
Biomimetic Non-Heme Iron-Catalyzed Epoxidation of Challenging Terminal Alkenes Using Aqueous H2O2 as an Environmentally Friendly Oxidant

 , and
, and 
Abstract
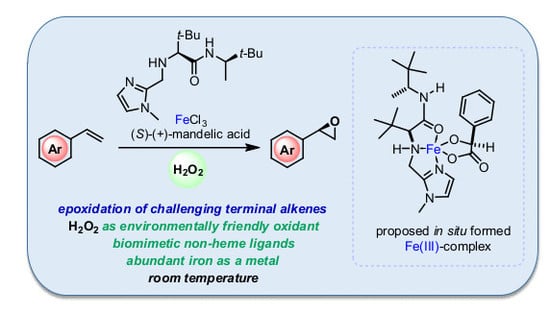
:
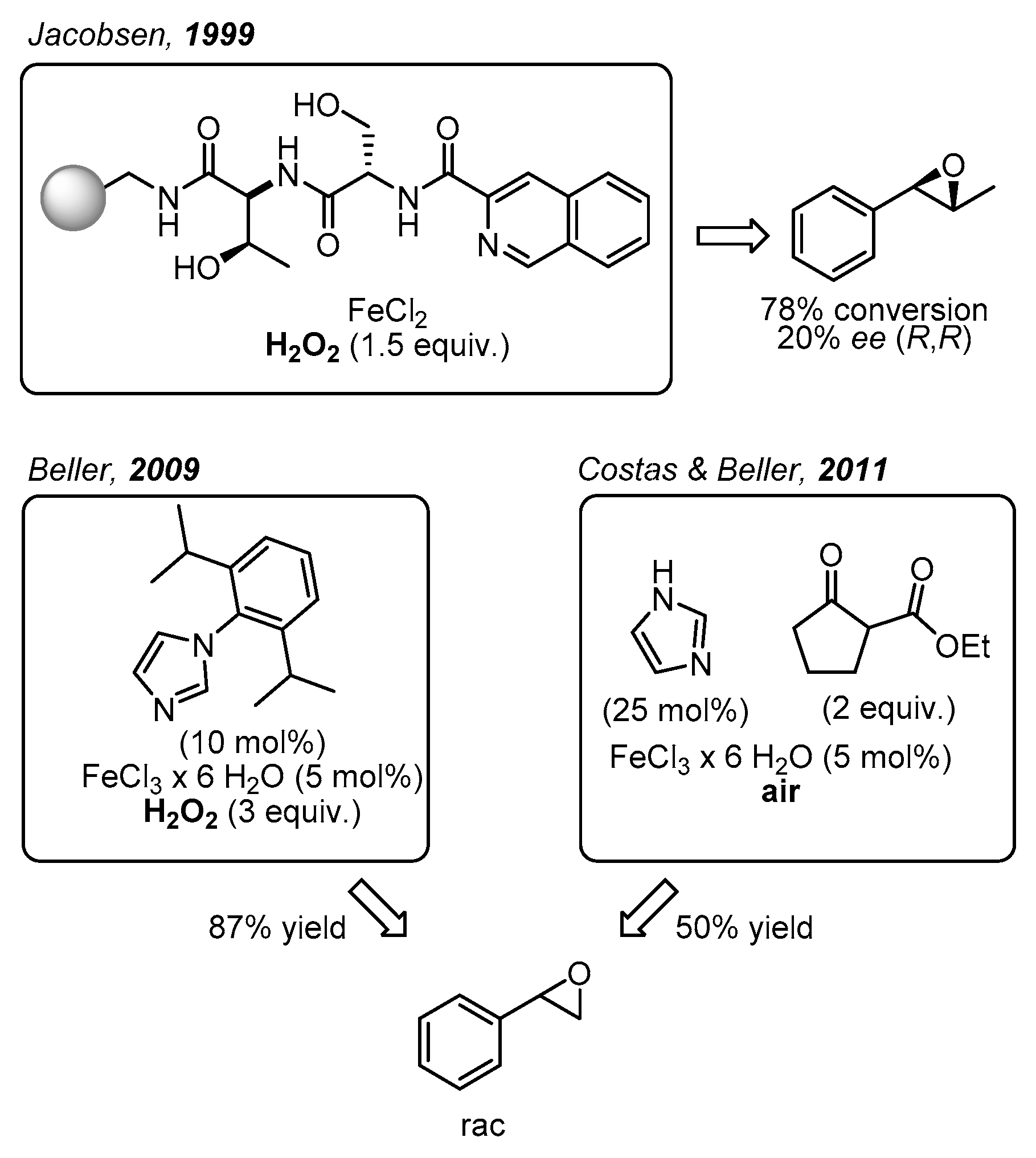
1. Introduction
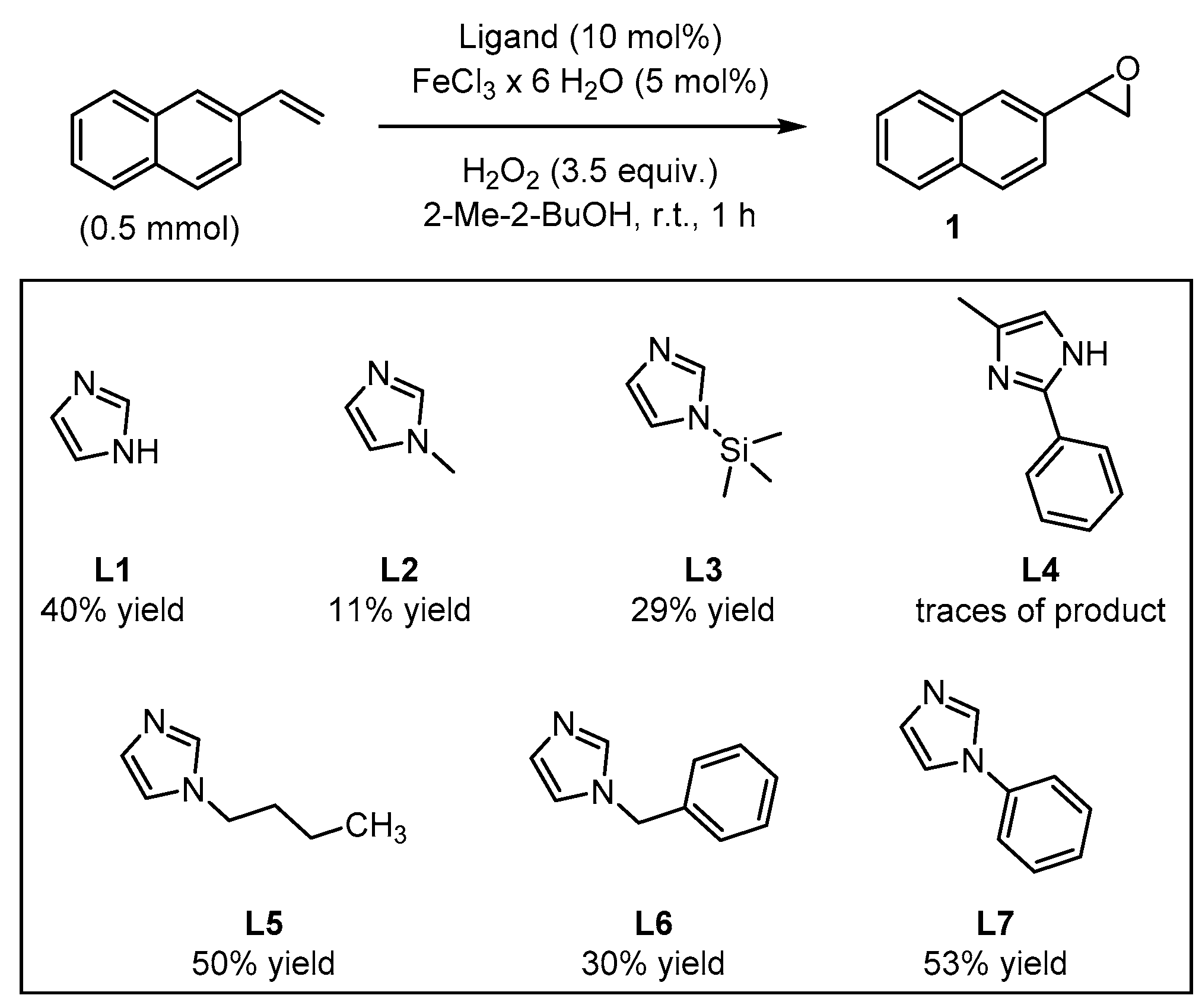
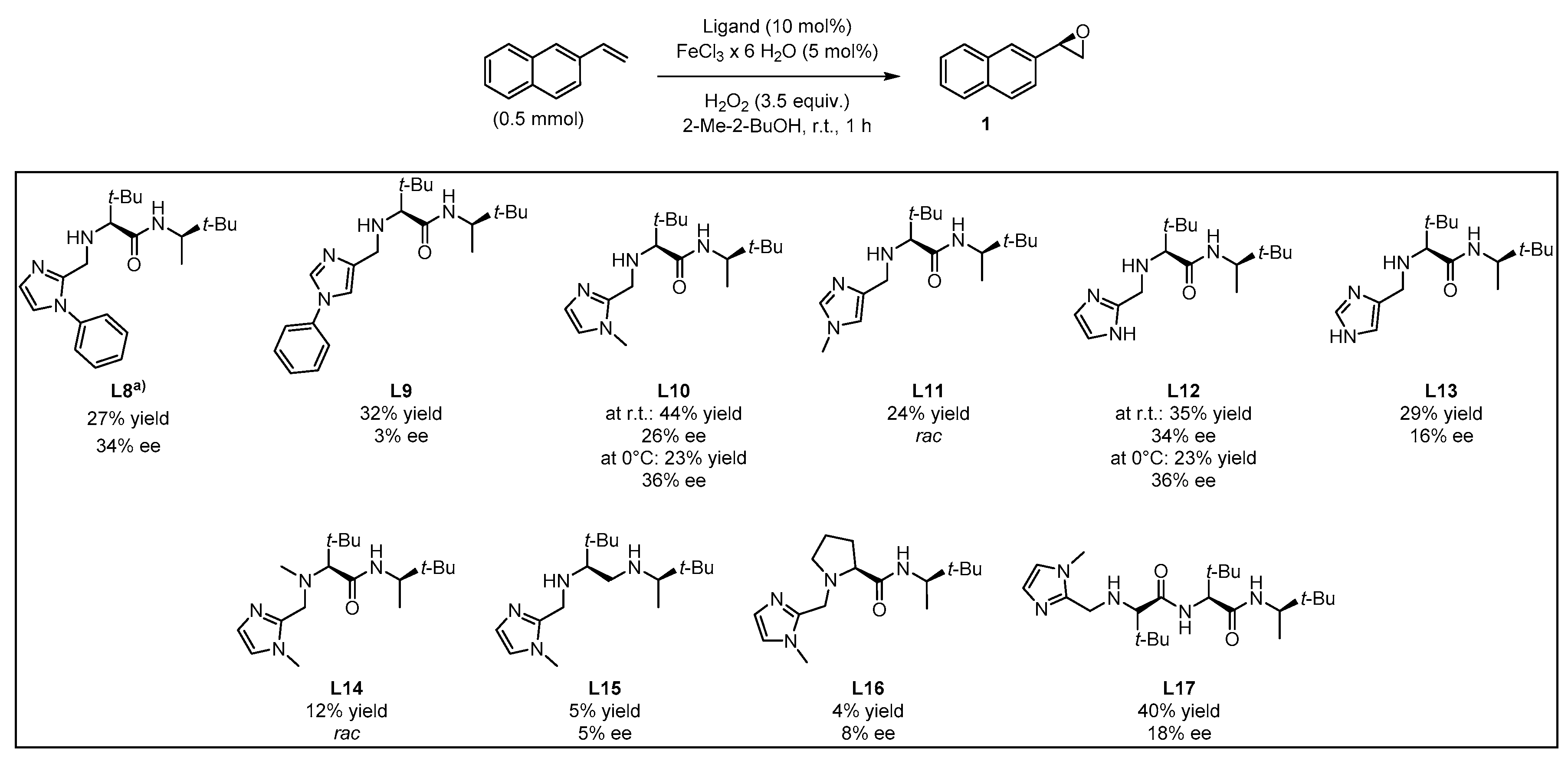
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Catalytic Procedure
3.2.1. Catalysis without Additive
3.2.2. Catalysis with (S)-(+)-Mandelic Acid
3.2.3. Substrate and Racemic Product References
3.3. General Procedure of Olefin Epoxidation with m-CPBA
3.3.1. Synthesis of Ligands
3.3.2. Synthesis of l-Proline or l-tert-Leucine Based Amines
3.3.3. Synthesis of Imidazole Based Aldehyde
3.3.4. Synthesis of Imidazole Based Peptide Like Ligand
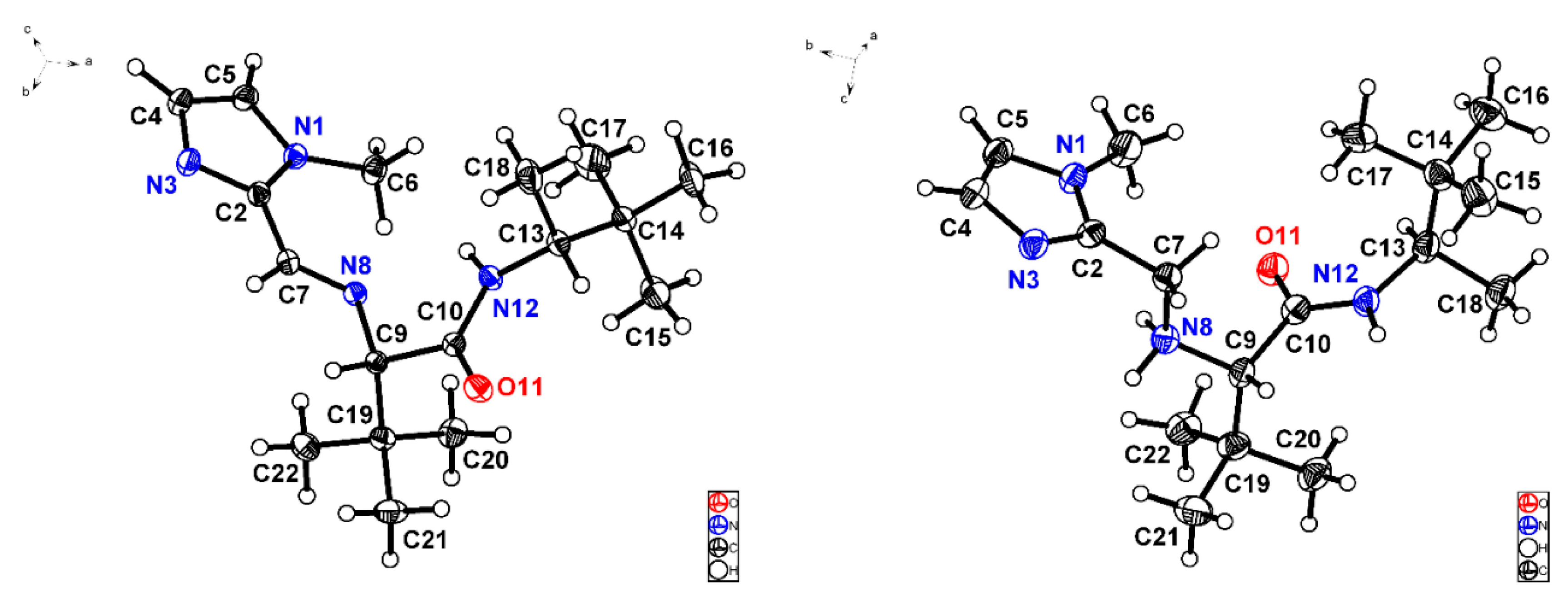
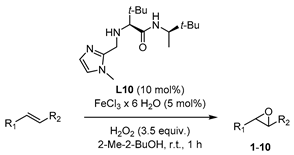
3.4. X-ray Crystallographic Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yudin, A.K. (Ed.) Aziridines and Epoxides in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Beauvais, B.; Sarfati, C.; Challier, S.; Derouin, F. In vitro model to assess effect of antimicrobial agents on Encephalitozoon cuniculi. Antimicrob. Agents Chemother. 1994, 38, 2440–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, J.L.; Kevin, N.J.; Kirk, B.A.; Chapman, K.T.; Schleif, W.A.; Olsen, D.B.; Stahlhut, M.; Rutkowski, C.A.; Kuo, L.C.; Jin, L.; et al. Synthesis and activity of novel HIV protease inhibitors with improved potency against multiple PI-resistant viral strains. Bioorg. Med. Chem. 2002, 12, 2423–2426. [Google Scholar] [CrossRef]
- Nachbauer, L.; Brückner, R. Synthesis of a Cn–Cn+6 Building Block Common to Important Polyol, Polyene Antibiotics from a Divinylcarbinol by a Desymmetrizing Sharpless Epoxidation. Eur. J. Org. Chem. 2013, 2013, 6545–6562. [Google Scholar] [CrossRef]
- Nakazawa, M.; Uehara, T.; Nomura, Y. Koningic Acid (a Potent Glyceraldehyde-3-Phosphate Dehydrogenase Inhibitor)-Induced Fragmentation and Condensation of DNA in NG108-15 Cells. J. Neurochem. 1997, 68, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
- Sunohara, K.; Mitsuhashi, S.; Shigetomi, K.; Ubukata, M. Discovery of N-(2,3,5-triazoyl)mycophenolic amide and mycophenolic epoxyketone as novel inhibitors of human IMPDH. Bioorg. Med. Chem. Lett. 2013, 23, 5140–5144. [Google Scholar] [CrossRef] [PubMed]
- Piontek, A.; Bisz, E.; Szostak, M. Iron-catalyzed cross-couplings in the synthesis of pharmaceuticals: In pursuit of sustainability. Angew. Chem. Int. Ed. 2018, 57, 11116–11128. [Google Scholar] [CrossRef] [PubMed]
- Enthaler, S.; Junge, K.; Beller, M. Sustainable Metal Catalysis with Iron: From Rust to a Rising Star? Angew. Chem. Int. Ed. 2008, 47, 3317–3321. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T.; Myers, R.S. Catalytic asymmetric epoxidations with chiral iron porphyrins. J. Am. Chem. Soc. 1983, 105, 5791–5796. [Google Scholar] [CrossRef]
- Groves, J.T.; Viski, P. Asymmetric hydroxylation by a chiral iron porphyrin. J. Am. Chem. Soc. 1989, 111, 8537–8538. [Google Scholar] [CrossRef]
- Collman, J.P.; Wang, Z.; Straumanis, A.; Quelquejeu, M.; Rose, E. An Efficient Catalyst for Asymmetric Epoxidation of Terminal Olefins. J. Am. Chem. Soc. 1999, 121, 460–461. [Google Scholar] [CrossRef]
- Darwish, M.; Wills, M. Asymmetric catalysis using iron complexes—‘Ruthenium Lite’? Catal. Sci. Technol. 2012, 2, 243–255. [Google Scholar] [CrossRef]
- Schomburg, D.; Schomburg, I.; Chang, A. (Eds.) Class 1 Oxidoreductases X: EC 1.9–1.13; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Karlsson, A.; Parales, J.V.; Parales, R.E.; Gibson, D.T.; Eklund, H.; Ramaswamy, S. Crystal Structure of Naphthalene Dioxygenase: Side-on Binding of Dioxygen to Iron. Science 2003, 299, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Bruijnincx, P.C.; Buurmans, I.L.; Gosiewska, S.; Moelands, M.A.; Lutz, M.; Spek, A.L.; van Koten, G.; Klein Gebbink, R.J. Iron(II) Complexes with Bio-Inspired N,N,O Ligands as Oxidation Catalysts: Olefin Epoxidation and cis-Dihydroxylation. Chem. Eur. J. 2008, 14, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Join, B.; Amali, A.J.; Junge, K.; Ribas, X.; Costas, M.; Beller, M. A Biomimetic Iron Catalyst for the Epoxidation of Olefins with Molecular Oxygen at Room Temperature. Angew. Chem. Int. Ed. 2011, 50, 1425–1429. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Enthaler, S.; Bitterlich, B.; Schulz, T.; Spannenberg, A.; Tse, M.K.; Junge, K.; Beller, M. Design of and Mechanistic Studies on a Biomimetic Iron–Imidazole Catalyst System for Epoxidation of Olefins with Hydrogen Peroxide. Chem. Eur. J. 2009, 15, 5471–5481. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.B.; Jacobsen, E.N. Discovery of Novel Catalysts for Alkene Epoxidation from Metal-Binding Combinatorial Libraries. Angew. Chem. Int. Ed. 1999, 38, 937–941. [Google Scholar] [CrossRef]
- Hasan, K.; Brown, N.; Kozak, C.M. Iron-catalyzed epoxidation of olefins using hydrogen peroxide. Green Chem. 2011, 13, 1230–1237. [Google Scholar] [CrossRef]
- Jiao, M.; Matsunaga, H.; Ishizuka, T. A simple, Iron-Catalyzed, Pyridine-Assisted Hydrogen Peroxide Epoxidation System. Chem. Pharm. Bull. 2011, 59, 799–801. [Google Scholar] [CrossRef]
- Gelalcha, F.G.; Bitterlich, B.; Anilkumar, G.; Tse, M.K.; Beller, M. Iron-Catalyzed Asymmetric Epoxidation of Aromatic Alkenes Using Hydrogen Peroxide. Angew. Chem. Int. Ed. 2007, 46, 7293–7296. [Google Scholar] [CrossRef]
- Gelalcha, F.G.; Anilkumar, G.; Tse, M.K.; Bruckner, A.; Beller, M. Biomimetic Iron-Catalyzed Asymmetric Epoxidation of Aromatic Alkenes by Using Hydrogen Peroxide. Chem. Eur. J. 2008, 14, 7687–7698. [Google Scholar] [CrossRef]
- Bitterlich, B.; Schröder, K.; Tse, M.K.; Beller, M. An Improved Iron-Catalyzed Epoxidation of Aromatic and Aliphatic Olefins with Hydrogen Peroxide as Oxidant. Eur. J. Org. Chem. 2008, 4867–4870. [Google Scholar] [CrossRef]
- Schröder, K.; Tong, X.; Bitterlich, B.; Tse, M.K.; Gelalcha, F.G.; Brückner, A.; Beller, M. Novel biomimetic iron-catalysts for environmentally benign epoxidations of olefins. Tetrahedron Lett. 2007, 48, 6339–6342. [Google Scholar] [CrossRef]
- Schröder, K.; Junge, K.; Spannenberg, A.; Beller, M. Design of a bio-inspired imidazole-based iron catalyst for epoxidation of olefins: Mechanistic insights. Catal. Today 2010, 157, 364–370. [Google Scholar] [CrossRef]
- Costas, M.; Que, J.L. Ligand Topology Tuning of Iron-Catalyzed Hydrocarbon Oxidations. Angew. Chem. Int. Ed. 2002, 41, 2179–2181. [Google Scholar] [CrossRef]
- Mas-Balleste, R.; Costas, M.; van den Berg, T.; Que, L., Jr. Ligand Topology Effects on Olefin Oxidations by Bio-Inspired [FeII(N2Py2)] Catalysts. Chem. Eur. J. 2006, 12, 7489–7500. [Google Scholar] [CrossRef] [PubMed]
- Cusso, O.; Garcia-Bosch, I.; Ribas, X.; Lloret-Fillol, J.; Costas, M. Asymmetric Epoxidation with H2O2 by Manipulating the Electronic Properties of Non-heme Iron Catalysts. J. Am. Chem. Soc. 2013, 135, 14871–14878. [Google Scholar] [CrossRef] [PubMed]
- Cusso, O.; Ribas, X.; Lloret-Fillol, J.; Costas, M. Synergistic Interplay of a Non-Heme Iron Catalyst and Amino Acid Coligands in H2O2 Activation for Asymmetric Epoxidation of α-Alkyl-Substituted Styrenes. Angew. Chem. Int. Ed. 2015, 54, 2729–2733. [Google Scholar] [CrossRef]
- Cusso, O.; Giuliano, M.W.; Ribas, X.; Miller, S.J.; Costas, M. A bottom up approach towards artificial oxygenases by combining iron coordination complexes and peptides. Chem. Sci. 2017, 8, 3660–3667. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Miao, C.-X.; Wang, S.; Hu, X.; Xia, C.; Kühn, F.E.; Sun, W. Chiral Bioinspired Non-Heme Iron Complexes for Enantioselective Epoxidation of α,β-Unsaturated Ketones. Adv. Synth. Catal. 2011, 353, 3014–3022. [Google Scholar] [CrossRef]
- Wang, B.; Wang, S.; Xia, C.; Sun, W. Highly Enantioselective Epoxidation of Multisubstituted Enones Catalyzed by Non-Heme Iron Catalysts. Chem. Eur. J. 2012, 18, 7332–7335. [Google Scholar] [CrossRef]
- Lyakin, O.Y.; Ottenbacher, R.V.; Bryliakov, K.P.; Talsi, E.P. Asymmetric Epoxidations with H2O2 on Fe and Mn Aminopyridine Catalysts: Probing the Nature of Active Species by Combined Electron Paramagnetic Resonance and Enantioselectivity Study. ACS Catal. 2012, 2, 1196–1202. [Google Scholar] [CrossRef]
- Lancaster, M. Green Chemistry: An Introductory Text, 3rd ed.; RSC Internet Services: London, UK, 2016. [Google Scholar]
- Lane, B.S.; Burgess, K. Metal-Catalyzed Epoxidations of Alkenes with Hydrogen Peroxide. Chem. Rev. 2003, 103, 2457–2473. [Google Scholar] [CrossRef] [PubMed]
- Held, F.E.; Wei, S.; Eder, K.; Tsogoeva, S.B. One-pot route to β-adrenergic blockers via enantioselective organocatalysed epoxidation of terminal alkenes as a key step. RSC Adv. 2014, 4, 32796–32801. [Google Scholar] [CrossRef]
- Fingerhut, A.; Serdyuk, O.V.; Tsogoeva, S.B. Non-heme iron catalysts for epoxidation and aziridination reactions of challenging terminal alkenes: Towards sustainability. Green Chem. 2015, 17, 2042–2058. [Google Scholar] [CrossRef]
- Yeung, H.L.; Sham, K.C.; Tsang, C.S.; Lau, T.C.; Kwong, H.L. A chiral iron-sexipyridine complex as a catalyst for alkene epoxidation with hydrogen peroxide. Chem. Commun. 2008, 3801–3803. [Google Scholar] [CrossRef] [PubMed]
- Hieke, M.; Greiner, C.; Thieme, T.M.; Schubert-Zsilavecz, M.; Werz, O.; Zettl, H. A novel class of dual mPGES-1/5-LO inhibitors based on the α-naphthyl pirinixic acid scaffold. Bioorg. Med. Chem. Lett. 2011, 21, 1329–1333. [Google Scholar] [CrossRef]
- Shukla, M.R.; Chaudhari, V.D.; Sayyed, M.B.; Phadtare, R.D.; Walke, N.B.; Kulkarni, S.A.; Palle, V.P.; Kamboj, R.K. Substituted Morpholines as Modulators for the Calcium Sensing Receptor. Patent Number WO2012120476 A1, 13 Septemper 2012. [Google Scholar]
- Bitterlich, B.; Anilkumar, G.; Gelalcha, F.G.; Spilker, B.; Grotevendt, A.; Jackstell, R.; Tse, M.K.; Beller, M. Development of a General and Efficient Iron-Catalyzed Epoxidation with Hydrogen Peroxide as Oxidant. Chem. Asian J. 2007, 2, 521–529. [Google Scholar] [CrossRef]
- Stingl, K.A.; Weiß, K.M.; Tsogoeva, S.B. Asymmetric vanadium- and iron-catalyzed oxidations: New mild (R)-modafinil synthesis and formation of epoxides using aqueous H2O2 as a terminal oxidant. Tetrahedron 2012, 68, 8493–8501. [Google Scholar] [CrossRef]
- Muñiz, K. Asymmetrische Katalyse mit Metall-Komplexen. Bild oder Spiegelbild? Chem. Unserer Zeit 2006, 40, 112–124. [Google Scholar] [CrossRef]
- White, M.C.; Doyle, A.G.; Jacobsen, E.N. A Synthetically Useful, Self-Assembling MMO Mimic System for Catalytic Alkene Epoxidation with Aqueous H2O2. J. Am. Chem. Soc. 2001, 123, 7194–7195. [Google Scholar] [CrossRef]
- Dubois, G.; Murphy, A.; Stack, T.D. Simple Iron Catalyst for Terminal Alkene Epoxidation. Org. Lett. 2003, 5, 2469–2472. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Que, L., Jr. In situ Formation of Peracetic Acid in Iron-Catalyzed Epoxidations by Hydrogen Peroxide in the Presence of Acetic Acid. Adv. Synth. Catal. 2004, 346, 190–194. [Google Scholar] [CrossRef]
- Yeori, A.; Gendler, S.; Groysman, S.; Goldberg, I.; Kol, M. Salalen: A hybrid Salan/Salen tetradentate [ONNO]-type ligand and its coordination behavior with group IV metals. Inorg. Chem. Commun. 2004, 7, 280–282. [Google Scholar] [CrossRef]
- Matsumoto, K.; Saito, B.; Katsuki, T. Asymmetric catalysis of metal complexes with non-planar ONNO ligands: Salen, salalen and salan. Chem. Commun. 2007, 3619–3627. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Mitra, A.W.; Hoveyda, A.H.; Snapper, M.L. Kinetic Resolution of 1,2-Diols through Highly Site- and Enantioselective Catalytic Silylation. Angew. Chem. Int. Ed. 2007, 46, 8471–8474. [Google Scholar] [CrossRef]
- Rodrigo, J.M.; Zhao, Y.; Hoveyda, A.H.; Snapper, M.L. Regiodivergent Reactions through Catalytic Enantioselective Silylation of Chiral Diols. Synthesis of Sapinofuranone, A. Org. Lett. 2011, 13, 3778–3781. [Google Scholar] [CrossRef] [PubMed]
- Manville, N.; Alite, H.; Haeffner, F.; Hoveyda, A.H.; Snapper, M.L. Enantioselective silyl protection of alcohols promoted by a combination of chiral and achiral Lewis basic catalysts. Nat. Chem. 2013, 5, 768–774. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Hoveyda, A.H.; Snapper, M.L. Catalytic Enantioselective Silylation of Acyclic and Cyclic Triols: Application to Total Syntheses of Cleroindicins D., F., and C. Angew. Chem. Int. Ed. 2009, 48, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Loebach, J.L.; Wilson, S.R.; Jacobsen, E.N. Enantioselective epoxidation of unfunctionalized olefins catalyzed by salen manganese complexes. J. Am. Chem. Soc. 1990, 112, 2801–2803. [Google Scholar] [CrossRef]
- Hong, L.; Sun, W.; Yang, D.; Li, G.; Wang, R. Additive Effects on Asymmetric Catalysis. Chem. Rev. 2016, 116, 4006–4123. [Google Scholar] [CrossRef]
- Avila-Ortiz, C.G.; López-Ortiz, M.; Vega-Peñaloza, A.; Regla, I.; Juaristi, E. Use of (R)-Mandelic Acid as Chiral Co-Catalyst in the Michael Addition Reaction Organocatalyzed by (1S,4S)-2-Tosyl-2,5-diazabicyclo [2.2.1]heptane under Solvent-Free Conditions. Asymmetric Catal. 2015, 2, 37–44. [Google Scholar] [CrossRef]
- Dicks, A.P.; Hent, A. Atom Economy and Reaction Mass Efficiency. In Green Chemistry Metrics: A Guide to Determining and Evaluating Process Grenness; Springer: Heidelberg, Germany, 2015; pp. 17–44. [Google Scholar]
- Zhang, C.P.; Wang, Z.L.; Chen, Q.Y.; Zhang, C.T.; Gu, Y.C.; Xiao, J.C. Generation of the CF3 radical from trifluoromethylsulfonium triflate and its trifluoromethylation of styrenes. Chem. Commun. 2011, 47, 6632–6634. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.Z.; Li, Q.Q.; Wang, M.M.; Ning, X.S.; Kang, Y.B. (E)-Specific direct Julia-olefination of aryl alcohols without extra reducing agents promoted by bases. Chem. Commun. 2015, 51, 7729–7732. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Inukai, S.; Asai, N.; Oyamada, M.; Ikegawa, S.; Sugiyama, Y.; Hamamoto, H.; Shioiri, T.; Matsugi, M. A comparative study of the asymmetric epoxidation of aromatic olefins using the first generation manganese salen epoxidation catalysts and their light fluorous variants: An interesting discovery on the use of benzotrifluoride as a cosolvent. Tetrahedron Asymmetry 2014, 25, 1209–1214. [Google Scholar] [CrossRef]
- Yadav, V.K.; Kapoor, K.K. 1,8-diazabicyclo [5.4.0] undec-7-ene: A remarkable base in the epoxidation of α,β-unsaturated-δ-lactones and other enones with anhydrous t-BuOOH. Tetrahedron 1995, 51, 8573–8584. [Google Scholar] [CrossRef]
- Zandbergen, P.; van den Niewendijk, A.M.C.H.; Brussee, J.; van der Gen, A.; Kruse, C.G. A one-pot reduction-transimition-reduction synthesis of N-substituted β-ethanolamines from cyanohydrins. Tetrahedron 1992, 48, 3977–3982. [Google Scholar] [CrossRef]
- Robinson, M.W.; Davies, A.M.; Buckle, R.; Mabbett, I.; Taylor, S.H.; Graham, A.E. Epoxide ring-opening and Meinwald rearrangement reactions of epoxides catalyzed by mesoporous aluminosilicates. Org. Biomol. Chem. 2009, 7, 2559–2564. [Google Scholar] [CrossRef] [Green Version]
- Pedragosa-Moreau, S.; Morisseau, C.; Zylber, J.; Archelas, A.; Baratti, J.; Furstoss, R. Microbiological Transformations. 33. Fungal Epoxide Hydrolases Applied to the Synthesis of Enantiopure Para-Substituted Styrene Oxides. A Mechanistic Approach. J. Org. Chem. 1996, 61, 7402–7407. [Google Scholar] [CrossRef]
- Grigg, R.D.; Rigoli, J.W.; Pearce, S.D.; Schomaker, J.M. Synthesis of Propargylic and Allenic Carbamates via the C–H Amination of Alkynes. Org. Lett. 2012, 14, 280–283. [Google Scholar] [CrossRef]
- Fristrup, P.; Dideriksen, B.B.; Tanner, D.; Norrby, P.O. Probing Competitive Enantioselective Approach Vectors Operating in the Jacobsen−Katsuki Epoxidation: A Kinetic Study of Methyl-Substituted Styrenes. J. Am. Chem. Soc. 2005, 127, 13672–13679. [Google Scholar] [CrossRef]
- Alvaro, G.; Décor, A.; Fontana, S.; Hamprecht, D.; Large, C.; Marasco, A. Imidazolidinedione Derivatives. Patent Number WO2011069951 A1, 16 June 2011. [Google Scholar]
- Huy, P.; Neudorfl, J.M.; Schmalz, H.G. A Practical Synthesis of Trans-3-Substituted Proline Derivatives through 1,4-Addition. Org. Lett. 2011, 13, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Gebert, U.; Kerékjártó, B.V. Modellreaktionen für die enzymatische Katalyse, IV1) Untersuchungen zur Struktur-Wirkung-Beziehung neuer Transaminatoren mit Imidazol-, Thiazol- und Benzimidazolgerüst. Justus Liebigs Ann. Chem. 1974, 1974, 644–654. [Google Scholar] [CrossRef]
- Kodera, M.; Terasako, N.; Kita, T.; Tachi, Y.; Kano, K.; Yamazaki, M.; Koikawa, M.; Tokii, T. Synthesis, Crystal Structures, and Catalytic Activity of Dicopper(II) Complexes with Dinucleating Tetraimidazole Ligands [Cu2(RO)(mbipl)](ClO4)2 (R = Me, Et, 2-Pr; Hmbipl = 1,5-Bis[bis[(1-methyl-4-imidazolyl)methyl]amino]-3-pentanol) and [Cu2(MeO)(pbipl)](ClO4)2 (Hpbipl = 1,5-Bis[bis[(1-isopropyl-4-imidazolyl)methyl]amino]-3-pentanol). Inorg. Chem. 1997, 36, 3861–3868. [Google Scholar]
- Zhao, Y.; Rodrigo, J.; Hoveyda, A.H.; Snapper, M.L. Enantioselective silyl protection of alcohols catalysed by an amino-acid-based small molecule. Nature 2006, 443, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the ligands (S,R)-I, L10 and L17 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Ligand L10 (mol%) | FeCl3∙ 6H2O (mol%) | (S)-(+)-mandelic acid (mol%) | Yield 1 (%) | ee 2 (%) |
| 1 | 10 | 5 | 5 | 27 | 42 |
| 2 | 5 | 5 | 15 | 20 | 37 |
| 3 | 20 | 10 | 10 | 43 | 38 |
 | |||
|---|---|---|---|
| Entry | Product | Yield 1,2 | ee 3 |
| 1 | 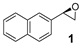 | 44 | 26 (R) |
| 2 | 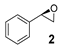 | 30 | 27 (R) |
| 3 | 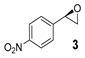 | 14 | 29 (S) |
| 4 | 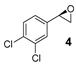 | 25 | 25 (R) 4 |
| 5 |  | 5 4 | 14 (R) 4 |
| 6 | 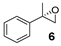 | 22 | 16 (S) |
| 7 |  | <5 5 | n.d. 6 |
| 8 |  | 32 | 50 |
| 9 | 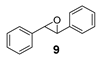 | 27 | 16 (R) |
| 10 | 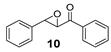 | <5 | 27 (2R,3S) |
| 11 7 |  | 7 | 51 (2R,3S) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fingerhut, A.; Vargas-Caporali, J.; Leyva-Ramírez, M.A.; Juaristi, E.; Tsogoeva, S.B. Biomimetic Non-Heme Iron-Catalyzed Epoxidation of Challenging Terminal Alkenes Using Aqueous H2O2 as an Environmentally Friendly Oxidant. Molecules 2019, 24, 3182. https://doi.org/10.3390/molecules24173182
Fingerhut A, Vargas-Caporali J, Leyva-Ramírez MA, Juaristi E, Tsogoeva SB. Biomimetic Non-Heme Iron-Catalyzed Epoxidation of Challenging Terminal Alkenes Using Aqueous H2O2 as an Environmentally Friendly Oxidant. Molecules. 2019; 24(17):3182. https://doi.org/10.3390/molecules24173182
Chicago/Turabian StyleFingerhut, Anja, Jorge Vargas-Caporali, Marco Antonio Leyva-Ramírez, Eusebio Juaristi, and Svetlana B. Tsogoeva. 2019. "Biomimetic Non-Heme Iron-Catalyzed Epoxidation of Challenging Terminal Alkenes Using Aqueous H2O2 as an Environmentally Friendly Oxidant" Molecules 24, no. 17: 3182. https://doi.org/10.3390/molecules24173182