137.7, 127.7, 130.1, 115.4 (d, 2J = 21 Hz), 79.5, 73.8, 52.0, 51.7, 51.5, 51.0, 44.1, 43.8, 31.9, 31.1, 29.8, 28.5, 22.8.
tert-Butyl (R)-4-(5-fluoro-4-(3-fluorophenoxy)pyrimidin-2-yl)-3-methylpiperazine-1-carboxylate (pre-20a): Yield: 74%; 1H-NMR (400MHz, CDCl3) δ 8.13 (d, J = 2.7 Hz, 1H), 7.39–7.33 (m, 1H), 6.99–6.94 (m, 3H), 4.44 (m, 1H), 4.05–3.80 (m, 3H), 3.06–3.2.99 (m, 2H), 2.99–2.82 (m, 1H), 1.45 (s, 9H), 1.04 (d, J = 6.6 Hz, 3H).
tert-Butyl (R)-4-(5-fluoro-4-(4-fluorophenoxy)pyrimidin-2-yl)-3-methylpiperazine-1-carboxylate (pre-20b): Yield: 61%; 1H-NMR (400MHz, CDCl3) δ 8.11 (d, J = 2.7 Hz, 1H), 7.15–7.12 (m, 2H), 7.11–7.06 (m, 2H), 4.50–4.30 (m, 1H), 4.03–3.80 (m, 3H), 3.04–2.97 (m, 2H), 2.97–2.81 (m, 1H), 1.45 (s, 9H), 1.03 (d, J = 6.6 Hz, 3H).
3.3.4. General Procedure for Preparing Compounds 10a and 10f
To a solution of pyrimidine 17 (0.115 mmol) in toluene (0.250 mL) was added 1,4-diazepane (0.230 mmol) and triethylamine (0.230 mmol). The reaction mixture was allowed to stir at 90 °C for 12 h. After completion of the reaction (monitored by TLC), it was quenched with saturated aqueous NH4Cl, extracted with EtOAc, and washed with brine. The organic layers were dried over anhydrous MgSO4 and concentrated in vacuo. The resulting residue was purified by flash column chromatography on silica gel (DCM/MeOH = 10:1) to afford 2,4-disubstituted pyrimidine 10a and 10f.
(R)-1-(5-fluoro-4-(1-(3-fluorophenyl)ethoxy)pyrimidin-2-yl)-1,4-diazepane (10a): Yield: 68%; 1H-NMR (400 MHz, CD3OD) δ 8.06 (d, J = 3.2 Hz, 1H), 7.23 (d, J = 7.7 Hz, 1H), 7.15 (d, J = 9.8 Hz, 1H), 7.00 (td, J = 2.1, 8.5 Hz, 1H), 6.11 (q, J = 6.5 Hz, 1H), 3.90–3.77 (m, 4H), 3.26–3.03 (m, 4H), 2.07–1.94 (m, 2H), 1.66 (d, J = 6.6 Hz, 3H). 13C-NMR (100 MHz, CD3OD) δ 163.0 (d, 1J = 244 Hz), 157.7 (d, 2J = 11 Hz), 155.9, 145.5 (d, 3J = 7 Hz), 142.5 (d, 2J = 21 Hz), 140.3 (d, 1J = 246 Hz), 130.3 (d, 3J = 8 Hz), 121.1 (d, 4J = 3 Hz), 114.1 (d, 2J = 21 Hz), 112.0 (d, 2J = 22 Hz), 74.7, 45.5, 45.3, 45.0, 43.3, 25.3, 22.0. LRMS-EI (m/z): [M+H]+ calcd for C17H21F2N4O: 335.17, found: 335.10.
(R)-1-(5-fluoro-4-(1-(4-fluorophenyl)ethoxy)pyrimidin-2-yl)-1,4-diazepane (10f): Yield: 79%; 1H-NMR (400 MHz, CD3OD) δ 8.05 (d, J = 3.2 Hz, 1H), 7.45 (dd, J = 5.4, 8.4 Hz, 1H), 7.11–7.07 (m, 2H), 6.16 (q, J = 6.5 Hz, 1H), 4.10–3.72 (m, 4H), 3.42–3.15 (m, 4H), 2.21–1.98 (m, 2H), 1.66 (d, J = 6.5 Hz, 3H). 13C-NMR (100 MHz, CD3OD) δ 162.3 (d, 1J = 243 Hz), 158.0 (d, 2J = 12 Hz), 155.60, 141.7 (d, 2J = 20 Hz), 140.3 (d, 1J = 246 Hz), 138.3 (d, 4J = 3 Hz), 127.4 (d, 3J = 8 Hz), 115.0 (d, 2J = 22 Hz), 75.0, 45.6, 45.4, 45.0, 43.4, 25.3, 22.1. LRMS-EI (m/z): [M+H]+ calcd for C17H21F2N4O: 335.17, found: 335.10.
3.3.5. General Procedure for Preparing Compounds 10b–e and 10g–j
To a solution of carboxylate 17 (0.144 mmol) in CH3CN (1.45 mL) was added 4M HCl in dioxane (1.44 mmol) at 0 °C. The reaction mixture was allowed to stir at the same temperature for 1 h. After completion of the reaction (monitored by TLC), the mixture was diluted with saturated aqueous NaHCO3 and extracted with EtOAc. The organic layers were dried over anhydrous MgSO4 and concentrated in vacuo. The resulting residue was purified by flash column chromatography on silica gel (DCM/MeOH = 10:1) to afford compound 10.
2-(5-fluoro-4-((R)-1-(3-fluorophenyl)ethoxy)pyrimidin-2-yl)-2,7-diazaspiro[4.4]nonane (10b): Yield: 66%; 1H-NMR (400 MHz, CD3OD, a mixture of rotamers) δ 8.00 (d, J = 3.4 Hz, 1H), 7.35–7.30 (m, 1H), 7.21 (d, J = 7.7 Hz, 1H), 7.13 (d, J = 9.8 Hz, 1H), 6.97 (td, J = 1.7, 8.4 Hz, 1 H), 6.23–6.16 (m, 1H), 3.59–3.37 (m, 6H), 3.30–3.23 (m, 2H), 2.11–1.98 (m, 4H), 1.64 (d, J = 6.6 Hz, 3H). 13C-NMR (100 MHz, CD3OD, a mixture of rotamers) δ 162.9 (d, 1J = 243 Hz), 158.3 (d, 2J = 12 Hz), 154.39, 144.7 (d, 3J = 9 Hz), 140.0 (d, 2J = 21 Hz), 139.8 (d, 1J = 246 Hz), 130.1 (d, 3J = 8 Hz), 121.5 (d, 4J = 3 Hz), 114.3 (d, 2J = 21 Hz), 112.5 (d, 2J = 22 Hz), 75.0, 74.9, 55.3, 55.5, 46.0, 44.8, 44.7, 34.3, 34.2, 33.8, 33.7, 21.5, 21.5. LRMS-EI (m/z): [M+H]+ calcd for C19H23F2N4O: 361.18, found: 361.10.
(3aR,6aS)-2-(5-fluoro-4-((R)-1-(3-fluorophenyl)ethoxy)pyrimidin-2-yl)octahydropyrrolo[3,4-c]pyrrole (10c): Yield: 92%; 1H-NMR (400 MHz, CD3OD, a mixture of rotamers) δ 8.26 (d, J = 4.4 Hz, 1H), 7.44–7.38 (m, 1H), 7.32 (d, J = 7.7 Hz, 1H), 7.24 (dd, J = 1.9, 9.7 Hz, 1H), 7.06 (td, J = 2.3, 8.4 Hz, 1H), 3.91 (bs, 1H), 3.80 (bs, 2H), 3.68–3.58 (m, 3H), 3.37–3.28 (m, 4H), 1.75 (d, J = 6.5 Hz, 3H). 13C-NMR (100 MHz, CD3OD, a mixture of rotamers) δ 162.9 (d, 1J = 244 Hz), 157.8 (d, 2J = 12 Hz), 152.12, 149.9, 143.9 (d, 3J = 7 Hz), 139.6 (d, 1J = 248 Hz), 135.6 (d, 2J = 26 Hz), 130.3 (d, 3J = 8 Hz), 121.7 (d, 4J = 3 Hz), 114.6 (d, 2J = 21 Hz), 112.6 (d, 2J = 22 Hz), 79.6, 50.9, 50.8, 50.7, 49.6, 49.4, 41.6, 41.5, 21.3. LRMS-EI (m/z): [M+H]+ calcd for C18H21F2N4O: 347.17, found: 347.05.
5-fluoro-4-((R)-1-(3-fluorophenyl)ethoxy)-N-((R)-pyrrolidin-3-yl)pyrimidin-2-amine (10d): Yield: 71%; 1H-NMR (400 MHz, CD3OD, a mixture of rotamers) δ 7.97 (d, J = 3.1 Hz, 1H), 7.36–7.29 (m, 1H), 7.18 (d, J = 7.7 Hz, 1H), 7.10 (d, J = 9.8 Hz, 1H), 6.96 (td, J = 2.3, 8.5 Hz, 1H), 6.17 (q, J = 6.5 Hz, 1H), 4.38–4.33 (m, 1H), 3.49–3.30 (m, 1H), 3.26–3.25 (m, 3H), 2.30–2.14 (m, 1H), 2.09–1.93 (m, 1 H), 1.61 (d, J = 6.6 Hz, 3H). 13C-NMR (100 MHz, CD3OD, a mixture of rotamers) δ 163.0 (d, 1J = 243 Hz), 158.0 (d, 2J = 12 Hz), 156.9, 144.9 (d, 3J = 7 Hz), 142.2 (d, 2J = 17 Hz), 140.6 (d, 1J = 247 Hz), 130.2 (d, 3J = 8 Hz), 121.2 (d, 4J = 3 Hz), 114.1 (d, 2J = 21 Hz), 112.1 (d, 2J = 22 Hz), 74.1, 51.0, 50.3, 50.1, 44.1, 29.7, 29.5, 21.74, 21.66. LRMS-EI (m/z): [M+H]+ calcd for C16H19F2N4O: 321.15, found: 321.05.
5-fluoro-4-((R)-1-(3-fluorophenyl)ethoxy)-N-((S)-pyrrolidin-3-yl)pyrimidin-2-amine (10e): Yield: 42%; 1H-NMR (400 MHz, CD3OD, a mixture of rotamers) δ 7.93–7.92 (m, 1H), 7.34–7.28 (m, 1H), 7.17 (d, J = 7.7 Hz, 1H), 7.09 (d, J = 9.9 Hz, 1H), 6.97–6.92 (m, 1H), 6.15 (q, J = 6.5 Hz, 1H), 4.34–4.29 (m, 1H), 3.49–3.38 (m, 2H), 3.35–3.25 (m, 2H), 2.98–2.11 (m, 1H), 2.10–1.89 (m, 1 H), 1.59 (d, J = 6.6 Hz, 3H). 13C-NMR (100 MHz, CD3OD, a mixture of rotamers) δ 162.9 (d, 1J = 243 Hz), 157.7 (d, 2J = 11 Hz), 157.3 (d, 4J = 3 Hz), 145.0 (d, 3J = 7 Hz), 143.0 (d, 2J = 20 Hz), 140.7 (d, 1J = 246 Hz), 130.1 (d, 3J = 8 Hz), 121.3 (d, 4J = 3 Hz), 114.1 (d, 2J = 21 Hz), 112.1 (d, 2J = 22 Hz), 73.8, 51.0, 50.2, 44.1, 29.7, 29.6, 21.8, 21.7. LRMS-EI (m/z): [M+H]+ calcd for C16H19F2N4O: 321.15, found: 321.05.
2-(5-fluoro-4-((R)-1-(4-fluorophenyl)ethoxy)pyrimidin-2-yl)-2,7-diazaspiro[4.4]nonane (10g): Yield: 58%; 1H-NMR (400 MHz, CD3OD, a mixture of rotamers) δ 7.90 (d, J = 3.3 Hz, 1H), 7.44–7.40 (m, 2H), 7.03 (t, J = 8.8 Hz, 2H), 6.22–6.15 (m, 1H), 3.59–3.35 (m, 6H), 3.30–3.19 (m, 2H), 2.09–1.96 (m, 4H), 1.61 (d, J = 6.6 Hz, 3H). 13C-NMR (100 MHz, CD3OD, a mixture of rotamers) δ 162.3 (d, 1J = 243 Hz), 157.5 (d, 2J = 11 Hz), 155.9, 142.7 (d, 2J = 20 Hz), 140.0 (d, 1J = 245 Hz), 138.3 (d, 4J = 7 Hz), 127.8 (d, 3J = 8 Hz), 114.8 (d, 2J = 21 Hz), 74.03, 73.97, 55.2, 52.7, 45.7, 44.8, 34.4, 34.3, 33.9, 33.8, 21.7, 21.6. LRMS-EI (m/z): [M+H]+ calcd for C19H23F2N4O: 361.18, found: 361.10.
(3aR,6aS)-2-(5-fluoro-4-((R)-1-(4-fluorophenyl)ethoxy)pyrimidin-2-yl)octahydropyrrolo[3,4-c]pyrrole (10h): Yield: 53%; 1H-NMR (400 MHz, CD3OD, a mixture of rotamers) δ 7.96 (d, J = 2.8 Hz, 1H), 7.27 (bs, 2H), 6.86 (t, J = 8.0 Hz, 1H), 6.11 (bs, 1H), 3.66–3.54 (m, 3H), 3.43–3.36 (m, 3H), 3.07–3.03 (m, 4H), 1.48 (d, J = 3.0 Hz, 3H). 13C-NMR (100 MHz, CD3OD, a mixture of rotamers) δ 162.8 (d, 1J = 244 Hz), 161.1 (d, 2J = 12 Hz), 150.0, 139.3 (d, 1J = 250 Hz), 136.4 (d, 4J = 3 Hz), 131.6 (d, 2J = 20 Hz), 128.4 (d, 3J = 8 Hz), 115.2 (d, 2J = 21 Hz), 78.2, 51.4, 51.2, 49.5, 41.7, 41.6, 21.2. LRMS-EI (m/z): [M+H]+ calcd for C18H21F2N4O: 347.17, found: 347.05.
5-fluoro-4-((R)-1-(4-fluorophenyl)ethoxy)-N-((R)-pyrrolidin-3-yl)pyrimidin-2-amine (10i): Yield: 51%; 1H-NMR (400 MHz, CD3OD, a mixture of rotamers) δ 7.93 (d, J = 3.3 Hz, 1H), 7.49–7.45 (m, 2H), 7.10 (t, J = 8.7 Hz, 2H), 6.25–6.22 (m, 1H), 4.31–4.30 (m, 1H), 3.56–3.40 (m, 3H), 3.15–3.06 (m, 1H), 2.23–2.16 (m, 1H), 1.94–1.90 (m, 1 H), 1.66 (d, J = 6.6 Hz, 3H). LRMS-EI (m/z): [M+H]+ calcd for C16H19F2N4O: 321.15, found: 321.00.
5-Fluoro-4-((R)-1-(4-fluorophenyl)ethoxy)-N-((S)-pyrrolidin-3-yl)pyrimidin-2-amine (10j): Yield: 27%; 1H-NMR (400 MHz, CD3OD, a mixture of rotamers) δ 7.92–7.91 (m, 1H), 7.42–7.39 (m, 2H), 7.06–7.01 (m, 2H), 6.18 (td, J = 4.8, 6.5 Hz, 1H), 4.39–4.30 (m, 1H), 3.50–3.39 (m, 2H), 3.36–3.28 (m, 2H), 2.39–2.14 (m, 1H), 2.10–1.90 (m, 1 H), 1.59 (d, J = 6.6 Hz, 3H). 13C-NMR (100 MHz, CD3OD, a mixture of rotamers) δ 162.3 (d, 1J = 243 Hz), 157.8 (d, 2J = 11 Hz), 157.3 (d, 4J = 3 Hz), 143.0 (d, 2J = 20 Hz), 140.8 (d, 1J = 233 Hz), 138.1, 127.7 (d, 3J = 8 Hz), 114.9 (d, 2J = 22 Hz), 73.9, 51.0, 50.4, 50.3, 44.2, 29.7, 29.6, 21.8, 21.7. LRMS-EI (m/z): [M+H]+ calcd for C16H19F2N4O: 321.15, found: 321.05.
(R)-4-(5-Fluoro-4-(3-fluorophenoxy)pyrimidin-2-yl)-3-methylpiperazin-1-ium·hydrochloride (20a): Yield: 58%; 1H-NMR (400MHz, CDCl3) δ 8.13 (s, 1H), 7.37–7.31 (m, 1H), 7.00–6.93 (m, 3H), 4.39–4.38 (m, 1H), 4.07–4.03 (m, 1H), 2.98–2.86 (m, 3H), 2.80–2.77 (m, 1H), 2.70–2.62 (m, 1H), 1.12 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CD3OD) δ 164.4 (d, 1J = 245 Hz), 158.7 (d, 2J = 11 Hz), 157.2 (d, 3J = 8 Hz), 154.4 (d, 3J = 11 Hz), 146.7 (d, 3J = 20 Hz), 141.4 (d, 1J = 248 Hz), 131.7 (d, 3J = 9 Hz), 118.8 (d, 4J = 3 Hz), 113.7 (d, 2J = 21 Hz), 110.6 (d, 2J = 24 Hz), 45.7, 44.3, 36.6, 13.5. LRMS-EI (m/z): [M-Cl]+ calcd for C15H17F2N4O: 306.32, found: 306.32.
(R)-4-(5-Fluoro-4-(4-fluorophenoxy)pyrimidin-2-yl)-3-methylpiperazin-1-ium·hydrochloride (20b): Yield: 65%; 1H-NMR (400MHz, CDCl3): δ 8.08 (d, J = 2.6 Hz, 1H), 7.13 (dd, J = 9.0, 4.5 Hz, 2H), 7.09–7.03 (m, 2H), 4.50–4.30 (s, 1H), 4.03–4.00 (m, 1H), 2.96–2.83 (m, 3H), 2.77–2.74 (m, 1H), 2.68–2.60 (m, 1H), 1.09 (d, J = 6.6 Hz, 3H). 13C-NMR (100 MHz, CD3OD): δ 161.6 (d, 1J = 241 Hz), 159.2 (d, 2J = 11 Hz), 149.3 (d, 4J = 22 Hz), 146.4 (d, 2J = 20 Hz), 141.5 (d, 1J = 248 Hz), 124.5 (d, 3J = 8 Hz), 117.1 (d, 2J = 23 Hz), 48.0, 45.6, 44.2, 36.5, 13.5. LRMS-EI (m/z): [M-Cl]+ calcd for C15H17F2N4O: 306.32, found: 306.32
3.4. Molecular Docking Study
Discovery studio 2019 software were used for modeling study [34]. Crystal structures of 5-HT2B (PDB ID = 4IB4) and 5-HT2C (PDB ID= 6BQH) proteins were downloaded from protein data bank. Ligand 10a and 8 were prepared at 7.4 pH. Proteins and ligands were prepared and minimized using CHARm forcefield. Binding sites were created over co-crystal ligands binding position. Ligands were docked using CDOCKER scoring function. Twenty binding poses were generated for each ligands. Obtained docked poses were analyzed based on docking scores and binding site interactions. Images were rendered using PyMOL software (www.pymol.org).
3.5. In Vitro Assay
3.5.1. Serotonin Receptor Cell-Based Functional Assays
The synthesized compounds were tested in agonist mode with the 5-HT2C receptor assay using recombinant human HEK-293 cells serviced by either Eurofins Cerep or DGMIF (Daegu-Gyeongbuk Medical Innovation Foundation). All the experiments were duplicated. The detailed assay protocols refer to the literature procedure [35].
3.5.2. Serotonin Receptor Binding Affinity Assays
Eleven dilutions (5 x assay concentration) of the test and reference compounds (Table S1) were prepared in standard binding buffer (50 mM tris(hydroxymethyl)-aminomethane–HCl (Tris-HCl), 10 mM MgCl2, 1 mM ethylenediaminetetraacetate (EDTA), pH 7.4) by serial dilution: 0.05 nM, 0.5 nM, 1.5 nM, 5 nM, 15 nM, 50 nM, 150 nM, 500 nM, 1.5 μM, 5 μM, and 50 μM. The radioligand (Table S1) was diluted to five times the assay concentration in standard binding buffer. Aliquots (50 μL) of the radioligand were dispensed into the wells of a 96-well plate containing 100 μL of standard binding buffer. Triplicate aliquots (50 μL) of the test and reference compound dilutions were then added. Finally, crude membrane fractions (50 μL) of cells (HEK293 or CHO) expressing human recombinant receptors were dispensed into each well. A total 250 μL of the reaction mixtures was incubated at room temperature and shielded from light for 1.5 h, then harvested by rapid filtration onto Whatman GF/B glass fiber filters presoaked with 0.3% polyethyleneimine, by using a 96-well Brandel harvester.
Four rapid washes were performed with chilled standard binding buffer (500 μL) to decrease nonspecific binding. Filters were placed in 6 mL scintillation tubes and allowed to dry overnight. The next day, 4 ml of EcoScint scintillation cocktail (National Diagnostics) was added to each tube. The tubes were capped, labeled, and counted by liquid scintillation counting. The filter mats were dried, and the scintillant was melted onto the filters, then the radioactivity retained on the filters was counted in a Microbeta scintillation counter. The IC50 values were obtained by using the Prism 4.0 program (GraphPad Software, San Diego, CA) and converted into Ki values. Each compound was tested in triplicate at least.
3.6. Drug-Like Properties
3.6.1. Plasma Stability
Human plasma in each culture tube, treated with test compound (10 μM), was incubated 37 °C for 0, 30, and 120 min, respectively. In a determined period of time, an internal standard solution of chlorpropamide in acetonitrile was added into each culture tube, which was shaken with a vortex mixer for 5 min and centrifuged for additional 5 min (14,000 rpm, 4 °C). The supernatant was then injected into thr LC-MS/MS system to analyze the plasma stability of compound 10a.
3.6.2. Microsomal Stability
To human liver microsomes (0.5 mg/mL) were added 0.1 M phosphate buffer (pH 7.4) and tested compounds (1 μM). After incubation at 37 °C for 5 min, NADPH generation system solution was also added and incubated at 37 °C for 30 min again. To terminate reaction, acetonitrile including internal standard (chlorpropamide) was added, and the solution was centrifuged for 5 min (14,000 rpm, 4 °C). The supernatant was then injected into the LC-MS/MS system to analyze the microsomal stability of compound 10a.
3.6.3. CYP Inhibition
To human liver microsomes (0.25 mg/mL), 0.1 M phosphate buffer (pH 7.4), a cocktail of five probe substrates (Phenacetin 50 μM, Diclofenac 10 μM, S-mephenytoin 100 μM, Dextromethorphan 5 μM, and Midazolam 2.5 μM), and tested compounds were added at concentrations of 0 μM (as a control) and 10 μM. After incubation at 37 °C for 5 min, NADPH generation system solution was also added and incubated at 37 °C for 15 min again. To terminate the reaction, acetonitrile including internal standard (Terfenadine) was added, and the solution was centrifuged for 5 min (14,000 rpm, 4 °C). The supernatant was then injected into the LC-MS/MS system to simultaneously analyze the metabolites of the probe substrates and evaluate the % CYP inhibition of the tested compound 10a.
4. Conclusions
Thus, we synthesized two series of disubstituted pyrimidine derivatives as potential 5-HT2C selective agonists. Initially, a cell-based assay of 2,5-disubstituted pyrimidines revealed that most compounds did not have good agonistic effects towards the 5-HT2C receptor, although 2-piperazinyl-5-(3-fluorobenzyloxy)pyrimidine 9b showed effective biological activity, with 61% activation. In vitro evaluation of the second series of pyrimidines, possessing different cyclic amines, demonstrated that four compounds showed greater than 50% activation against 5-HT2C in the primary cell-based assay. In the secondary binding affinity assay, compounds 10a and 10f, with 1,4-diazepane at the 2-position of pyrimidine, were identified as the most potent 5-HT2C ligands, with excellent Ki values of 7.9 nM and 19.0 nM, respectively. Compound 10a showed high selectivity profiles for other 5-HT receptor subtypes, with a greater than 10-fold difference in potency. Additional in vitro stability experiments indicated that compound 10a has excellent plasma and microsomal stability, along with low CYP inhibition. Based on these results, 2,4-disubstituted pyrimidine 10a could be considered a potential lead compound as a 5-HT2C selective agonist for further application in chemical probes such as a 5-HT2C PET radiotracer.
Supplementary Materials
The following are available online at https://www.mdpi.com/1420-3049/24/18/3234/s1, (NMR spectral data of compounds 9a–9r, 10a–10j, and 20a/b; Table S1: A list of 5-HT receptor radioligands and reference compounds for binding assay).
Author Contributions
S.-J.M. initiated the project and designed the experiments; J.K., and Y.J.K. performed the experiments; S.-J.M. and J.K. analyzed the data; H.C. and H.J.K. contributed to analyze drug-like properties; A.M.L. and A.N.P. performed molecular docking studies; S.-J.M., J.K., and Y.J.K. wrote the paper.
Acknowledgments
This work was financially supported by the Korea Health Industry Development Institute (KHIDI, HI16C1677, HI17C1037), the National Research Foundation of Korea (NRF-2019R1A2C1008186) and the research fund of Hanyang University (HY-2015-N). Binding affinity data were generously provided by the US National Institute of Mental Health (NIMH) Psychoactive Drug Screening Program (HHSN-271-2008-00025-C).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brummelte, S.; MC Glanaghy, E.; Bonnin, A.; Oberlander, F. Developmental changes in serotonin signaling: Implications for early brain function, behavior and adaptation. Neuroscience 2017, 342, 212–231. [Google Scholar] [CrossRef]
- Amato, D. Serotonin in antipsychotic drugs action. Behav. Brain Res. 2015, 277, 125–135. [Google Scholar] [CrossRef]
- Meltzer, H.Y.; Li, Z.; Kaneda, Y.; Ichikawa, J. Serotonin receptors: Their key role in drugs to treat schizophrenia. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2003, 27, 1159–1172. [Google Scholar] [CrossRef]
- Hoyer, D.; Clarke, D.E.; Fozard, J.R.; Hartig, P.R.; Martin, G.R.; Mylecharane, E.J.; Saxena, P.R.; Humphrey, P.P.A. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin). Pharmacol. Rev. 1994, 46, 157–203. [Google Scholar]
- McCorvy, J.D.; Roth, B.L. Structure and function of serotonin G protein-coupled receptors. Pharmacol. Ther. 2015, 150, 129–142. [Google Scholar] [CrossRef] [Green Version]
- Chou-Green, J.M.; Holscher, T.D.; Dallman, M.F.; Akana, S.F. Compulsive behavior in the 5-HT2C receptor knockout mouse. Physiol. Behav. 2003, 78, 641–649. [Google Scholar] [CrossRef]
- Lee, M.A.; Jayathilake, K.; Sim, M.Y.; Meltzer, H.Y. Decreased serotonin2C receptor responses in male patients with schizophrenia. Psychiatry Res. 2015, 226, 308–315. [Google Scholar] [CrossRef]
- Chagraoui, A.; Thibaut, F.; Skiba, M.; Thuillez, C.; Bourin, M. 5-HT2C receptors in psychiatric disorders: A review. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2016, 66, 120–135. [Google Scholar] [CrossRef]
- Barnes, N.M.; Sharp, T.A. Review of central 5-HT receptors and their function. Neuropharmacology 1999, 38, 1083–1152. [Google Scholar] [CrossRef]
- Berg, K.A.; Stout, B.D.; Cropper, J.D.; Maayani, S.; Clarke, W.P. Novel Actions of Inverse Agonists on 5-HT2C Receptor Systems. Mol. Pharmacol. 1999, 55, 863–872. [Google Scholar]
- Thomsen, W.J.; Grottick, A.J.; Menzaghi, F.; Reyes-Saldana, H.; Espitia, S.; Yuskin, D.; Whelan, K.; Martin, M.; Morgan, M.; Chen, W.; et al. Lorcaserin, a Novel Selective Human 5-Hydroxytryptamine2C Agonist: In Vitro and in Vivo Pharmacological Characterization. JPET 2008, 325, 577–587. [Google Scholar] [CrossRef]
- Dunlop, J.; Watts, S.W.; Barrett, J.E.; Coupet, J.; Harrison, B.; Mazandarani, H.; Nawoschik, S.; Pangalos, M.N.; Ramamoorthy, S.; Schechter, L.; et al. Characterization of Vabicaserin (SCA-136), a Selective 5-Hydroxytryptamine 2C Receptor Agonist. JPET 2011, 337, 673–680. [Google Scholar] [CrossRef]
- Jensen, A.A.; Plath, N.; Pedersen, M.H.F.; Isberg, V.; Krall, J.; Wellendorph, P.; Stensboel, T.B.; Gloriam, D.E.; Krogsgaard-Larsen, P.; Froelund, B. Design, Synthesis, and Pharmacological Characterization of N- and O-Substituted 5,6,7,8-Tetrahydro-4H-isoxazolo[4,5-d]azepin-3-ol Analogues: Novel 5-HT2A/5-HT2C Receptor Agonists with Pro-Cognitive Properties. J. Med. Chem. 2013, 56, 1211–1227. [Google Scholar] [CrossRef]
- Cheng, J.; Giguere, P.M.; Lv, W.; Roth, B.L.; Kozikowski, A.P. Design and synthesis of (2-(5-chloro-2,2-dimethyl-2,3-dihydrobenzofuran-7-yl)cyclopropyl)methanamine as a selective serotonin 2C agonist. Tetrahedron Lett. 2015, 56, 3420–3422. [Google Scholar] [CrossRef] [Green Version]
- Siuciak, J.A.; Chapin, D.S.; McCarthy, S.A.; Guanowsky, V.; Brown, J.; Chiang, P.; Marala, R.; Patterson, T.; Seymour, P.A.; Swick, A.; et al. CP-809,101, a selective 5-HT2C agonist, shows activity in animal models of antipsychotic activity. Neuropharmacology 2007, 52, 279–290. [Google Scholar] [CrossRef]
- Cheng, J.; Giguere, P.M.; Onajole, O.K.; Lv, W.; Gaisin, A.; Gunosewoyo, H.; Schmerberg, C.M.; Pogorelov, V.M.; Rodriguiz, R.M.; Vistoli, G.; et al. Optimization of 2-Phenylcyclopropylmethylamines as Selective Serotonin 2C Receptor Agonists and Their Evaluation as Potential Antipsychotic Agents. J. Med. Chem. 2015, 58, 1992–2002. [Google Scholar] [CrossRef] [Green Version]
- Rouquet, G.; Moore, D.E.; Spain, M.; Allwood, D.M.; Battilocchio, C.; Blakemore, D.C.; Fish, P.V.; Jenkinson, S.; Jessiman, A.S.; Ley, S.V.; et al. Design, Synthesis, and Evaluation of Tetra substituted Pyridines as Potent 5-HT2C Receptor Agonists. ACS Med. Chem. Lett. 2015, 6, 329–333. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Cho, S.J.; Jensen, N.H.; Allen, J.A.; Svennebring, A.M.; Roth, B.L. HTS and Rational Drug Design to Generate a Class of 5-HT2C-Selective Ligands for Possible Use in Schizophrenia. ChemMedChem 2010, 5, 1221–1225. [Google Scholar] [CrossRef]
- Green, M.P.; McMurray, G.; Storer, R.I. Selective 5-HT2C receptor agonists: Design and synthesis of pyridazine-fused azepines. Bioorg. Med. Chem. Lett. 2016, 26, 4117–4121. [Google Scholar] [CrossRef]
- Granda, M.L.; Carlin, S.M.; Moseley, C.K.; Neelamegam, R.; Mandeville, J.B.; Hooker, J.M. Synthesis and Evaluation of Methylated Arylazepine Compounds for PET Imaging of 5-HT2c Receptors. ACS Chem. Neurosci. 2013, 4, 261–265. [Google Scholar] [CrossRef]
- Kamlet, A.S.; Neumann, C.N.; Lee, E.; Carlin, S.M.; Moseley, C.K.; Stephenson, N.; Hooker, J.M.; Ritter, T. Application of palladium-mediated 18F-fluorination to PET radiotracer development: Overcoming hurdles to translation. PLoS ONE 2013. [Google Scholar] [CrossRef]
- Kim, J.; Jo, H.; Lee, H.; Choo, H.; Kim, H.-J.; Pae, A.N.; Cho, Y.S.; Min, S.-J. Identification of Optically Active Pyrimidine Derivatives as Selective 5-HT2C Modulators. Molecules 2017, 22, 1416. [Google Scholar] [CrossRef]
- Kim, j.; Moon, B.S.; Lee, B.C.; Lee, H.Y.; Kim, H.-J.; Choo, H.; Pae, A.N.; Cho, Y.S.; Min, S.-J. A Potential PET Radiotracer for the 5-HT2C Receptor: Synthesis and in Vivo Evaluation of 4-(3-[18F]fluorophenethoxy) pyrimidine. ACS Chem. Neurosci. 2017, 8, 996–1003. [Google Scholar] [CrossRef]
- Prabhakaran, J.; Underwood, M.D.; Kumar, J.S.D.; Simpson, N.R.; Kassir, S.A.; Bakalian, M.J.; Mann, J.J.; Arango, V. Synthesis and in vitro evaluation of [18F]FECIMBI-36: A potential agonist PET ligand for 5-HT2A/2C receptors. Bioorg. Med. Chem. Lett. 2015, 25, 3933–3936. [Google Scholar] [CrossRef]
- Neelamegam, R.; Hellenbrand, T.; Schroeder, F.K.A.; Wang, C.; Hooker, J.M. Imaging Evaluation of 5HT2C Agonists, [11C]WAY-163909 and [11C]Vabicaserin, Formed by Pictet−Spengler Cyclization. J. Med. Chem. 2014, 57, 1488–1494. [Google Scholar] [CrossRef]
- Zeng, F.; Nye, J.A.; Voll, R.J.; Howell, L.; Goodman, M.M. Synthesis and Evaluation of Pyridyloxypyridyl Indole Carboxamides as Potential PET Imaging Agents for 5-HT2C Receptors. ACS Med. Chem. Lett. 2018, 9, 188–192. [Google Scholar] [CrossRef]
- Heifetz, A.; Storer, R.I.; McMurray, G.; James, T.; Morao, I.; Aldeghi, M.; Bodkin, M.J.; Biggin, P.C. Application of an Integrated GPCR SAR-Modeling Platform To Explain the Activation Selectivity of Human 5-HT2C over 5-HT2B. ACS Chem. Biol. 2016, 11, 1372–1382. [Google Scholar] [CrossRef]
- Peng, Y.; McCorvy, J.D.; Harpsoe, K.; Lansu, K.; Yuan, S.; Popov, P.; Qu, L.; Pu, M.; Che, T.; Nikolajsen, L.F.; et al. 5-HT2C Receptor Structures Reveal the Structural Basis of GPCR Polypharmacology. Cell 2018, 172, 719–730. [Google Scholar] [CrossRef]
- Maiti, D.; Buchwald, S.L. Cu-Catalyzed Arylation of Phenols: Synthesis of Sterically Hindered and Heteroaryl Diaryl Ethers. J. Org. Chem. 2010, 75, 1791–1794. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Chen, X.; Zhou, W.; Fan, M.; Lai, Y.; Ma, D. Copper-Catalyzed Diaryl Ether Formation from (Hetero)aryl Halides at Low Catalytic Loadings. J. Org. Chem. 2017, 82, 4964–4969. [Google Scholar] [CrossRef]
- Wolter, M.; Nordmann, G.; Job, G.E.; Buchwald, S.L. Copper-Catalyzed Coupling of Aryl Iodides with Aliphatic Alcohols. Org. Lett. 2002, 4, 973–976. [Google Scholar] [CrossRef]
- Kim, J.; Cho, Y.S.; Min, S.-J. Facile synthesis of 2-amino-4-alkoxypyrimidines via consecutive nucleophilic aromatic substitution (SNAr) reactions. Bull. Korean Chem. Soc. 2016, 37, 1998–2008. [Google Scholar] [CrossRef]
- Wacker, D.; Wang, C.; Katritch, V.; Han, G.W.; Huang, X.P.; Vardy, E.; McCorvy, J.D.; Jiang, Y.; Chu, M.; Siu, F.Y.; et al. Structural features for functional selectivity at serotonin receptors. Science 2013, 340, 615–619. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment, Release 2019; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
- Porter, R.H.P.; Benwell, K.R.; Lamb, H.; Malcolm., C.S.; Allen, N.H.; Revell, D.F.; Adams, D.R.; Sheardown, M.J. Functional characterization of agonists at recombinant human 5-HT2A, 5-HT2B and 5-HT2C receptors in CHO-K1 cells. Brit. J. Pharmacol. 1999, 128, 13–20. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 9a–9r, 10a–10j, and 20a/b are available from the authors. |
Figure 1.
The representative 5-HT2C selective agonists and PET radioligands.

Figure 2.
Design of new pyrimidine derivatives as 5-HT2C agonists.
Scheme 1.
Synthesis of 2,5-disubstituted pyrimidine derivatives 9a–r. Reagents and conditions: (a) N-Boc-protected amines, K2CO3, CH3CN, 80 °C, 88–97%; (b) 3- or 4-fluorophenol/benzyl alcohol/phenethyl alcohol, CuI or CuO2, 14 or 15, Cs2CO3, toluene, 110 °C, 42–85%; (c) TFA, CH2Cl2, 0 °C, 52–67%.
Scheme 1.
Synthesis of 2,5-disubstituted pyrimidine derivatives 9a–r. Reagents and conditions: (a) N-Boc-protected amines, K2CO3, CH3CN, 80 °C, 88–97%; (b) 3- or 4-fluorophenol/benzyl alcohol/phenethyl alcohol, CuI or CuO2, 14 or 15, Cs2CO3, toluene, 110 °C, 42–85%; (c) TFA, CH2Cl2, 0 °C, 52–67%.
Scheme 2.
Synthesis of 2,4-disubstituted pyrimidine derivatives 10a–j and 20ab. Reagents and conditions: (a) (i) Vinyl acetate (0.4 eq), CAL-B, pyridine, hexane, rt, 66–67%; (ii) 1 M NaOH, MeOH, rt, 78%; (b) 2,4-dichloro-5-fluoropyrimidine, t-BuONa, toluene, 0 °C, 51–93%; (c) amines, Et3N, toluene (0.5 M), 100 °C, 12 h, 61–98%; (d) (i) N-Boc-protected amines, Et3N, DMSO (0.5 M), 100 °C, 12 h, 44–94%; (ii) HCl (4.0 M in dioxane), CH3CN, 0 °C or TFA, CH2Cl2, 0 °C.
Scheme 2.
Synthesis of 2,4-disubstituted pyrimidine derivatives 10a–j and 20ab. Reagents and conditions: (a) (i) Vinyl acetate (0.4 eq), CAL-B, pyridine, hexane, rt, 66–67%; (ii) 1 M NaOH, MeOH, rt, 78%; (b) 2,4-dichloro-5-fluoropyrimidine, t-BuONa, toluene, 0 °C, 51–93%; (c) amines, Et3N, toluene (0.5 M), 100 °C, 12 h, 61–98%; (d) (i) N-Boc-protected amines, Et3N, DMSO (0.5 M), 100 °C, 12 h, 44–94%; (ii) HCl (4.0 M in dioxane), CH3CN, 0 °C or TFA, CH2Cl2, 0 °C.

Figure 3.
Interactions of 10a and 8 inside the 5-HT2C and 5-HT2B receptors. Ligands 10a and 8 are shown in green and magenta color stick format. 5-HT2C (wheat color) and 5-HT2B (green color) protein structures are shown in cartoon format. Hydrogen bond, π-cationic, π–π stacking, and hydrophobic interactions are shown in green, orange, dark pink, and light pink dash line format. (A) Overlap of 10a and 8 inside 5-HT2C protein. (B) Interactions between ligand 10a and the 5-HT2C receptor. (C) Interactions between ligand 8 and the 5-HT2C receptor. (D) Overlap of 10a and 8 inside 5-HT2B protein. (E) Interactions between ligand 10a and the 5-HT2B receptor. (F) Interactions between ligand 8 and the 5-HT2B receptor.
Figure 3.
Interactions of 10a and 8 inside the 5-HT2C and 5-HT2B receptors. Ligands 10a and 8 are shown in green and magenta color stick format. 5-HT2C (wheat color) and 5-HT2B (green color) protein structures are shown in cartoon format. Hydrogen bond, π-cationic, π–π stacking, and hydrophobic interactions are shown in green, orange, dark pink, and light pink dash line format. (A) Overlap of 10a and 8 inside 5-HT2C protein. (B) Interactions between ligand 10a and the 5-HT2C receptor. (C) Interactions between ligand 8 and the 5-HT2C receptor. (D) Overlap of 10a and 8 inside 5-HT2B protein. (E) Interactions between ligand 10a and the 5-HT2B receptor. (F) Interactions between ligand 8 and the 5-HT2B receptor.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
In vitro agonistic activity of 2,5-disubstituted pyrimidine 9a–r against 5-HT2C.![Molecules 24 03234 i001]()

| Entry | Compds | n | R1 | R2 | %activation (10 μM) |
|---|---|---|---|---|---|
| 1 | 9a | 0 | 3-F |  | 7 |
| 2 | 9b | 1 | 3-F | 61 | |
| 3 | 9c | 2 | 3-F | 15 | |
| 4 | 9d | 0 | 4-F | 5 | |
| 5 | 9e | 1 | 4-F | 18 | |
| 6 | 9f | 2 | 4-F | 4 | |
| 7 | 9g | 0 | 3-F |  | 35 |
| 8 | 9h | 1 | 3-F | 48 | |
| 9 | 9i | 2 | 3-F | 40 | |
| 10 | 9j | 0 | 4-F | 26 | |
| 11 | 9k | 1 | 4-F | 48 | |
| 12 | 9l | 2 | 4-F | 32 | |
| 13 | 9m | 0 | 3-F |  | 16 |
| 14 | 9n | 1 | 3-F | −1 | |
| 15 | 9o | 2 | 3-F | 3 | |
| 16 | 9p | 0 | 4-F | 14 | |
| 17 | 9q | 1 | 4-F | 16 | |
| 18 | 9r | 2 | 4-F | −1 | |
| 19 | Lorcaserin (1) | 94a |
a %activation value at 1 μM, EC50 = 14 nM.
Table 2.
In vitro activity of 2,4-disubsitutued pyrimidine against 5-HT2C.![Molecules 24 03234 i002]()

| Entry | Compds | R1 | R2 | %activationa | %binding | Ki (nM) |
|---|---|---|---|---|---|---|
| 1 | 10a |  |  | 81.0 | 81.3 | 7.9 |
| 2 | 10b |  | 78.2 | 47.6 | ND | |
| 3 | 10c | 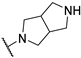 | 29.5 | 73.6 | 295.0 | |
| 4 | 10d | 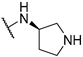 | 16.4 | 88.6 | 119.0 | |
| 5 | 10e |  | 7.7 | 61.4 | 1255.0 | |
| 6 | 10f | 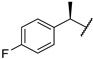 |  | 52.4 | 93.9 | 19.0 |
| 7 | 10g |  | ND | 78.1 | 232.0 | |
| 8 | 10h | 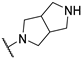 | 10.1 | 50.8 | 466.0 | |
| 9 | 10i | 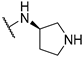 | ND | 85.9 | 120.0 | |
| 10 | 10j | 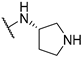 | 0.1 | 26.2 | ND | |
| 11 | 20a | 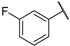 | 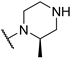 | 56.0 | 64.8 | 660.0 |
| 12 | 20b |  | 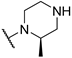 | 34.0 | 54.8 | 1107.0 |
| 13 | 8 | N/Ab | 89.0 | 5.9 |
a Serotonin was used as a reference compound (EC50 = 2.6 nM). b Not available.
Table 3.
Evaluation of binding affinity of 10a and 10f to 5-HT receptor subtypes a.
| 5-HT Subtypes (%Binding at 10 μM/Ki (nM)) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1A | 1B | 1D | 1E | 2A | 2B | 2C | 3 | 5A | 6 | 7 | |
| 10a | 74.1 /578.0 | 56.1 /3237.0 | 71.4 /679.0 | 43.1 /NDb | 75.5 /1284.0 | 98.0 /83.0 | 81.3 /7.9 | 89.0 /323.0 | 22.5 /ND | 50.7 /348.0 | 61.3 /1016.0 |
| SI | 73.2 | 409.7 | 85.9 | - | 162.5 | 10.5 | 1.0 | 40.9 | 23.4 | 44.1 | 128.6 |
| 10f | 67.6 /1066.0 | 19.2 /NDb | 81.3 /937.0 | 31.3 /NDb | 52.9 /2863.0 | 95.0 /118.0 | 98.9 /19.0 | 88.0 /415.0 | 15.2 /ND | 56.5 /467.0 | 55.4 /616.0 |
| SI | 56.1 | - | 49.3 | - | 150.7 | 6.2 | 1.0 | 21.8 | 10.3 | 24.6 | 32.4 |
| 8c | 89.1 /271.0 | 4.9 /NDb | 83.4 /1052.0 | 48.0 /NDb | 85.3 /511.0 | 98.1 /38.0 | 89.0 /5.9 | 83.4 /543.0 | 18.5 /NDb | 56.7 /30.0 | 63.4 /528.0 |
| SI | 45.9 | - | 178.3 | - | 86.6 | 6.4 | 1.0 | 92.0 | - | 5.1 | 89.5 |
a 5-HT receptor binding was determined by competitive binding assay using radioligands and reference compounds in Table S1. b Not determined due to low %binding. c Compound 8 was used as a reference compound. SI, selectivity index.
Table 4.
The results of plasma stability, microsomal stability, and CYP inhibition.
| Compd | Plasma Stability %Remaining @ 10 μM after 0.5 and 2 h | HLM %Remaining @ 1 μM after 0.5 h | CYP Isozymes (%Remaining @ 10 μM) | ||||
|---|---|---|---|---|---|---|---|
| 1A2 | 2D6 | 2C9 | 3A4 | 2C19 | |||
| 10a | 99.9/98.0 | 96.6 | 86.5 | 63.6 | >100 | 86.2 | 86.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, J.; Kim, Y.J.; Londhe, A.M.; Pae, A.N.; Choo, H.; Kim, H.J.; Min, S.-J. Synthesis and Biological Evaluation of Disubstituted Pyrimidines as Selective 5-HT2C Agonists. Molecules 2019, 24, 3234. https://doi.org/10.3390/molecules24183234
AMA Style
Kim J, Kim YJ, Londhe AM, Pae AN, Choo H, Kim HJ, Min S-J. Synthesis and Biological Evaluation of Disubstituted Pyrimidines as Selective 5-HT2C Agonists. Molecules. 2019; 24(18):3234. https://doi.org/10.3390/molecules24183234
Chicago/Turabian StyleKim, Juhyeon, Yoon Jung Kim, Ashwini M. Londhe, Ae Nim Pae, Hyunah Choo, Hak Joong Kim, and Sun-Joon Min. 2019. "Synthesis and Biological Evaluation of Disubstituted Pyrimidines as Selective 5-HT2C Agonists" Molecules 24, no. 18: 3234. https://doi.org/10.3390/molecules24183234